")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Inhibitor of IGF1 receptor alleviates the inflammation process in the diabetic kidney mouse model without activating SOCS2
Authors Li J , Dong R , Yu J, Yi S, Da J, Yu F, Zha Y
Received 19 April 2018
Accepted for publication 15 June 2018
Published 11 September 2018 Volume 2018:12 Pages 2887—2896
DOI https://doi.org/10.2147/DDDT.S171638
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Jiayu Li,1 Rong Dong,2 Jiali Yu,2 Sun Yi,2 Jingjing Da,2 Fuxun Yu,3 Yan Zha2
1Guizhou University School of Medicine, Guizhou University, 2Department of Nephrology, Guizhou Provincial People’s Hospital, 3Department of Central Laboratory, Guizhou Provincial People’s Hospital, Guiyang, China
Objective: To explore the anti-inflammatory mechanism of IGF1R inhibitor in diabetic nephropathy.
Methods: C57/BL6 mice were reared with high-fat diet for 8 weeks, then were injected 30 mg/kg streptozotocin intraperitoneally to induce type 2 diabetes. After 8 weeks, the type 2 diabetes nephropathy model was successfully set up the different drugs were administrated to mice with diabetes (insulin 1–2 U/day, benazepril 10 mg/kg per day intragastrically, IGF-1R inhibitor 30 mg/kg per day intragastrically). After 8 weeks drugs administration, all mice were collected the kidney tissue, measured levels of inflammatory factor (F4/80, TLR4and CD68) and fibrosis markers(αSMA, E-cadherin and SR) using immunohistochemistry and in situ hybridization.
Results: The type 2 diabetes nephropathy model was built successfully, which along with increased urinary protein excretion rate and increased inflammatory infiltration, and the correlation was characterized by increased CD68+, F4/80+ cells and increased TLR4, αSMA, SR expression. IGF-1R inhibitors reversed this changes, but benazepril and insulin were without significant changes. The insulin decreased the expression level of IGF-1, and increased the levels of suppressor of cytokine signaling 2 (SOCS2). Benazepril and IGF-1R inhibitor were no significant changes like insulin.
Conclusion: Inhibition of IGF1R was a more effective choice for inflammation treatment than Ben or Ins in diabetic kidney disease (DKD). The IGF1R inhibitor blocked pathological changes induced by the over-expression of IGF1 in DKD without up-regulating SOCS2 protein levels.
Keywords: diabetes kidney disease, IGF1R inhibitor, insulin, SOCS2
Introduction
Diabetes affects around 425 million people worldwide, and China has the highest number of patients of any nation.1 About 40% of diabetes patients eventually die of diabetic kidney disease (DKD), which is a leading cause of end-stage KD (ESKD).2 There are few therapeutic drugs for DKD, principally blood pressure and blood glucose control drugs, such as blockers of the renin–angiotensin–aldosterone system and insulin (Ins).3,4 Angiotensin-converting enzyme inhibitors like benazepril (Ben) can reduce blood pressure and improve the blood flow in the kidneys. Studies have revealed that Ben alone or combined with rhein or leflunomide has a good effect on relieving the fibrosis process of diabetic nephropathy.5–8 Studies have also shown that Ins mildly reduces inflammatory infiltration and fibrosis of tissue through regulating the activity of IGF1R by SOCS signaling.9 However, control of blood glucose and blood pressure by Ins and blockers of the renin–angiotensin–aldosterone system cannot completely relieve inflammatory and fibrosis progress in DKD, microalbuminuria still occurs in 16%–26% of diabetes patients, and patients progress relentlessly to ESKD. As such, more strategies are needed for more effective treatment of DKD.4
Inflammation processes play a vital role in the pathogenesis of DKD, driven by multiple factors like lipotoxicity, glucotoxicity, endoplasmic reticulum stress, oxidative stress, formation of amyloid deposits in islets, and alterations in gut microbiota caused by Ins resistance.10 Anti-inflammation treatment can instead act on the dysfunctional pathway that causes several changes associated with DKD.11 Treatments addressing inflammation could be used to prevent DKD progression.
IGF1 is a powerful regulatory factor in various cell types, including glomerular and tubular cells.12 It is an important growth factor for keeping the nephritic structure and function. It also plays a key role in the pathological process of DKD.13 Many studies have shown that IGF1 overexpression causes many histopathology changes, such as kidney tissue hyperplasia, renal cell proliferation, nephromegaly, mesangial expansion, and increased expression of inflammation cytokines and extracellular matrix proteins.12–14 An IGF1R inhibitor is a drug commonly used in the treatment of tumors and cancer, as it has a significant inhibitory effect on tumor growth.15 In our previous report, we also found that inhibition of IGF1R could alleviate inflammation in DKD more efficiently.10 The mechanism of the IGF1R inhibitor alleviating inflammation needs to be addressed.
To investigate the effect of IGF1R inhibitors on DKD further and address the mechanisms involved therein, we developed a DKD mouse model, treating them with an IGF1R inhibitor and comparing this to Ins and Ben treatment. Factors involved in macrophage infiltration and fibrosis were examined. We found that the IGF1R inhibitor alleviated the inflammation process more efficiently than Ins or Ben by avoiding the activation of the SOCS pathway.
Methods
Animals
A total of 30 specific pathogen-free male C57/BL6 mice (aged 6–8 weeks and weighing 20±3 g) were purchased from the Animal Experiment Center of Guizhou Medical University (Guiyang, China). Mouse experiments were conducted in accordance with the policies of the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and China animal welfare legislation. Our research was approved by the ethics committee of Guizhou People’s Hospital. The study protocol was approved by the institutional animal care and welfare committee. All mice were kept under standard temperature (21°C±2°C), standard humidity (55%±2%), and a 12-hour light–dark cycle. All mice had free access to a standard rodent diet and drinking water. All animal experiments were approved by Guizhou Medical University.
Sample size calculation
We calculated a sample size with the formula n=2 ([α + β]2 × δ2)/(μ1 − μ2)2 reported by Dekker et al, where μ1 and μ2 were the population means in treatment groups 1 and 2, μ1−μ2 the minimal clinically relevant difference, δ2 the population variance (SD), α the conventional multiplier for α, and β the conventional multiplier for power.36 According to this formula, we determined the sample size to be six for each group.
Experimental protocol and sample preparation
After 1 week of adaptive feeding, all mice were randomly assigned to five groups (six mice per group):36 control (Con), DKD model, Ins-DKD (Wanbang Biochemical Pharmaceuticals, Xuzhou, China), Ben-DKD (Novartis, Basel, Switzerland), and IGF1R inhibitor (DKD-IGF; selective IGF1R inhibitor [GSK4529;19 Selleck Chemicals, Houston, TX]). Except for Con, the other groups were fed a high-fat diet (D12492) for 8 weeks to establish an Ins resistance model. All Ins resistance model mice were converted to DKD mice with a single intraperitoneal injection of streptozotocin (Stz; 30 mg/kg mouse weight diluted in 0.1 M precooled citrate buffer, pH 4.5). The Con group were fed normal food (4% calories from fat) for 8 weeks and then intraperitoneally injected with the same dose of normal saline (pH 7.3, 0.154 mol/L). At 72 hours after Stz injection, blood glucose levels were measured randomly to confirm the success of the diabetes model. A random blood glucose level of >16.7 mmol/L after Stz injection was the indicator of establishment of the model.37,38 All mice in the model groups successfully developed type two diabetes mellitus. Mice in the DKD-Ins group were injected subcutaneously with 1–2 U/day Ins, mice in the DKD-Ben group administered Ben 10 mg/kg per day intragastrically, and mice in the DKD-IGF group administered IGF1R inhibitor 30 mg/kg per day intragastrically, while mice in the Con and DKD groups received equivalent doses of normal saline. This experiment lasted for 16 weeks.
Body weight and random blood glucose levels were monitored weekly in all mice. All mice were housed in special metabolic cages for collecting 24-hour urine at the end of the 16th week and then killed under chloral hydrous anesthesia. Data for random blood glucose levels and body weight were compared among all groups. Blood samples were acquired by cardiac puncture at the time of death. Serum was then separated by centrifugation and stored at −20°C for subsequent experiments. Kidney tissue samples were also excised, weighed, and stored in liquid nitrogen or fixed in 4% paraformaldehyde. The kidney weight-to-body weight ratio was calculated for each mouse.
Assessment of renal biochemical markers
Creatinine and 24-hour urinary protein concentration were determined using a creatinine assay kit and a protein estimation kit, respectively, according to the protocols provided by the manufacturers.
Histopathological examination of kidney tissue
Portions of renal tissue fixed in 4% paraformaldehyde were embedded in paraffin and sliced into 4 μm-thick sections. The sections were stained with HE for assessment under light microscopy (Leica, Wetzlar, Germany). Renal sections 4 μm thick were stained with Sirius red to evaluate the area occupied by collagen fibrils. From each kidney, ten random interstitial cortical fields were captured at 40× magnification using a high-resolution video camera (Leica) connected to a light microscope (Leica DM 300 LED). The area occupied by collagen was measured using a computerized image analysis system (Image-Pro Plus).
Immunohistochemistry for αSMA, E-cadherin, SOCS2, F4/80, TLR4, and CD68 expression in kidney tissue
Following deparaffinization and hydration, 4 μm-thick renal cortical tissue sections were treated with 3% H2O2 for 10 minutes to inactivate endogenous enzymes. After incubation with appropriate primary antibodies (αSMA, E-cadherin, SOCS2, F4/80, TLR4, IGF1, and CD68 [Abcam, Cambridge, UK]) overnight at 4°C, the sections were incubated with a horseradish peroxidase-labeled goat antirabbit or goat antimouse polyclonal antibody for 30 minutes at room temperature. The sections were then counterstained with hematoxylin, dehydrated, cleared, and mounted with mounting solution. Ten fields were observed at 400× magnification with the Leica microscope. F4/80+ or CD68+ cells number and the positive integrated optical density value of each field were calculated and analyzed with Image-Pro Plus, and the mean values were compared among the experimental groups.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). For overall data, one-way ANOVA and post hoc analysis were used for multiple comparisons and two-tailed Student’s t-test for single two-sample comparisons, data from which are presented as means ± SD. P<0.05 was considered statistically significant.
Results
Establishment of DKD model
To develop the DKD mouse model, C57/BL6 mice were fed a high-fat diet for 8 weeks, then injected with Stz intraperitoneally (30 mg/kg mice weight).16,17 To determine whether the DKD model had been successfully established, blood glucose and urinary albumin levels were tested. In our model, the fasting blood glucose level of the Con group was 7.33 mmol/L, and that of the DKD group was 23.14 mmol/L. Blood glucose levels increased significantly in DKD mice and reached the diagnosis standard of diabetes (Figure 1A). Urinary albumin excretion rate increased significantly at the same time (Figure 1B). HE staining showed morphological glomerular changes in diabetic kidney tissue (Figure 1C). Sirius red staining showed renal interstitium fiber levels which had changed accentuation in diabetic kidney tissue (Figure 1C and D). These results demonstrated that the DKD mouse model had been developed successfully.
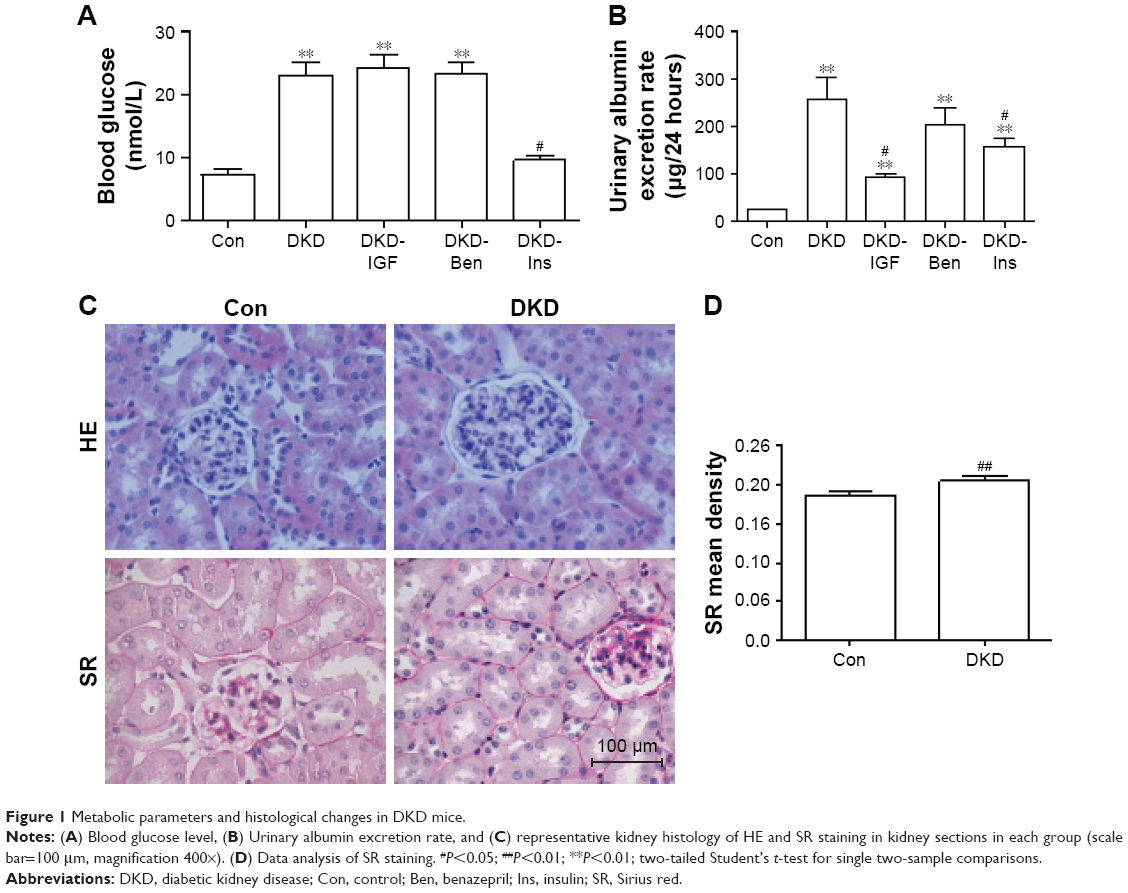 | Figure 1 Metabolic parameters and histological changes in DKD mice. |
Inflammation and fibrosis of renal tissue in DKD mice
In order to detect inflammatory infiltration and fibrosis changes in DKD, immunohistochemical staining was applied to determine the expression levels of inflammation markers (CD68, F4/80, and TLR4) and fibrosis markers (E-cadherin and αSMA). CD68 and F4/80 expression indicated the infiltration of macrophages, a sign of inflammation.18 TLR4 expression is an indicator of consistent inflammatory cell infiltration. Compared with the Con group, obvious increases in CD68+ and F4/80+ cells were found in the renal interstitia of DKD mice (Figure 2A–C). TLR4 expression in DKD mice also showed a significant increase (Figure 2A and D). DKD mice also showed significant progress in fibrosis, with decreased E-cadherin and increased αSMA protein expression (Figure 3A–C).
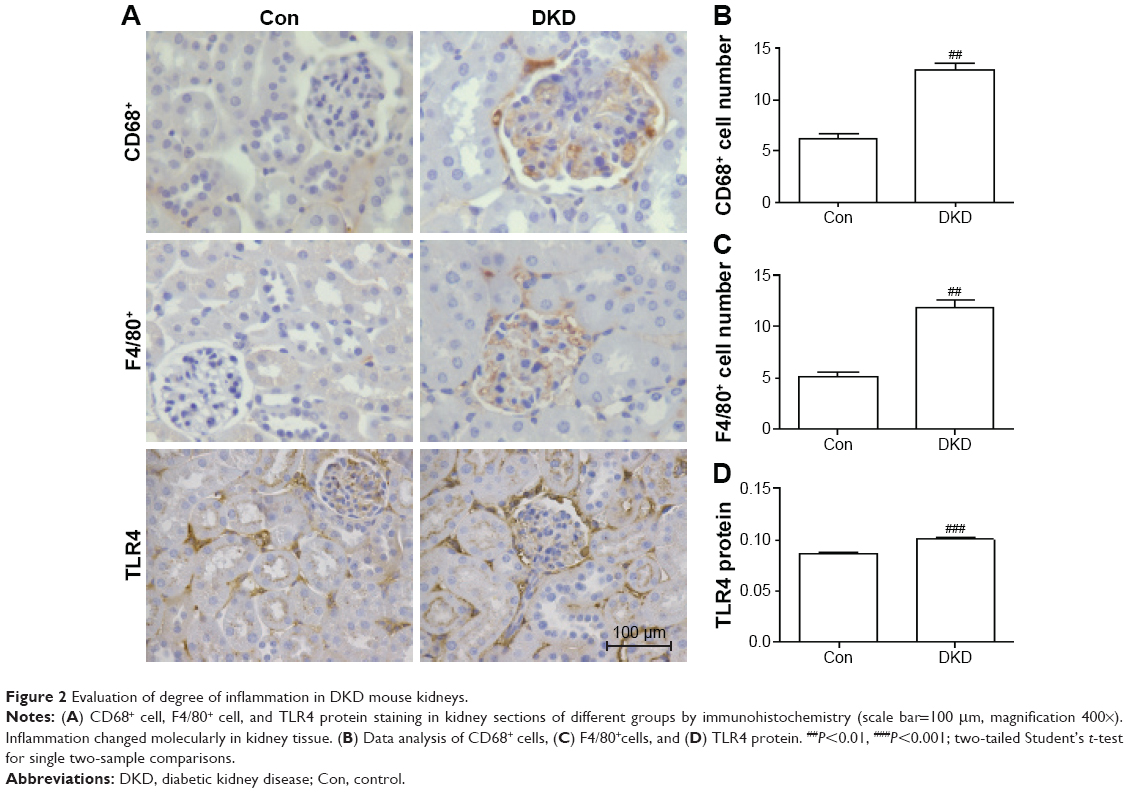 | Figure 2 Evaluation of degree of inflammation in DKD mouse kidneys. |
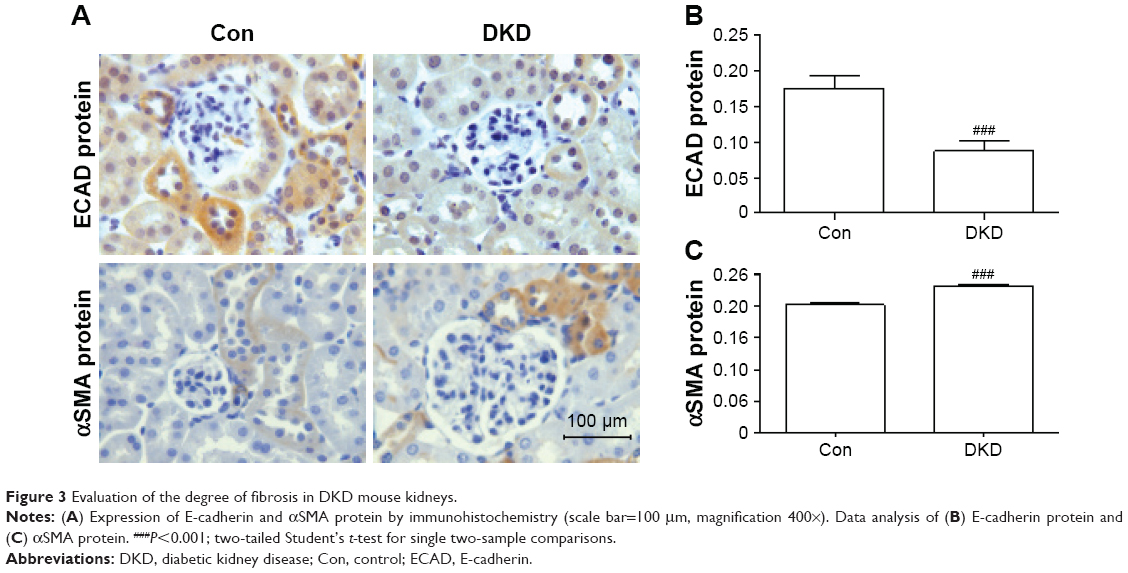 | Figure 3 Evaluation of the degree of fibrosis in DKD mouse kidneys. |
Abnormal expression of IGF1 and SOCS2 in DKD mice
To investigate the protein and mRNA expression levels of IGF1 and SOCS2 in renal tissue of DKD mice, immunohistochemical staining and in situ hybridization were conducted for IGF1 and SOCS2. IGF1 expression levels were significantly increased and SOCS2 significantly reduced in DKD mice (Figure 4A–E).
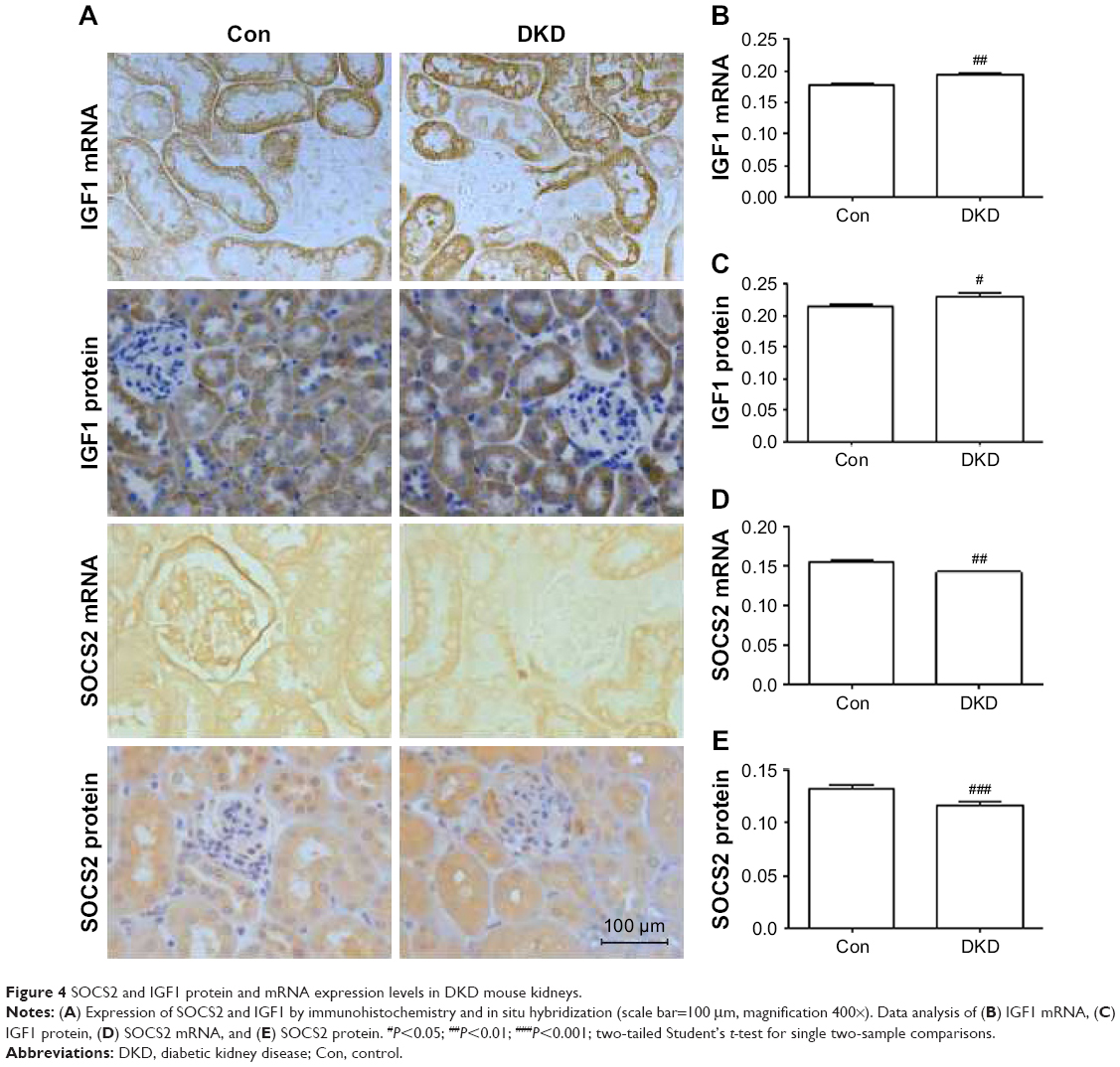 | Figure 4 SOCS2 and IGF1 protein and mRNA expression levels in DKD mouse kidneys. |
IGF1R inhibitor alleviated inflammation and fibrosis in DKD
To ascertain the effect of the IGF1R inhibitor on inflammatory infiltration and fibrosis in DKD mice, we used Ins, Ben, and the IGF1R inhibitor (GSK4529)19 for intervention in the DKD model. The results showed that blood glucose levels decreased with Ins, but not with the IGF1R inhibitor or Ben (Figure 1A). The urinary albumin excretion rate decreased significantly in all interventions, with the IGF1R inhibitor having a better effect than Ins or Ben (Figure 1B). HE and immunohistochemical staining showed that the degree of inflammatory cell filtration was different in all interventions. The decrease in CD68+ and F4/80+ cells was more significant with the IGF1R inhibitor, closely followed by Ben, while the effect of Ins was not statistically significant (Figure 5A–C). TLR4 expression in DKD renal tissue was reduced by the IGF1R inhibitor and Ins, but not by Ben, and there was no difference between the IGF1R inhibitor and Ins (Figure 5A and D).
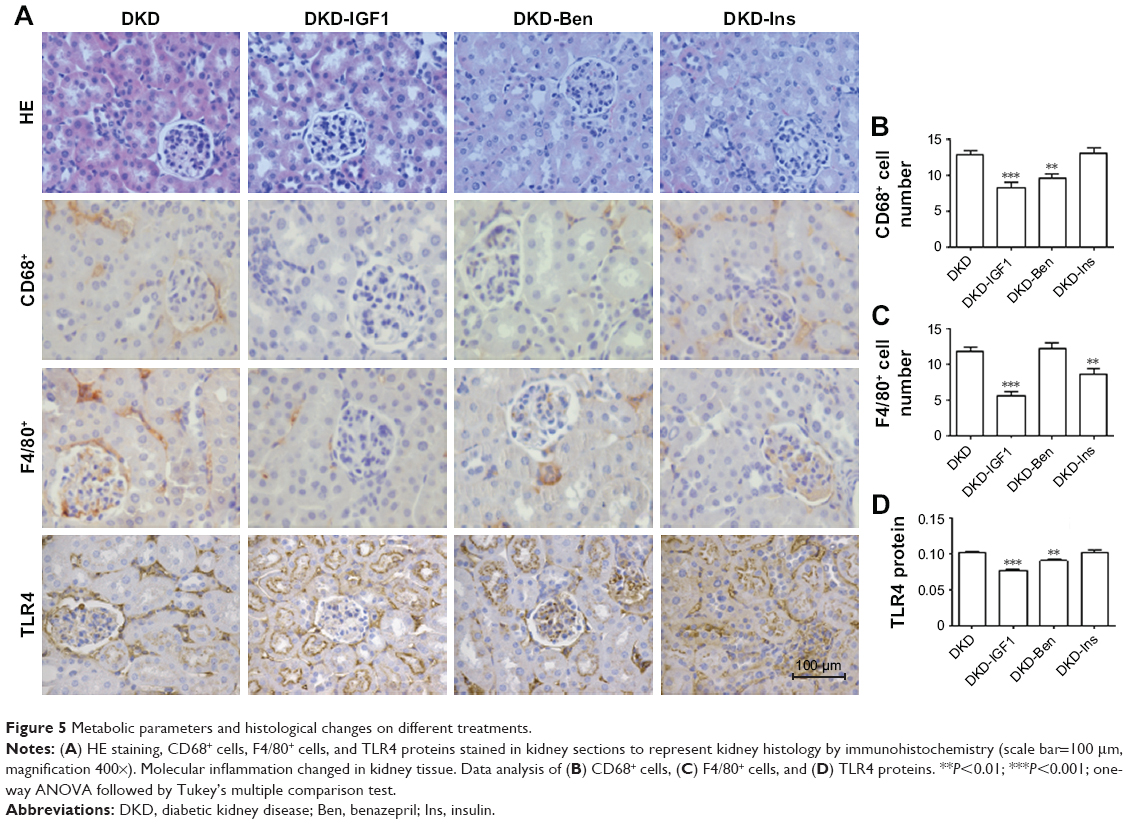 | Figure 5 Metabolic parameters and histological changes on different treatments. |
According to Sirius red staining results, renal fibrosis was alleviated by the IGF1R inhibitor and Ben, but not by Ins, and the IGF1R inhibitor had the best effect on improving the fibrosis process (Figure 6A and B). Furthermore, the IGF1R inhibitor reversed the expression of both E-cadherin and αSMA, while Ben reduced only αSMA expression without any effect on E-cadherin (Figure 6A, C, and D).
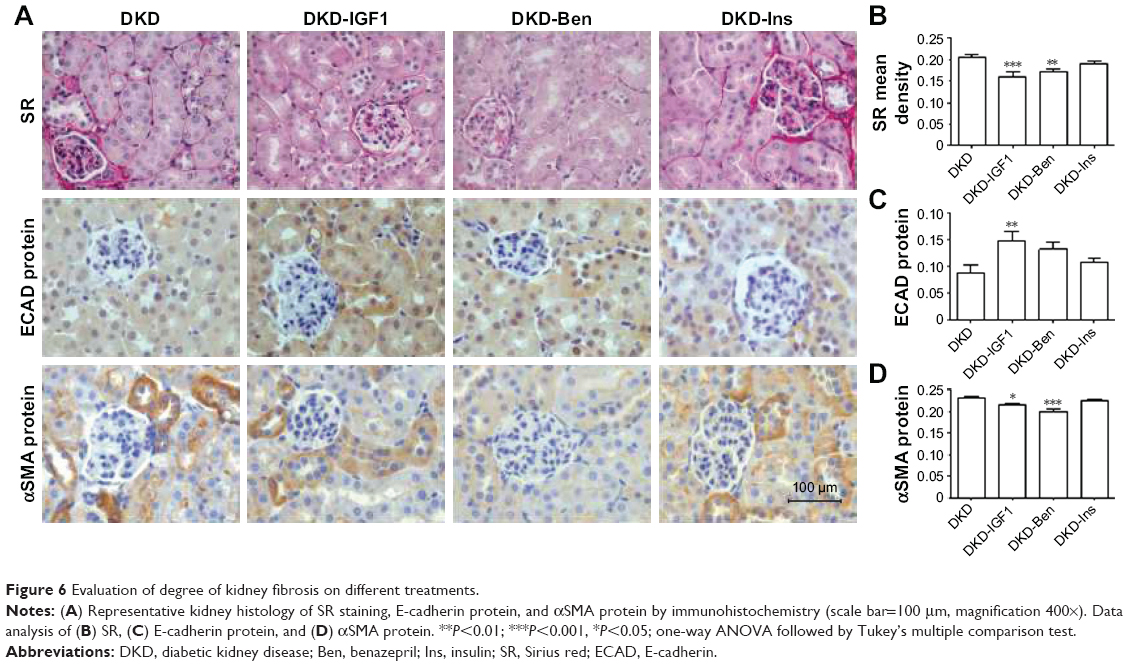 | Figure 6 Evaluation of degree of kidney fibrosis on different treatments. |
IGF1R inhibitor alleviated inflammation without activating SOCS2
Next, we tried to explore the reason that the IGF1R inhibitor improved inflammatory infiltration by investigating IGF1 and SOCS2 expression changes after different treatments. As can been seen in Figure 7, IGF1 protein and mRNA levels were decreased by Ins, but not by the IGF1R inhibitor or Ben (Figure 7A–C). The IGF1R inhibitor did not change the expression of SOCS2, while Ben up regulated SOCS2 mRNA but had no effect on the protein. Conversely, SOCS2 mRNA and protein levels showed a significant increase after Ins treatment (Figure 7A, D, and E).
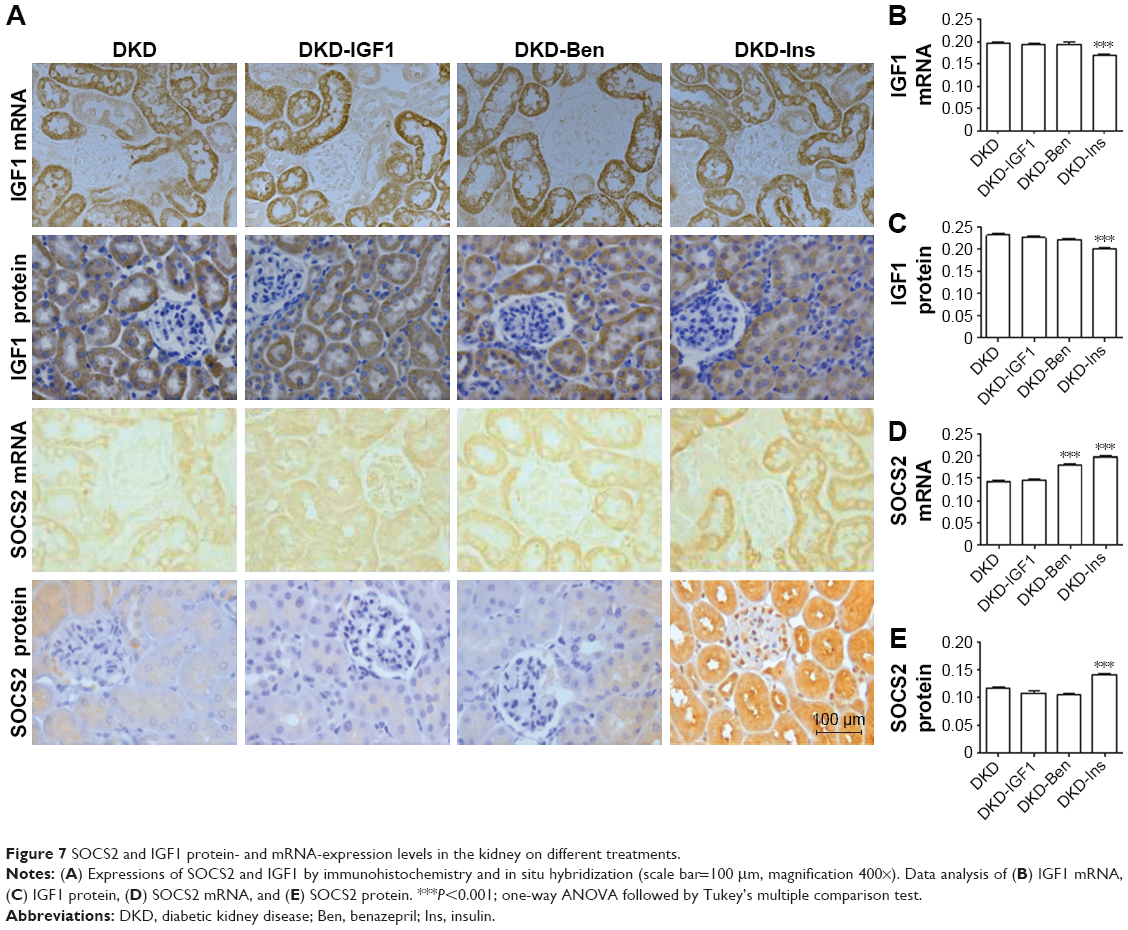 | Figure 7 SOCS2 and IGF1 protein- and mRNA-expression levels in the kidney on different treatments. |
Discussion
We used a high-fat diet with a low-dose Stz C57 mouse model, which is a good animal model to study the regulation of dietary intake and nutrient metabolism like obesity and Ins resistance in humans. It is the most commonly used model in type 2 diabetes research.16,17,20,21 In our model, the fasting blood glucose level of the Con group was 7.33 mmol/L and that of the DKD group was 23.14 mmol/L (Figure 1A), similar to other reports using the same model.16,17 This is a simple and effective mouse model for DKD study. Limitations of this model were Stz having potential for aspecific toxicity in the kidney and liver and Stz diabetic mice exhibiting significant weight loss, which may significantly affect mouse survival, and thus more experience is required to manage this model.22,23
In this report, the results showed that in DKD mice IGF1 was upregulated, accompanied by urinary albumin excretion rate. DKD mice showed inflammatory infiltration marked by CD68+ and F4/80+ cell increases and TLR4 overexpression, and fibrosis marked by fibrogenesis downregulation of E-cadherin and αSMA upregulation. These changes were reversed by the IGF1R inhibitor much better than Ben or Ins.
Currently, the treatment strategy for DKD consists mainly of antihypertensive measures by Ben and antihyperglycemia by Ins. Ben has a slight anti-inflammation effect through reducing the stimulation of inflammatory cytokines by lowering glomerular perfusion pressure and stimulating the secretion of the anti-inflammatory factor IL10.5,7 On the other hand, IL10 overexpression had the effect of stimulating inflammation, so the anti-inflammatory effect of Ben was not ideal. In this study, although Ben reduced αSMA effectively, its anti-inflammatory and antifibrotic effects were not as effective as with the IGF1 inhibitor (Figure 6).
Ins is another drug commonly used for DKD treatment by upregulating SOCS2 and downregulating IGF1.24–27 Several studies have shown that Ins can regulate the activity of IGF1 by regulating the expression of SOCS2 proteins,25,27 so as to reduce the inflammatory infiltration and fibrosis of tissue, thus protecting the tissue.26 At the same time, Ins can also lead to an increased expression of SOCS3 and IGF1R, which are likely to aggravate the inflammatory response.24 In vivo, high levels of Ins will increase SOCS protein expression, including SOCS1, SOCS2, SOCS3, and CIS.24,25 Complex regulation patterns in the body may be one of the factors behind Ins only delaying the progression of diabetic nephropathy, but not preventing ESKD.28–33
IGF1 has been reported to be related to local tissue hyperplasia, typical glomerular hypertrophy, vascular dilatation, and extracellular matrix protein increase in diabetic nephropathy.13 Our previous report demonstrated that IGF1R inhibitors regulated HMGN1 and TIL4 pathways in DKD.11 As such, we used an IGF1R inhibitor to confirm the effect on DKD further. In this study, the IGF1R inhibitor was more effective than Ins or Ben in reducing the progression of inflammation (Figures 5 and 6). Qu et al reported that IGF1R inhibitors had an anticancer effect without aggravating the imbalance of the IGF–GH axis.34 Suzuki et al reported that IGF1R inhibitors directly controlled abnormal IGF1R expression on the cell surface.35 In this report, we also found that SOCS2 expression was upregulated by Ins, but not by the IGF1R inhibitor (Figure 7). There have been no reports on the direct link between IGF1R inhibitors and SOCS protein. Isshiki et al reported that Ins reduced inflammation by upregulating SOCS2 which inhibited IGF1R activation.9 It might be that both IGF1R inhibitors and SOCS protein can inhibit IGF1R activation in vivo. We propose that the IGF1R inhibitor directly reduced the pathological effects of IGF1 in DKD mice without upregulating the SOCS pathway, and hence avoided the proinflammatory effect of SOCS, thus signifying potential long-term kidney protection.
Conclusion
In this study, we found that inhibition of IGF1R was a more effective choice for inflammation treatment than Ben or Ins in DKD. The IGF1R inhibitor blocked pathological changes induced by the overexpression of IGF1 in DKD without upregulating SOCS2 protein levels. This strategy may lead to new therapy to relieve DKD.
Acknowledgments
We extend our sincerest appreciation to Dr Yan Xiao for her expert input and technical assistance. This work was partly supported by grants from the National Natural Science Foundation of China (2017, 81760134), the Collaborative Fund of the Science and Technology Department of Guizhou Province and Guizhou Provincial People’s Hospital (20167169 and 20167170), and the Youth Foundation of Guizhou Provincial People’s Hospital (GZSYQN 201610).
Author contributions
JYL, YJL, YS, JJD and RD performed the experiments. All authors participated in experimental design and data analysis. JYL, RD, and FXY wrote the paper. YZ edited the final version. All authors contributed to revising the paper and agree to be accountable for all aspects of the work
Disclosure
The authors report no conflicts of interest in this work.
References
International Diabetes Federation. IDF Diabetes Atlas. 8th ed. Brussels: IDF; 2017. | ||
Feng X, Tang H, Leng J, Jiang Q. Suppressors of cytokine signaling (SOCS) and type 2 diabetes. Mol Biol Rep. 2014;41(4):2265–2274. | ||
Elrggal ME, Ahmed SM, el Nahas M. Renin-angiotensin-aldosterone system blockade in diabetic kidney disease: a critical and contrarian point of view. Saudi J Kidney Dis Transpl. 2016;27(6):1103. | ||
Lin YC, Chang YH, Yang SY, Wu KD, Chu TS. Update of pathophysiology and management of diabetic kidney disease. J Formos Med Assoc. Epub 2018 Feb 24. | ||
Niu H, Nie L, Liu M, et al. Benazepril affects integrin-linked kinase and smooth muscle α-actin expression in diabetic rat glomerulus and cultured mesangial cells. BMC Nephrol. 2014;15:135. | ||
Jia ZH, Liu ZH, Zheng JM, Zeng CH, Li LS. Combined therapy of rhein and benazepril on the treatment of diabetic nephropathy in db/db mice. Exp Clin Endocrinol Diabetes. 2007;115(9):571–576. | ||
Peng T, Wang J, Zhen JH, Hu Z, Yang XD. Effect of benazepril on the transdifferentiation of renal tubular epithelial cells from diabetic rats. Biomed Rep. 2014;2(4):490–494. | ||
Jin H, Piao SG, Jin JZ, et al. Synergistic effects of leflunomide and benazepril in streptozotocin-induced diabetic nephropathy. Nephron Exp Nephrol. 2014;126(3):148–156. | ||
Isshiki K, He Z, Maeno Y, et al. Insulin regulates SOCS2 expression and the mitogenic effect of IGF-1 in mesangial cells. Kidney Int. 2008;74(11):1434–1443. | ||
Donath MY. Targeting inflammation in the treatment of type 2 diabetes. Diabetes Obes Metab. 2013;15(Suppl 3):193–196. | ||
Yu JL, Da JJ, Dong R, et al. IGF-1R inhibitor ameliorates diabetic nephropathy with suppressed HMGN1/TLR4 pathway. Endocr Metab Immune Disord Drug Targets. 2018;18(3):241–250. | ||
Nambam B, Schatz D. Growth hormone and insulin-like growth factor-I axis in type 1 diabetes. Growth Horm IGF Res. 2018;38:49–52. | ||
Bach LA, Hale LJ. Insulin-like growth factors and kidney disease. Am J Kidney Dis. 2015;65(2):327–336. | ||
Rajpathak SN, Gunter MJ, Wylie-Rosett J, et al. The role of insulin-like growth factor-I and its binding proteins in glucose homeostasis and type 2 diabetes. Diabetes Metab Res Rev. 2009;25(1):3–12. | ||
Yee D. Anti-insulin-like growth factor therapy in breast cancer. J Mol Endocrinol. 2018;61(1):T61–T68. | ||
Wang HJ, Jin YX, Shen W, et al. Low dose streptozotocin (STZ) combined with high energy intake can effectively induce type 2 diabetes through altering the related gene expression. Asia Pac J Clin Nutr. 2007;16(Suppl 1):412–417. | ||
Islam MS, Choi H. Nongenetic model of type 2 diabetes: a comparative study. Pharmacology. 2007;79(4):243–249. | ||
Han HI, Skvarca LB, Espiritu EB, Davidson AJ, Hukriede NA. The role of macrophages during acute kidney injury: destruction and repair. Pediatr Nephrol. Epub 2018 Jan 30. | ||
Sabbatini P, Rowand JL, Groy A, et al. Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin-like growth factor-I receptor tyrosine kinase. Clin Cancer Res. 2009;15(9):3058–3067. | ||
Zhang F, Ye C, Li G, et al. The rat model of type 2 diabetic mellitus and its glycometabolism characters. Exp Anim. 2003;52(5):401–407. | ||
Reed MJ, Meszaros K, Entes LJ, et al. A new rat model of type 2 diabetes: the fat-fed, streptozotocin-treated rat. Metabolism. 2000;49(11):1390–1394. | ||
Zheng G, Mo F, Ling C, et al. Portulaca oleracea L. alleviates liver injury in streptozotocin-induced diabetic mice. Drug Des Devel Ther. 2018;12:47–55. | ||
Breyer MD, Bottinger E, Brosius FC, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16(1):27–45. | ||
Sadowski CL, Choi TS, Le M, Wheeler TT, Wang LH, Sadowski HB. Insulin induction of SOCS-2 and SOCS-3 mRNA expression in C2C12 skeletal muscle cells is mediated by Stat5. J Biol Chem. 2001;276(23):20703–20710. | ||
Galic S, Sachithanandan N, Kay TW, Steinberg GR. Suppressor of cytokine signalling (SOCS) proteins as guardians of inflammatory responses critical for regulating insulin sensitivity. Biochem J. 2014;461(2):177–188. | ||
Zhou Y, Lv C, Wu C, et al. Suppressor of cytokine signaling (SOCS) 2 attenuates renal lesions in rats with diabetic nephropathy. Acta Histochem. 2014;116(5):981–988. | ||
Lebrun P, Cognard E, Gontard P, et al. The suppressor of cytokine signalling 2 (SOCS2) is a key repressor of insulin secretion. Diabetologia. 2010;53(9):1935–1946. | ||
Sheng M, Huang Z, Pan L, et al. SOCS2 exacerbates myocardial injury induced by ischemia/reperfusion in diabetic mice and H9c2 cells through inhibiting the JAK-STAT-IGF-1 pathway. Life Sci. 2017;188:101–109. | ||
Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol. 2002;2(6):410–416. | ||
al-Shanti N, Stewart CE. Inhibitory effects of IL-6 on IGF-1 activity in skeletal myoblasts could be mediated by the activation of SOCS-3. J Cell Biochem. 2012;113(3):923–933. | ||
Metcalf D, Greenhalgh CJ, Viney E, et al. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature. 2000;405(6790):1069–1073. | ||
Farquharson C, Ahmed SF. Inflammation and linear bone growth: the inhibitory role of SOCS2 on GH/IGF-1 signaling. Pediatr Nephrol. 2013;28(4):547–556. | ||
Othman EM, Altabaa T, Hintzsche H, Stopper H. IR and IGF-1R expression affects insulin induced proliferation and DNA damage. Toxicol In Vitro. 2017;39:68–74. | ||
Qu X, Wu Z, Dong W, et al. Update of IGF-1 receptor inhibitor (ganitumab, dalotuzumab, cixutumumab, teprotumumab and figitumumab) effects on cancer therapy. Oncotarget. 2017;8(17):29501–29518. | ||
Suzuki S, Morimoto S, Fujishiro M, et al. Inhibition of the insulin-like growth factor system is a potential therapy for rheumatoid arthritis. Autoimmunity. 2015;48(4):251–258. | ||
Dekker FW, Jager KJ, Noordzij M, et al. Sample size calculations. Nephron Clin Pract. 2011;118(4):319–323. | ||
Thibodeau JF, Holterman CE, Burger D, et al. A novel mouse model of advanced diabetic kidney disease. PLoS One. 1932;9(12):e113459. | ||
Baribault H. Mouse models of type II diabetes mellitus in drug discovery. Methods Mol Biol. 2013;602:135–155. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.