")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Investigating isoquinoline derivatives for inhibition of inhibitor of apoptosis proteins for ovarian cancer treatment
Authors Chen C, Wu J, Zhu P, Xu C, Yao L
Received 21 March 2017
Accepted for publication 30 June 2017
Published 11 September 2017 Volume 2017:11 Pages 2697—2707
DOI https://doi.org/10.2147/DDDT.S137608
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Chen Chen,1,* Jie Wu,2,* Pengfei Zhu,3 Congjian Xu,1 Liangqing Yao1
1Obstetrics and Gynecology Hospital and Shanghai Key Laboratory of Female Reproductive Endocrine Related Diseases, 2Department of Chemistry, Fudan University, Shanghai, 3Department of Obstetrics and Gynecology, Shangyu City Hospital, Shangyu, Zhejiang Province, People’s Republic of China
*These authors contributed equally to this work
Objective: To discover novel isoquinoline derivatives for inhibition of inhibitor of apoptosis proteins (IAP) for the treatment of ovarian cancer.
Methods: We first synthesized 533 isoquinoline derivatives, and screened them using CCK-8 to measure their antiproliferative activity. These compounds were further tested by Hoechst staining and flow cytometric analysis to assess proapoptotic activity. The in vivo antitumor efficacy and safety of the screened compounds were evaluated on the xenograft mouse model. Ki-67 staining and TUNEL assay were used to evaluate proliferation and apoptosis in the resected tumors, respectively. Western blot and polymerase chain reaction (PCR) were conducted to evaluate the levels of proliferating cell nuclear antigen (PCNA), caspase-3, PARP, and IAP in resected tumors.
Results: Compound B01002 and C26001 displayed antiproliferative and proapoptotic activity on SKOV3 ovarian cancer with an IC50 of 7.65 and 11.68 µg/mL, respectively. Both compounds inhibited tumor growth in a xenografted mouse model with good safety profiles, and tumor growth inhibition (TGI) of B01002 and C26001 was 99.53% and 84.23%, respectively. Resected tumors showed that both compounds inhibited tumor cell proliferation and induced apoptosis in vivo. Caspase-3 and PARP were activated, whereas IAP proteins were downregulated at the protein level.
Conclusion: Compound B01002 and C26001 could inhibit ovarian tumor growth and promote tumor apoptosis, partly by downregulating the IAPs, and, thus, might be promising candidates for treatment of ovarian cancer.
Keywords: isoquinoline derivatives, target-oriented syntheses, IAP inhibitor, chemotherapy, ovarian cancer
Introduction
Malignant tumors could fend off anticancer therapies in several ways. One strategy involves the evasion of apoptosis by upregulating the inhibitors of apoptosis proteins (IAP).1 IAP comprise a family of functionally and structurally related proteins that share the presence of one or more baculoviral IAP repeat (BIR) domains.2 The property of apoptosis inhibition has been described for at least four IAP family members – cIAP1, cIAP2, XIAP, and survivin.3–5 IAP subverts apoptosis through binding and inactivating caspases via one or more BIR domains,6 which are approximately 80-amino acid zinc-binding domains and are essential for the antiapoptotic functions of the IAP. For example, the second BIR domain (BIR2) of XIAP is essential for potent inhibition of caspase-3.
Elevated IAP have been identified in numerous neoplasms, and have been proved to be correlated with poor prognosis. In several recent reports, it has been demonstrated that IAP play an active role in promoting tumor maintenance through the inhibition of cellular death, thus enhancing tumor’s resistance to radiotherapy and chemotherapy.7,8 Furthermore, IAP participate in signaling pathways associated with tumor proliferation, invasion, metastasis, and angiogenesis.9–11 Therefore, agents targeting IAP might be an effective approach for the treatment of cancer.
Different IAP-targeting therapies have been evaluated in preclinical and clinical studies in a variety of tumors recently.12 Among those therapeutic strategies, one appealing approach is based on the occupation of the IAP–BIR peptide-binding pocket by certain reagents and, thus, interference with the IAP–caspases interactions.13 An endogenous inhibitor of the BIR peptide, the four amino-acid N-terminus of the second mitochondrial activator of caspases (Smac; AVPI), can antagonize IAPs, with high binding affinity against the BIR2/3 domains of XIAP, the BIR3 domains of cIAP1/2, and the single BIR domain of survivin. This AVPI tetrapeptide sequence has been regarded as the lead structure for designing small-molecule BIR inhibitors for antagonizing IAPs by many researchers. Moreover, efforts to design new compounds capable of inhibiting BIR domains have focused on high-throughput-screening bioactive Smac mimetics that are based on those AVPI tetrapeptides, and subsequently evaluate their anti-cancer activity and underlying mechanism. Preclinical studies have demonstrated the capacity of those small-molecule BIR inhibitors to inhibit tumor growth and to sensitize cells to conventional therapeutic agents, such as radiation or cisplatin, in multiple solid tumors. To date, at least six BIR inhibitors have entered human clinical trials. Preliminary data indicate good tolerance and predicted biomarker modulation, such as cIAP1 downregulation and increased levels of processed caspase-3. However, it appears that those reported BIR inhibitors either have low potency or poor metabolic stability. For instance, results from phase I clinical trials indicated that AT406 (Ascenta Therapeutics) did not show equivalent efficacy as previously tested in mice, and HGS1029 (Aegera Therapeutics/Human Genome Sciences) required intravenous administration owing to large molecular size.12,14 Therefore, efforts need to be made to develop more efficient BIR inhibitors with improved potency and better pharmacokinetics.
Molecules with an isoquinoline skeleton have been reported to have the capacity to antagonize IAP with a high binding affinity to the BIR2 domain, highlighting their utility as a potential drug lead.11 Compounds with an isoquinoline skeleton are reported to be capable of antagonizing XIAP. In this study, we firstly designed and synthesized series of small compounds, with an isoquinoline skeleton and different chemical groups, and then evaluated their antitumor activities on ovarian cancer cells in vivo. Two compounds screened out were further evaluated on the nude mouse xenograft model, and showed good antitumor efficacy and in vivo safety. They inhibited tumor cell proliferation mainly in an apoptotic way, as caspase-3 and PARP were activated, whereas IAP were downregulated at the protein level.
Materials and methods
Synthesis
We synthesized 533 molecules with an isoquinoline skeleton as described previously.15,16
Unless otherwise stated, all commercial reagents were used as received. All solvents were dried and distilled according to standard procedures. Flash column chromatography was performed using silica gel (60-Å pore size, 32–63 μm, standard grade). Analytical thin-layer chromatography was undertaken using glass plates pre-coated with 0.25 mm 230–400 mesh silica gel impregnated with a fluorescent indicator (254 nm). Thin-layer chromatography plates were visualized by exposure to ultraviolet light. Organic solutions were concentrated on rotary evaporators at ~20 Torr at 25°C–35°C. Nuclear magnetic resonance (NMR) spectra are recorded in parts per million from internal tetramethylsilane on the δ scale.
Cell culture and cell growth inhibition assay
The human ovarian cancer SKOV3 cells (purchased from Sigma-Aldrich Co., St Louis, MO, USA) were seeded in 96-well flat-bottomed cell culture plates at a density of 1×103 cells/well and grown overnight.17 Then, 5 mg compound stock in 100% dimethylsulfoxide (DMSO) was diluted in medium at concentrations of 0, 1, 2.5, 5, 7.5, and 10 μg/mL separately, filtered, serially added into a 96-well plate, and continuously incubated for 24 h. Viable cells were quantified by a CCK-8 assay in which the absorbance of the samples was measured at 450 nm.18 IC50 was calculated according to the cell growth curves. Experiments were repeated at least three times.
Apoptosis assay
Apoptotic nuclear changes were assessed by Hoechst staining, followed by fluorescent microscopic examination.19 SKOV3 cells were seeded in dish slides. After incubation with testing agents (5 μg/mL compounds and 1% DMSO; 5 μg/mL cisplatin [DDP] as positive control; phosphate buffered-saline [PBS] as negative control) for 24 h, cells were stained with 20 mg/mL Hoechst at 37°C for 20 min, washed three times with PBS, and fixed with 4% formaldehyde for 15 min at room temperature. Morphological changes of nuclei were observed under a fluorescent microscope. The percentage of apoptotic cells was calculated for each field and averaged for the treatment group. The cytotoxicity of cisplatin was also evaluated under the same experimental conditions for comparison. In each slide, at least 120 cells were assessed in three contiguous fields.
SKOV-3 cells were treated with different concentrations of each compound (5 μg/mL of compounds and 1% DMSO; 10 μg/mL of compounds and 1% DMSO) or PBS for 24 h. Cells were washed and resuspended in binding buffer at a concentration of 1×106 cells/mL; 100 μL of the solution (1×105 cells) was transferred to a 5 mL culture tube. Then, 5 μL of recombinant human annexin V-FITC conjugate (ANNEXINV01, Invitrogen) and 5 μL of propidium iodide solution (P4864, Sigma-Aldrich) were added, and cells were gently vortexed and incubated for 15 min at room temperature (25°C) in the dark. Thereafter, 400 μL binding buffer was added to each tube. Finally, the harvested cells were analyzed by flow cytometry.
Tumor xenograft study
This study was approved by the Animal Ethics Committee of Fudan University (approval number: 20140823), and all animals were maintained and used in accordance with the guidelines of the Institutional Animal Care and Use Committee of Fudan University. SKOV3 cells were suspended in PBS, and then implanted subcutaneously into the right oxter of 40 female nude mice (age 6–8 weeks).20 Tumor volumes were evaluated using the formula V = 0.5 × a × b2, where a and b are the largest and smallest perpendicular tumor diameters, respectively. Six mice with the appropriate mean tumor volume were assigned randomly to each of the four groups. The mean tumor volume for all four groups was 189±3 mm3 at the initiation of treatment (day 0). Mice (six per group) were treated with compound B01002, C26001, and DDP by intraperitoneal injection at a dose of 4 mg/kg every other day, with PBS as the control. Tumor volumes and mice body weights were measured every other day. Percent tumor growth inhibition (TGI) was calculated using the formula % TGI = 100 * (1 − tumor volumedose/tumor volumePBS). At the end of the experiment, the mice were sacrificed and the tumor tissues were harvested and weighed; the liver, spleen, small bowel, kidneys, and lungs were excised; and blood samples were collected.
Hematological and biochemical study
At the end of the experimental period, blood samples were collected from all animals from the retro-orbital venous plexus for safety evaluation. The effect of these compounds on blood parameters in mice was determined in the collected blood samples. Serum aspartate aminotransferase (AST), alanine transaminase (ALT), alkaline phosphatase (ALP), blood urea nitrogen (BUN), and creatinine (Cr) levels were determined using enzyme-linked immunosorbent assay kits (Shanghai Institute of Biological Products Co.).
TUNEL staining and immunofluorescence analysis
TUNEL assay was conducted using the In Situ Cell Death Detection Kit (Roche) according to the kit protocol. Briefly, the dissected tumors were fixed in 4% paraformaldehyde. Then, TUNEL staining was conducted in fixed tumor sections (5 μM) that were counterstained with 4,6-diamidino-2-phenylindole (DAPI). The numbers of TUNEL-positive cells and the total cells in tissue sections were counted.21
Immunohistochemical assessment of Ki-67
Moreover, 5 μm-thick sections were cut from the tissue blocks, and then dewaxed and rehydrated. MIB1 antibody (Abcam) at a dilution of 1:100 was used for Ki-67 immunostaining.22 Harris hematoxylin was used to counterstain Ki-67–immunostained sections. All sections were assessed blindly and independently by two observers. Preceding this, the percentage of positively staining cells was recorded and used as a quality control measure in our statistical analysis.
Western blot analysis
The resected tumors were washed with cold PBS, and prepared by homogenization in RIPA buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecylsulfate in PBS) supplemented with protease inhibitor cocktail. Then, 30 mg protein was resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with the respective antibodies.23
Gene expression analysis
We isolated RNA with the RNeasy Lipid Tissue kit (QIAGEN) and DNase digestion, synthesized cDNA with the High-capacity cDNA Reverse Transcription kit, and conducted quantitative real-time PCR (RT-PCR) with goTaq qPCR Master Mix in a Bio-Rad CDX96 Real-time PCR system. We calculated relative gene expression levels by the ΔΔCt method using cyclophilin A as the internal control.24
Statistical analysis
All data were presented as mean ± standard error. For relative gene expression, the mean value of the control group was defined as 100%. Two-tailed unpaired Student’s t-test and ANOVA were used for statistical evaluation of the data.24 The Sigma stat statistical analysis program was used for data analysis. P<0.05 was considered significant.
Results
Isoquinoline derivatives were synthesized to inhibit IAP
The synthetic routes for isoquinolines 1-2 are shown in Scheme 1. Isoquinoline 1 (B01002) was prepared by a silver triflate-catalyzed reaction of 2-alkynylbenzaldoxime with amine. Isoquinoline 2 (C26001) was synthesized by a silver triflate-catalyzed reaction of 2-alkynylbenzaldehyde with 2-isocyanoacetate.
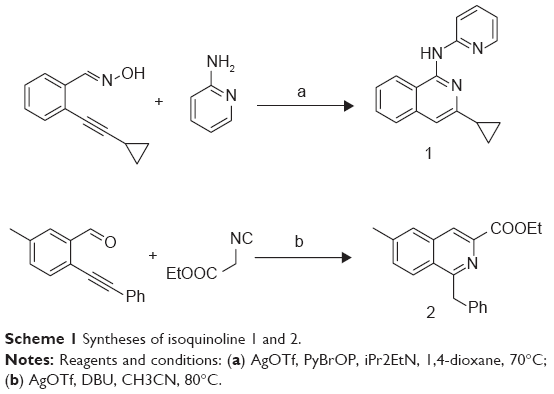 | Scheme 1 Syntheses of isoquinoline 1 and 2. |
Isoquinoline derivatives inhibit proliferation and promote apoptosis
Molecular structures of isoquinoline derivatives are shown in Supplementary materials. Firstly, we tested the antiproliferative activity of each compound in vitro on human SKOV3 ovarian cancer cells and, as shown in Figure 1A, CCK-8 assays revealed that two compounds – denoted as B01002 and C26001 – inhibited SKOV3 cell proliferation in a concentration-dependent manner with an IC50 of 7.65 and 11.68 μg/mL, respectively. The screened results of all 533 compounds are shown in Supplementary materials.
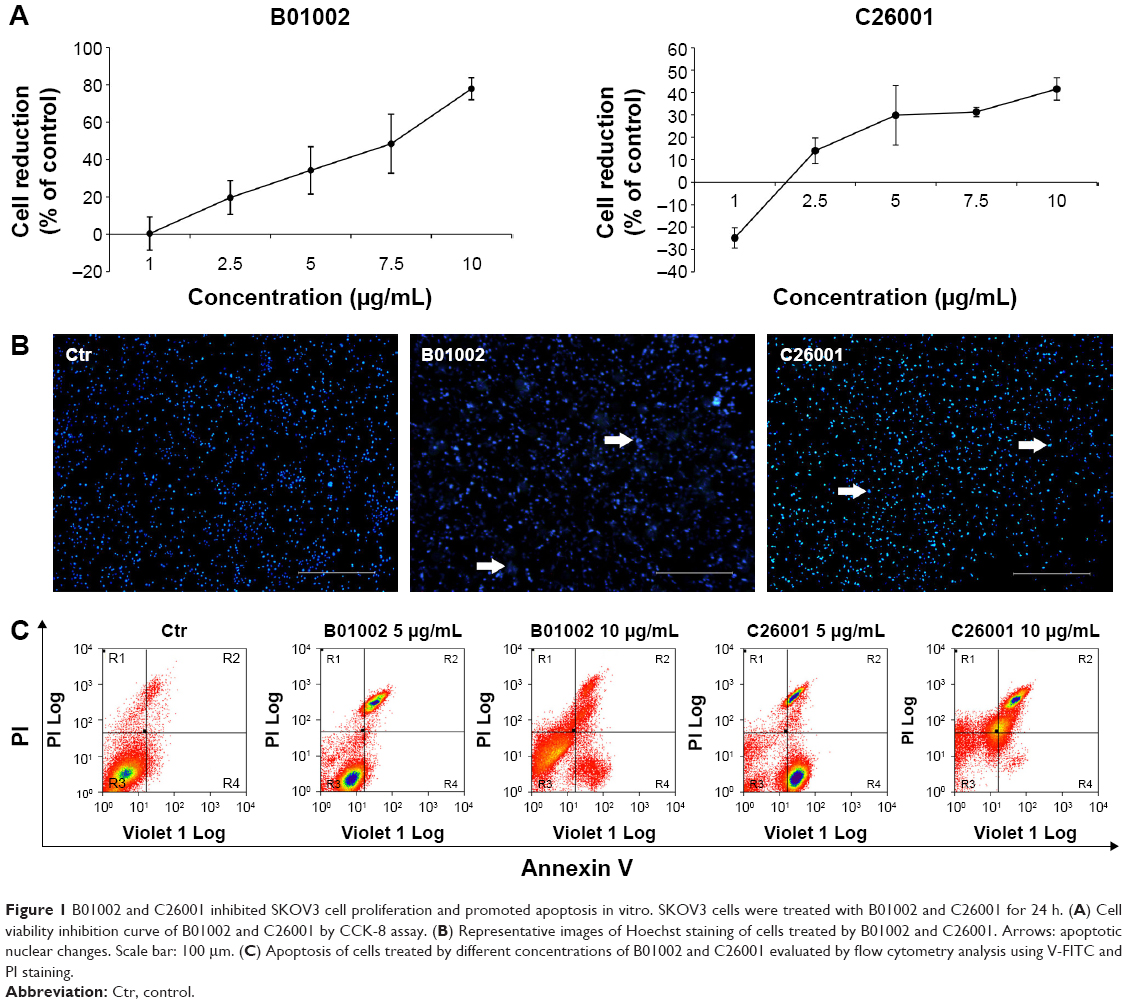 | Figure 1 B01002 and C26001 inhibited SKOV3 cell proliferation and promoted apoptosis in vitro. SKOV3 cells were treated with B01002 and C26001 for 24 h. (A) Cell viability inhibition curve of B01002 and C26001 by CCK-8 assay. (B) Representative images of Hoechst staining of cells treated by B01002 and C26001. Arrows: apoptotic nuclear changes. Scale bar: 100 μm. (C) Apoptosis of cells treated by different concentrations of B01002 and C26001 evaluated by flow cytometry analysis using V-FITC and PI staining. |
Spectroscopic data for B01002 are as follows: 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J=8.4 Hz, 1H) (Ar-H, the eighth position of isoquinoline), 8.28 (dd, J=1.2, 5.2 Hz, 1H) (Ar-H, the sixth position of pyridinyl), 8.08 (s, 1H) (ArNHAr’), 7.94 (d, J=8.4 Hz, 1H) (Ar-H, the fifth position of isoquinoline), 7.71 (t, J=8.0 Hz, 1H) (Ar-H, the fourth position of pyridinyl), 7.65 (d, J=8.0 Hz, 1H) (Ar-H, the third position of pyridinyl), 7.58 (t, J=8.0 Hz, 1H) (Ar-H, the sixth position of isoquinoline), 7.44 (t, J=7.6 Hz, 1H) (Ar-H, the seventh position of isoquinoline), 7.10 (s, 1H) (Ar-H, the fourth position of isoquinoline), 6.93 (t, J=6.0 Hz, 1H) (Ar-H, the fifth position of pyridinyl), 2.06–2.11 (m, 1H) (CH, cyclopropyl), 1.11–1.18 (m, 2H), (CH2, cyclopropyl), 0.97–0.99 (m, 2H) (CH2, cyclopropyl); 13C NMR (100 MHz, CDCl3) δ 154.00, 153.80, 150.75, 147.92, 147.55, 138.04, 130.20, 126.84, 125.66, 121.57, 117.56, 117.26, 113.13, 110.62, 17.40 (CH, cyclopropyl), 9.01 (CH2, cyclopropyl); IR (cm−1): 3446.23, 3048.23, 2927.25, 2852.63; HRMS (ESI) calculated for C17H15N3: 262.1344 (M + H+), found: 262.1396.
Spectroscopic data for C26001 are as follows: 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H) (Ar-H, the fourth position of isoquinoline), 8.00 (d, J=8.4 Hz, 1H) (Ar-H, the eighth position of isoquinoline), 7.67 (s, 1H) (Ar-H, the fifth position of isoquinoline), 7.40 (d, J=8.4 Hz, 1H) (Ar-H, the seventh position of isoquinoline), 7.22–7.12 (m, 5H) (Ar-H, phenyl), 4.73 (s, 2H) (CH2, Ar-CH2-Ph), 4.53–4.49 (m, 2H) (CH2, -COOCH2CH3), 2.49 (s, 3H) (CH3, Ar-CH3), 1.47 (t, J=6.8 Hz, 3H) (CH3, -COOCH2CH3); 13C NMR (100 MHz, CDCl3 ) δ 166.2 (carbonyl or the first position of isoquinoline), 160.4 (carbonyl or the first position of isoquinoline), 141.0, 139.3, 136.6, 131.7, 128.6, 128.5, 127.7, 127.0, 126.8, 126.3, 126.0, 125.0, 61.7 (-0-CH2-), 42.5 (Ar-CH2-Ph), 21.8 (CH3, Ar-CH3), 14.5 (CH3, -CH2-CH3); HRMS (ESI) calculated for C20H19NO2: 306.1494 (M + H+), found: 306.1525.
To gain insights into the underlying mechanisms, apoptotic nuclear changes were assessed by Hoechst staining. Figure 1B demonstrated that B01002 and C26001, both at a concentration of 5 μg/mL significantly induced apoptosis of SKOV3 cells. Then, a flow cytometric analysis with annexin V–FITC and PI staining was further performed. The level of apoptotic cells in SKOV3 was assessed by the percentage of annexin V-positive/PI-negative cells present after exposure of SKOV3 cells to different concentrations (5 or 10 μg/mL) of B01002 and C26001 for 24 h, as illustrated in Figure 1C. At the concentration of 5 μg/mL, B01002 and C26001 induced apoptosis, as indicated by a 21% and 9% increase in apoptotic cells (annexin V-positive/PI-negative), respectively. At a concentration of 10 μg/mL, B01002 and C26001 promoted cell death via both necrosis and apoptosis. For B01002, 46% of cells were necrotic (annexin V-positive/PI-positive), and 5% of cells were apoptotic, whereas for C26001, 20% cells were necrotic and 46% were apoptotic. As a comparison, 4% cells were apoptotic in the control group.
In vivo dosage experiment proved the safety of isoquinoline derivatives
B01002 and C26001 were selected for in vivo assays. The compounds B01002 and C26001 were injected into six animals, and DDP was administered as positive control, with PBS as negative control. Daily observation of clinical signs, physical examination, body weight, and food consumption with no abnormalities suggested good tolerance. A general observation of these organs treated with B01002 and C26001 showed no abnormal changes, whereas there were some lesions in the liver of the mice treated with DDP. Histopathological studies were carried out subsequently, and were also consistent. Abnormal findings were observed in the organs of the mice treated with DDP; no lesions were found in the organs of mice treated with B01002 or C26001. Blood samples were tested for blood count (RBC, WBC, hemoglobin, and platelets), and hepatorenal function (AST, ALT, ALP, BUN, and Cr). Supplementary materials show that the counts of blood components in mice treated with these two compounds were not altered significantly compared with the control. WBCs were slightly reduced by C26001, by 12%. Supplementary materials also show mean AST, ALT, ALP, BUN, and Cr values in animals treated with B01002 and C26001 were largely unchanged.
Isoquinoline derivatives decreased tumor size in a xenograft mouse model
The in vivo antitumor activity of B01002 and C26001 was determined in nude mice bearing ovarian xenografted tumors. The two compounds, as well as DDP and PBS, were slowly injected intraperitoneally at the dosages and the time intervals described earlier (Figure 2A). Both compounds induced tumor growth delay for a longer period of time when compared with PBS controls. Furthermore, B01002 and C26001 revealed a clear tumor reduction of 99.53% and 84.23%, respectively, compared with PBS controls (Figure 2B). In summary, these preliminary data demonstrate the potential of B01002 and C26001 for the treatment of ovarian cancer.
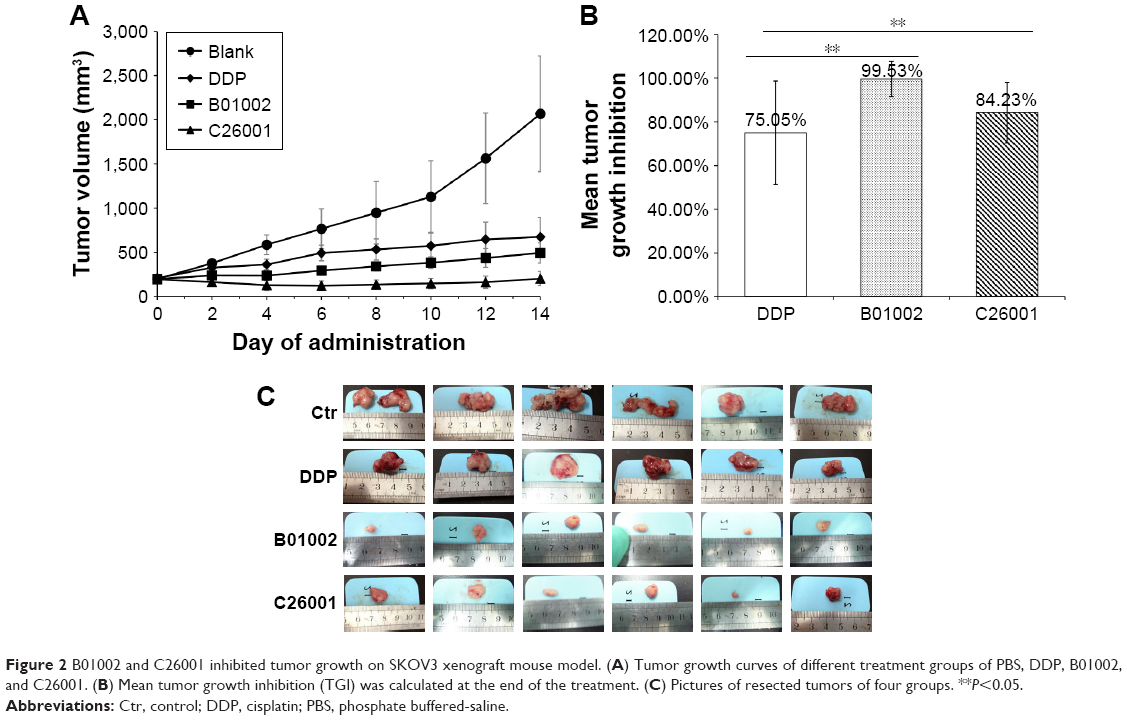 | Figure 2 B01002 and C26001 inhibited tumor growth on SKOV3 xenograft mouse model. (A) Tumor growth curves of different treatment groups of PBS, DDP, B01002, and C26001. (B) Mean tumor growth inhibition (TGI) was calculated at the end of the treatment. (C) Pictures of resected tumors of four groups. **P<0.05. |
Isoquinoline derivatives inhibited tumor cell proliferation mainly in an apoptotic way
To study the antiproliferative effects of B01002 and C26001 on xenografted tumors, we immunostained the resected tumors with a Ki-67 antibody and immunoblotted the resected tumor with a proliferating cell nuclear antigen (PCNA) antibody, respectively. The proliferation index was determined according to the Ki-67 expression level. We found that tumors treated with B01002 and C26001 had a significantly lower percentage of Ki-67 positivity, compared with tumors treated with PBS (33.2% and 68.5%, respectively; Figure 3A). To further verify these findings, we examined the expression level of a typical marker of cell proliferation, PCNA, which is abundant in SKOV3 cell lines. Figure 3B shows that PCNA (a typical marker of cell proliferation) was significantly decreased after treatment with C26001 (13.1%). However, PCNA was not decreased in tumors treated with B01002. Additionally, quantitative real-time PCR was conducted to assess mRNA levels of PCNA in resected tumors. Treatment with C26001 resulted in an evident rise (33%) in the mRNA levels of PCNA (Figure 3C). These results suggest that the antiproliferative potential of C26001 is associated with the elevation of mRNA levels of PCNA. Moreover, B01002 and C26001 caused tumor cell apoptosis (16.5% and 37.2%, respectively) in vitro as revealed by TUNEL assay (Figure 4), suggesting that these two compounds may predominantly kill cells via the apoptosis pathway.
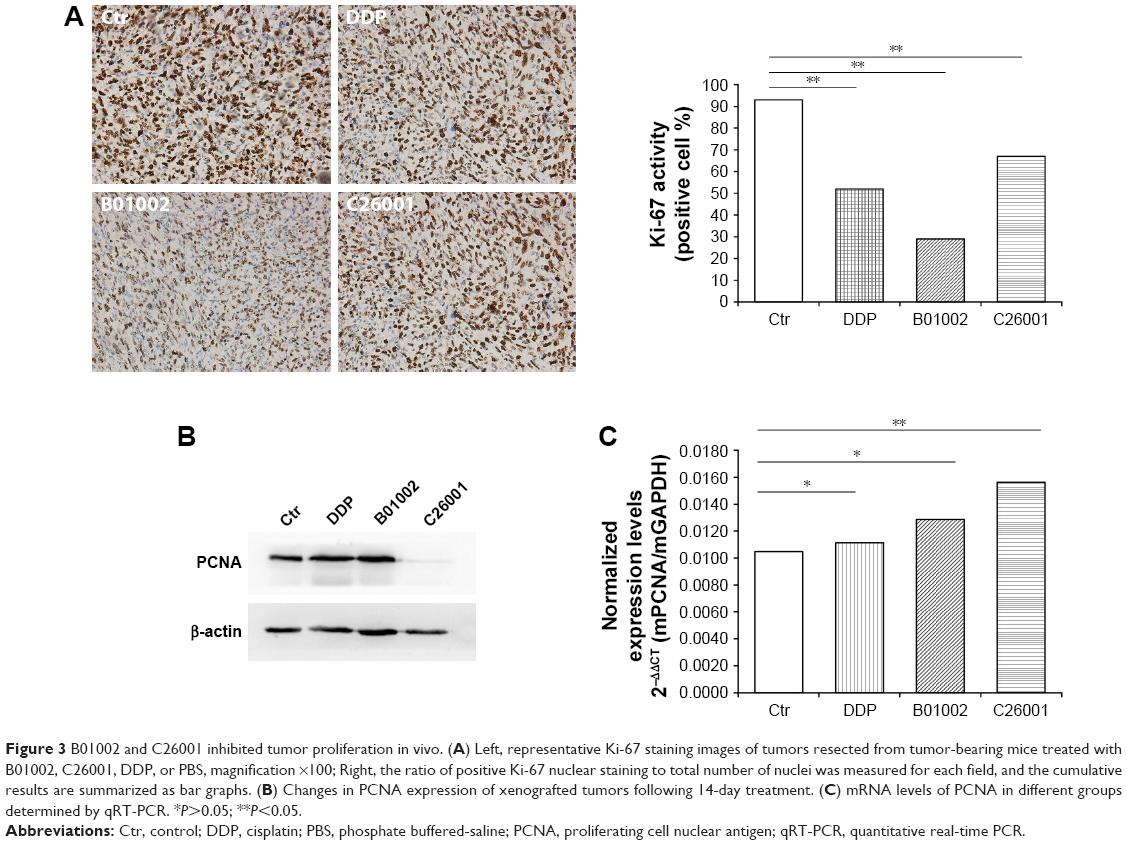 | Figure 3 B01002 and C26001 inhibited tumor proliferation in vivo. (A) Left, representative Ki-67 staining images of tumors resected from tumor-bearing mice treated with B01002, C26001, DDP, or PBS, magnification ×100; Right, the ratio of positive Ki-67 nuclear staining to total number of nuclei was measured for each field, and the cumulative results are summarized as bar graphs. (B) Changes in PCNA expression of xenografted tumors following 14-day treatment. (C) mRNA levels of PCNA in different groups determined by qRT-PCR. *P>0.05; **P<0.05. |
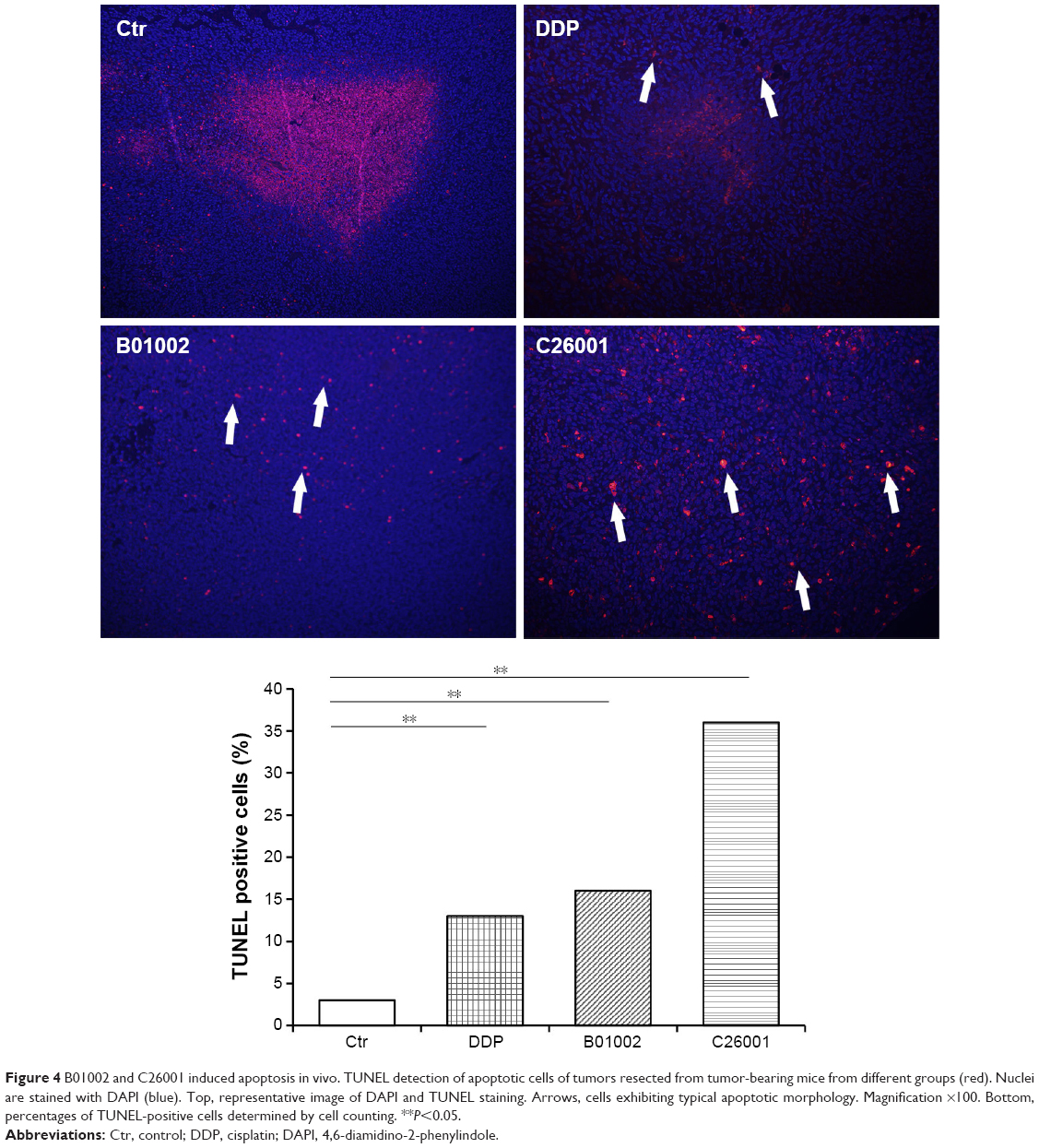 | Figure 4 B01002 and C26001 induced apoptosis in vivo. TUNEL detection of apoptotic cells of tumors resected from tumor-bearing mice from different groups (red). Nuclei are stained with DAPI (blue). Top, representative image of DAPI and TUNEL staining. Arrows, cells exhibiting typical apoptotic morphology. Magnification ×100. Bottom, percentages of TUNEL-positive cells determined by cell counting. **P<0.05. |
Isoquinoline derivatives inhibited IAP at the protein level and showed antagonistic effect through the caspase–IAP pathway
Levels of IAP including XIAP, cIAP, and survivin in resected tumors were measured using Western blotting assay, and the results were shown in Figure 5A. XIAP, cIAP-1, and survivin were all reduced in tumors treated with B01002 and C26001, compared with tumors treated with DDP or PBS. mRNA levels of XIAP, cIAP-1, and survivin were examined by quantitative RT-PCR (Figure 5B). For mRNA levels of XIAP, no significant differences were found between each group. However, an increased mRNA level of cIAP-1 in the B01002 group and an increased mRNA level of survivin in C26001 group were found.
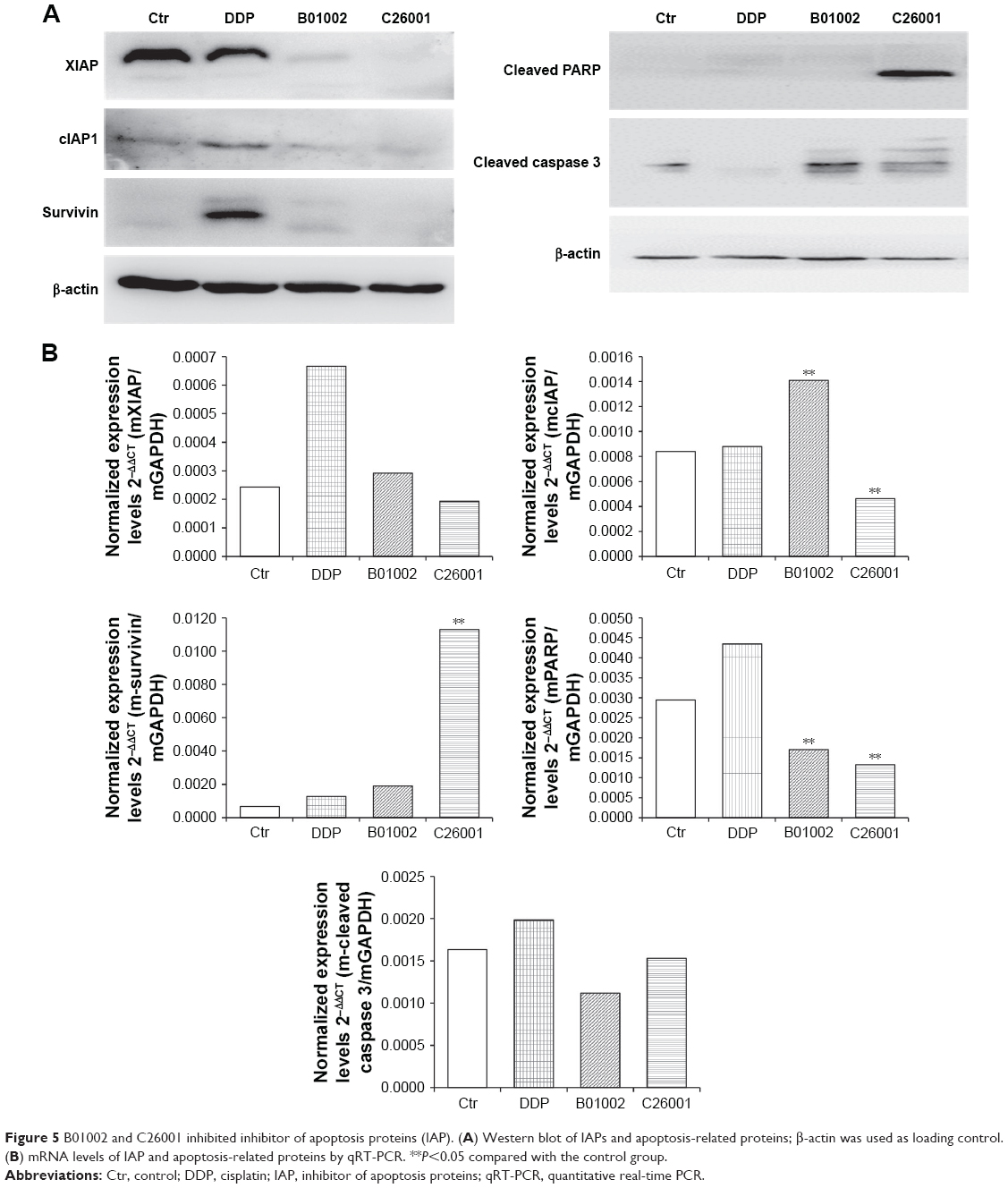 | Figure 5 B01002 and C26001 inhibited inhibitor of apoptosis proteins (IAP). (A) Western blot of IAPs and apoptosis-related proteins; β-actin was used as loading control. (B) mRNA levels of IAP and apoptosis-related proteins by qRT-PCR. **P<0.05 compared with the control group. |
Cleaved PARP and cleaved caspase-3 were also tested by Western blotting assay (Figure 5A). Cleaved PARP was elevated in cells treated by C26001, whereas both cleaved PARP and cleaved caspase-3 were elevated in cells treated by B01002, indicating that both of the compounds activated apoptosis through the caspase–IAP pathway. However, it is notable that the protein level of cleaved caspase-3 was correlated with that of XIAP (Pearson’s correlation 0.911, P<0.05). mRNA levels of PARP in the B01002 and C26001 group were decreased as compared with the control group. However, there were no significant differences in mRNA levels of caspase-3 between groups; instead, mRNA levels of all treatment groups were decreased as compared with the control group.
Discussion
In this study, we synthesized 533 new isoquinoline derivatives in the hope of inhibiting IAP and killing ovarian cancer. The CCK-8 assay showed that two of the isoquinoline derivatives could inhibit tumor cell proliferation. Hoechst staining, as well as fluorescence-activated cell sorting (FACS), results displayed that the two isoquinoline derivatives induced tumor cell apoptosis but not necrosis. Moreover, both of these new compounds were proved capable of decreasing tumor growth and have good safety profiles in the xenograft mouse model. Further analysis of the resected tumors revealed that the two compounds downregulated several IAP, including XIAP, cIAP-1, and survivin, thus promoting caspase-induced apoptosis, which may be one of the mechanisms of their antitumor activity.
As the incidence of ovarian cancer keeps rising and chemoresistance becomes increasingly common in the clinic, exploring new chemotherapeutic agents has become an important and urgent task, especially for patients with locally advanced, platinum-resistant ovarian cancer.25 Modern drug discovery often involves screening small molecules for their ability to bind to a preselected protein target. Drug discovery can also involve screening small molecules for their ability to modulate a biological pathway in cells or organisms, without regard for any particular protein target.26–28 In this study, we synthesized a collection of 533 molecules with an isoquinoline skeleton, aiming at perturbing the caspase–IAP pathway to induce apoptosis and kill cancer cells.
IAP block programmed cell death and are expressed at high levels in various human cancers. Moreover, IAP have been implied in the regulation of additional signaling cascades, such as mitogen-activated protein kinase (MAPK) pathway,29 TGFb signaling,30,31 as well as innate and adaptive immunity signaling pathways32,33 – making them attractive targets for cancer drug development.34 Therefore, many efforts have been made over the past decade to develop strategies to neutralize IAP, including antisense oligonucleotides and small-molecule inhibitors.35,36 Smac is a mitochondrial protein that is released into the cytosol upon the induction of programmed cell death and promotes apoptosis by neutralizing IAP.37 On this basis, a variety of small-molecule inhibitors have been developed that mimic the binding domain of the native Smac protein to IAP. So far, various Smac mimetics (monovalent or bivalent) are proved to inhibit Smac tetrapeptide binding to recombinant IAP, rescue IAP-bound caspase-3 activity, and show antiproliferative activities against malignant human cancer cells.38,39 Monovalent Smac mimetics are less potent than their corresponding bivalent Smac mimetics.40 However, because bivalent Smac mimetics have molecular weights exceeding 1,000, such compounds are expected to have very low oral bioavailability and will have to be administered intravenously – a potential disadvantage if the drug must be administered frequently.41 Therefore, smaller molecules with higher efficacy are still needed.
Isoquinoline derivatives have been reported to show high affinity for the BIR2 domain and demonstrated potent IAP inhibitory activities in biochemical and cellular assays.42 Therefore, we screened the 533 isoquinoline derivatives synthesized earlier to obtain compounds with effective antitumor activity on ovarian cancer.
Firstly, we tested their cytotoxicity in vitro using the SKOV3 ovarian cancer cell line, and found that both B01002 and C26001 could inhibit tumor cell proliferation effectively (IC50 =7.65 and 11.68 μg/mL, respectively). Moreover, we found that both compounds could induce apoptosis of tumor cell, which seems the main mechanism of the toxicity, as a very small percentage of necrotic cells was observed. To evaluate the translational potential of these new agents, we tested their in vivo efficacy and safety using a xenograft mouse model. Consistent with the in vitro results, our study showed that both compounds decreased the size of xenografted tumors. By Ki-67 staining and PCNA level, we showed that both compounds inhibited the tumor proliferation in vivo. By measuring body weight, food consumption, observing clinical signs, physical examination, blood routine testing, and biochemical tests, as well as necropsy of major organs, the mice showed good tolerance in various dosages to both compounds, indicating their clinical translational potential in future applications.
Our data also revealed that these two new isoquinoline derivatives can downregulate XIAP, cIAP-1, and survivin at the protein level, but not at the mRNA level, suggesting that they work on the translational or post-translational process. As IAP block apoptosis by neutralizing caspases, it is not surprising that our new isoquinoline derivatives exert a pro-apoptosis effect, as also evidenced by TUNEL staining. The fact that both compounds upregulated the cleaved caspase-3 and partly upregulated cleaved PARP at the protein level further verified that the two compounds inhibited the tumor cells through the IAP–caspases pathway.
In addition, it is worth noting that isoquinoline-skeleton compounds are reported to participate in several signaling ways associated with tumor proliferation, invasion, metastasis, and angiogenesis, indicating that the antitumor effect of isoquinoline derivatives is not only limited to downregulating IAP.43 We hope to explore more biological functions of these new compounds to fully elaborate their antitumor mechanisms.
In conclusion, we have synthesized two new isoquinoline derivatives that significantly inhibit ovarian tumor growth and promote tumor apoptosis, both in vivo and in vitro, partly by downregulating the IAP. Combined with their good safety profile, this makes it worthwhile to evaluate their efficacies in a clinical setting in the future.
Acknowledgment
This study was supported by Science and Technology Commission of Shanghai Municipality (124119a2900, 114119a2100 to L Yao), and New Hundred Talents Program of Health and Family Planning Commission of Shanghai (13B122 to L Yao).
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11(2):109–124. | ||
Gill C, Dowling C, O’Neill AJ, Watson RW. Effects of cIAP-1, cIAP-2 and XIAP triple knockdown on prostate cancer cell susceptibility to apoptosis, cell survival and proliferation. Mol Cancer. 2009;8:39. | ||
LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene. 2008;27(48):6252–6275. | ||
Song Z, Yao X, Wu M. Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J Biol Chem. 2003;278(25):23130–23140. | ||
Fulda S. Regulation of cell migration, invasion and metastasis by IAP proteins and their antagonists. Oncogene. 2014;33(6):671–676. | ||
Fulda S. Inhibitor of apoptosis (IAP) proteins as therapeutic targets for radiosensitization of human cancers. Cancer Treat Rev. 2012;38(6):760–766. | ||
Wen X, Lin ZQ, Liu B, Wei YQ. Caspase-mediated programmed cell death pathways as potential therapeutic targets in cancer. Cell Prolif. 2012;45(3):217–224. | ||
Vucic D, Fairbrother WJ. The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin Cancer Res. 2007;13(20):5995–6000. | ||
Yang X, Xing H, Gao Q, et al. Regulation of HtrA2/Omi by X-linked inhibitor of apoptosis protein in chemoresistance in human ovarian cancer cells. Gynecol Oncol. 2005;97(2):413–421. | ||
Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17(1):53–64. | ||
González-López M, Welsh K, Finlay D, et al. Design, synthesis and evaluation of monovalent Smac mimetics that bind to the BIR2 domain of the anti-apoptotic protein XIAP. Bioorg Med Chem Lett. 2011;21(14):4332–4336. | ||
Boulahjar R, Ouach A, Matteo C, et al. Novel tetrahydropyrido[1,2-a]isoindolone derivatives (valmerins): potent cyclin-dependent kinase/glycogen synthase kinase 3 inhibitors with antiproliferative activities and antitumor effects in human tumor xenografts. J Med Chem. 2012;55(22):9589–9606. | ||
Morris GF, Mathews MB. Regulation of proliferating cell nuclear antigen during the cell cycle. J Biol Chem. 1989;264(23):13856–13864. | ||
Zheng D, Chen Z, Liu J, Wu J. An efficient route to 1-aminoisoquinolines via AgOTf-catalyzed reaction of 2-alkynylbenzaldoxime with amine. Org Biomol Chem. 2011;9(13):4763–4765. | ||
Zheng D, Li S, Wu J. An unexpected silver triflate catalyzed reaction of 2-alkynylbenzaldehyde with 2-isocyanoacetate. Org Lett. 2012;14(11):2655–2657. | ||
Fani S, Kamalidehghan B, Lo KM, Hashim NM, Chow KM, Ahmadipour F. Synthesis, structural characterization, and anticancer activity of a monobenzyltin compound against MCF-7 breast cancer cells. Drug Des Devel Ther. 2015;9:6191–6201. | ||
Yao Z, Sun B, Hong Q, et al. PACE4 regulates apoptosis in human prostate cancer cells via endoplasmic reticulum stress and mitochondrial signaling pathways. Drug Des Devel Ther. 2015;9:5911–5923. | ||
Jin R, Xia Y, Chen Q, et al. Da0324, an inhibitor of nuclear factor-kappaB activation, demonstrates selective antitumor activity on human gastric cancer cells. Drug Des Devel Ther. 2016;10:979–995. | ||
Jin Z, Niu H, Wang X, Zhang L, Wang Q, Yang A. Preclinical study of CC223 as a potential anti-ovarian cancer agent. Oncotarget. Epub 2017 May 10. | ||
Jiang S, Chen X. HMGB1 siRNA can reduce damage to retinal cells induced by high glucose in vitro and in vivo. Drug Des Devel Ther. 2017;11:783–795. | ||
Pala Ş, Atilgan R, Kuloğlu T, et al. Protective effects of vitamin C and vitamin E against hysterosalpingography-induced epithelial degeneration and proliferation in rat endometrium. Drug Des Devel Ther. 2016;10:4079–4089. | ||
Pandey S. Aldose reductase inhibitor fidarestat as a promising drug targeting autophagy in colorectal carcinoma: a pilot study. Asian Pac J Cancer Prev. 2015;16(12):4981–4985. | ||
Kadivar A, Kamalidehghan B, Akbari Javar H, Karimi B, Sedghi R, Noordin MI. Antiproliferation effect of imatinib mesylate on MCF7, T-47D tumorigenic and MCF 10A nontumorigenic breast cell lines via PDGFR-beta, PDGF-BB, c-Kit and SCF genes. Drug Des Devel Ther. 2017;11:469–481. | ||
Ren F, Shen J, Shi H, Hornicek FJ, Kan Q, Duan Z. Novel mechanisms and approaches to overcome multidrug resistance in the treatment of ovarian cancer. Biochim Biophys Acta. 2016;1866(2):266–275. | ||
Huang R, Leung IK. Protein-directed dynamic combinatorial chemistry: a guide to protein ligand and inhibitor discovery. Molecules. 2016;21(7). pii:E910. | ||
Narayana KKC, Aswathanarayan JB, Vittal RR. Endophytic peptides – a source of therapeutic agents. Curr Protein Pept Sci. 2017;18(3):284–290. | ||
Schreiber SL. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science. 2000;287(5460):1964–1969. | ||
Varfolomeev E, Goncharov T, Maecker H, et al. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and MAPK activation by members of the TNF family of receptors. Sci Signal. 2012;5(216):ra22. | ||
Birkey Reffey S, Wurthner JU, Parks WT, Roberts AB, Duckett CS. X-linked inhibitor of apoptosis protein functions as a cofactor in transforming growth factor-beta signaling. J Biol Chem. 2001;276(28):26542–26549. | ||
Hofer-Warbinek R, Schmid JA, Stehlik C, Binder BR, Lipp J, de Martin R. Activation of NF-kappa B by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1. J Biol Chem. 2000;275(29):22064–22068. | ||
Beug ST, Cheung HH, LaCasse EC, Korneluk RG. Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol. 2012;33(11):535–545. | ||
Lopez J, Meier P. To fight or die – inhibitor of apoptosis proteins at the crossroad of innate immunity and death. Curr Opin Cell Biol. 2010;22(6):872–881. | ||
Fulda S. Promises and challenges of Smac mimetics as cancer therapeutics. Clin Cancer Res. 2015;21(22):5030–5036. | ||
LaCasse EC. Pulling the plug on a cancer cell by eliminating XIAP with AEG35156. Cancer Lett. 2013;332(2):215–224. | ||
Straub CS. Targeting IAPs as an approach to anti-cancer therapy. Curr Top Med Chem. 2011;11(3):291–316. | ||
Fulda S. Molecular pathways: targeting death receptors and smac mimetics. Clin Cancer Res. 2014;20(15):3915–3920. | ||
Talbott RL, Borzilleri RM, Chaudhry C, et al. Pharmacology of smac mimetics; chemotype differentiation based on physical association with caspase regulators and cellular transport. Exp Cell Res. 2015;338(2):251–260. | ||
Wang S. Design of small-molecule Smac mimetics as IAP antagonists. Curr Top Microbiol Immunol. 2011;348:89–113. | ||
Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305(5689):1471–1474. | ||
Sun H, Nikolovska-Coleska Z, Lu J, et al. Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent Smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J Am Chem Soc. 2007;129(49):15279–15294. | ||
Kim KS, Zhang L, Williams D, et al. Discovery of tetrahydroisoquinoline-based bivalent heterodimeric IAP antagonists. Bioorg Med Chem Lett. 2014;24(21):5022–5029. | ||
Pandey MK, Prasad S, Tyagi AK, et al. Targeting cell survival proteins for cancer cell death. Pharmaceuticals (Basel). 2016;9(1). pii:E11. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.