")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Inhibition of sialidase activity as a therapeutic approach
Authors Glanz VY , Myasoedova VA , Grechko AV, Orekhov AN
Received 4 June 2018
Accepted for publication 21 August 2018
Published 10 October 2018 Volume 2018:12 Pages 3431—3437
DOI https://doi.org/10.2147/DDDT.S176220
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Qiongyu Guo
Victor Yu Glanz,1 Veronika A Myasoedova,2 Andrey V Grechko,3 Alexander N Orekhov2,4
1Department of Genetics, Cytology and Bioengineering, Faculty of Biology and Medicine, Voronezh State University, Voronezh, Russia; 2Laboratory of Angiopathology, Institute of General Pathology and Pathophysiology, Moscow, Russia; 3Federal Research and Clinical Center of Intensive Care Medicine and Rehabilitology, Moscow, Russia; 4Institute for Atherosclerosis Research, Skolkovo Innovative Center, Moscow, Russia
Abstract: The demand for novel anti-influenza drugs persists, which is highlighted by the recent pandemics of influenza affecting thousands of people across the globe. One of the approaches to block the virus spreading is inhibiting viral sialidase (neuraminidase). This enzyme cleaves the sialic acid link between the newly formed virions and the host cell surface liberating the virions from the cell and maintaining the cycle of infection. Viral neuraminidases appear therefore as attractive therapeutic targets for preventing further spread of influenza infection. Compared to ion channel blockers that were the first approved anti-influenza drugs, neuraminidase inhibitors are well tolerated and target both influenza A and B viruses. Moreover, neuraminidase/sialidase inhibitors may be useful for managing some other human pathologies, such as cancer. In this review, we discuss the available knowledge on neuraminidase or sialidase inhibitors, their design, clinical application, and the current challenges.
Keywords: sialidase, neuraminidase, neuraminidase inhibitor, influenza, drug design
Introduction
Sialic acids are frequently present in the cell surface glycoconjugates as terminal residues. In mammals, the two most frequent types of these monosaccharides are N-acetylneuraminic acid (Neu5Ac) and its hydroxylated version Neu5Gc. Sialic acids convey the negative electric charge to the cell surface and are vitally involved in many biological processes at the cell membrane, including transmembrane signaling.1 Sialic acid residues can be removed from the polysaccharide chains by the enzymes called sialidases (also known as neuraminidases). There are four types of mammalian neuraminidases: NEU1, NEU2, NEU3, and NEU4. Neuraminidases are also frequently present in microorganisms and viruses that need them for interaction with host mammalian cells and tissues and propagation of infection. Mammalian and microbial and viral neuraminidases have different sensitivities to neuraminidase inhibitors. For instance, anti-influenza drugs Tamiflu (oseltamivir) and Relenza (zanamivir), which are characterized by demonstrated clinical efficacy, have almost no effect on human neuraminidases in vitro. Although general neuraminidase inhibitors are known, such as sialic acid analog 2-deoxy-2,3-dehydro-N-acetylneuraminic acid (DANA or Neu5Ac2en), in most cases, the difference in sensitivity to inhibitors between host and pathogen neuraminidases exceeds 10 times. This difference makes it possible to use viral and bacterial neuraminidase inhibitors for therapeutic purposes to fight infections.2
Mammalian neuraminidases are involved in many cellular processes and may serve as potential therapeutic targets. Modulation or inhibition of neuraminidase activity may prove useful for the development of novel therapeutic and diagnostic strategies. For instance, various neoplasms share a common feature of increased level of sialylation of surface glycoconjugates, which is explained by reduced sialidase activity and increased sialyltransferase activity in cancer cells.3 More studies are needed, however, to understand the mechanism of the sialylation-related changes in different cancers and to use these changes for diagnostic and therapeutic purposes.
The role of neuraminidases in viral and bacterial life cycles is currently characterized in more detail. Because of their tight involvement in the host–pathogen interaction and infection progression and their distinction from mammalian neuraminidases, these enzymes became targets of choice for developing novel drugs. In influenza virus, two of at least 10 viral proteins encoded in a segmented RNA genome are hemagglutinin and neuraminidase, which are crucial for virus entry and release, and also determine the virus subtype.4 Influenza neuraminidase is necessary for the release of the newly formed virions from the host cell and therefore for the continuation of the infection cycle. Blocking of this viral enzyme allows restraining the ongoing infection and is a promising therapeutic strategy, thanks to relatively high sensitivity of viral neuraminidase to available inhibitors. Despite its susceptibility to mutations, influenza neuraminidase currently remains the most promising drug target for treatment of influenza infection.
Design and discovery of sialidase inhibitors for drug development
First attempts to design virus-specific neuraminidase inhibitors were based on the available knowledge of viral enzyme functions and mechanisms of substrate interaction.5 The first generation of inhibitory assays developed for searching for potential inhibitors was using phenyl-α-ketoside of Neu5Ac or fetuin as substrates for measuring the enzyme activity. These inhibitory assays allowed discovering specific synthetic neuraminidase inhibitors such as DANA and 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid (FANA). More recent studies used computer-aided structure-based design that provided unparalleled computational power and broadened the opportunities for drug discovery.6 This method helped identifying anti-influenza drugs that could later be introduced in clinical practice: a highly active N-acetyl-α-D-neuraminic acid (NANA) derivative zanamivir (Relenza) and oseltamivir (Tamiflu). Neuraminidase inhibitory assays based on fluorescence or chemiluminescence are also widely used for identifying novel inhibiting compounds.7
Owing to the high rate of mutations occurring during viral genome replication, viral enzymes are characterized by relatively high variability that can convey drug resistance. Viral neuraminidase inhibitors aimed to overcome such mutation-induced resistance target, the so-called 150-cavity of the enzyme, which is adjacent to the drug-binding site. This approach and identification of novel inhibitors have been described recently.8 The design of novel inhibitors implemented a virtual screening of the National Cancer Institute (NCI) database compounds followed by establishing a site-moiety map for studying the binding site of dual H274Y/I222R mutant neuraminidase. Overall study framework consisted of virtual screening for inhibitors, modeling consensus interactions between docked compounds and amino acids of the enzyme, building site-moiety map to identify potential binding pockets, and listing the potential inhibitors. The identified potential inhibitors were validated using cellular and enzymatic assays. The authors concluded that exploiting the 150-cavity to form an open conformation of the enzyme is beneficial for neuraminidase inhibition.8
All currently used viral neuraminidase inhibitors contain an anomeric carboxy group, which forms electrostatic bonds with amino acids in the active center of the enzyme. It has been suggested that sialic acid anomeric sulfonic acid analogs may prove to be more potent inhibiting compounds. This suggestion was based on the prediction that sulfo groups due to their high acidity and electronegativity should form strong bonds with the amino acids in the neuraminidase active center.9 Modeling of the interactions of newly designed compounds with the enzyme active center was performed using the Molecular Operating Environment software. The sulfo-sialic acid analogs were synthesized via oxidation of a mixture of acetylthio intermediates. As a result, 2-decarboxy-2-deoxy-2-sulfo-N-acetylneuraminic acid and its 4-deoxy-3,4-dehydrogenated pseudoglycal were developed and tested on influenza A/Anhui/1/2005 (H5N1) NA (N1), A/RI/5+/1957 (H2N2) NA (N2), Clostridium perfringens NanJ NA (CpNA), and Streptococcus 6646K NA (StrepNA). These compounds demonstrated a superior inhibitory activity on influenza and bacterial neuraminidases. Moreover, they may potentially be active already at sub-nanomolar levels, while the neuraminidase inhibitors currently used in clinical practice inhibit influenza neuraminidase at a low nanomolar level.9
Effective concentration is one of the most important characteristics of newly designed neuraminidase-inhibiting compounds. Bicyclic analog of sialic acid was designed to mimic the conformation of the neuraminidase active site corresponding to the enzymatic cleavage of the substrate. However, the compounds initially failed to demonstrate convincing results due to flaws in the synthesis process. This led to the development of a novel, simplified synthetic route, which started from cyclopentenone cyclopropanation followed by aziridination to achieve the common precursor that was then functionalized with various ether side chains. Despite the achieved advances, none of these new compounds was efficient against influenza A neuraminidase at concentrations <2 mM.10
Currently, such methods as in silico screening, computational docking studies, and machine-learning algorithms are the methods of choice for identifying and characterizing novel neuraminidase-inhibiting compounds. It is important to characterize the novel neuraminidase inhibitors in terms of synergistic effect between them and marketed drugs to achieve a prolonged inhibitory action, which would greatly expand their application.11 Synergetic effects can take place when neuraminidase inhibitors are used with some other antiviral drugs. An example of such synergy was observed between nitazoxanide and oseltamivir applied against A/Puerto Rico/8/1934 (H1N1) and A/WSN/1933 (H1N1) influenza A viruses in vitro.12
Machine-learning algorithms are widely and successfully used in different aspects of pharmaceutical research. Recently, a machine-learning-based scoring function (RF-NA-Score) was used for virtual screening of lead compounds targeting the viral neuraminidase tested on a dataset of 281 known neuraminidase inhibitors and 322 non-inhibitors. The algorithm used 67 viral neuraminidase–ligand complexes and their experimental binding affinities obtained from the PDBbind database as the training input. As a result, two compounds, AH-034/11365875 and AH-262/08373040, were identified and proposed as lead components for developing novel anti-influenza drugs. This approach appears to be promising, despite its known limitations.13
Another strategy in the search for novel neuraminidase inhibitors is screening the compounds of natural origin, many of which demonstrate some potency.7,14,15 Development and optimization of screening methods allow for rapid analysis of large numbers of substances. For instance, a recent study proposed an innovative approach of using magnetic beads with immobilized neuraminidase for screening compound libraries and natural extracts for potential neuraminidase inhibitors and identified a number of inhibitors using this method.16 The first step in the search for novel potential drugs is often a simple screening of some promising sources, such as traditional medicinal plants.17,18 The identified compounds that often possess only limited neuraminidase-blocking activity can be further used as base molecules for design of novel potent inhibitors.19,20 However, the occurrence of naturally present neuraminidase inhibitors is not limited to plants. A specific neuraminidase inhibitor was found in mouse saliva, which can possibly explain the insusceptibility of the species to influenza infection in the wild. This finding may provide some insight into a novel antiviral defense mechanism.21 Moreover, the very existence of natural neuraminidase inhibitors might be regarded as an evidence highlighting the significance of this strategy to prevent viral infection.
Neuraminidase inhibitors for controlling of influenza infection
Influenza viruses belong to Orthomyxoviridae family. The influenza A virus genome encodes, apart from hemagglutinin and neuraminidase, ion channel (M2), matrix protein (M), nucleoprotein (NP), RNA polymerase components, and nonstructural proteins. Influenza viruses can be classified based on different antigens of M and NP into A, B, and C types. Influenza A viruses are the most dangerous for humans. The genetic variability of influenza viruses is caused by the frequent occurrence of point mutations in the hemagglutinin and neuraminidase genes and by occasional genetic rearrangements between different viruses from humans and animals. Such variability is an important mechanism of drug resistance and can also increase the virulence of the pathogen resulting in pandemics.22
As from May 2018, according to the World Health Organization (WHO), a total of 1,567 laboratory-confirmed cases of human infection with avian influenza A (H7N9) viruses have been reported, with at least 615 lethal cases.23 The infection is heavily dependent on viral hemagglutinin and neuraminidase proteins that are necessary for binding to the cell surface via terminal sialic acid residues of cellular glycoproteins and glycolipids (Figure 1). Neuraminidase then cleaves the sialic acid linking the viral hemagglutinin and cell surface glycans, liberating newly formed virions and maintaining the cycle of infection.24 Viruses with a low neuraminidase enzymatic activity have a low virulence and are not effectively transmitted by respiratory droplets.25
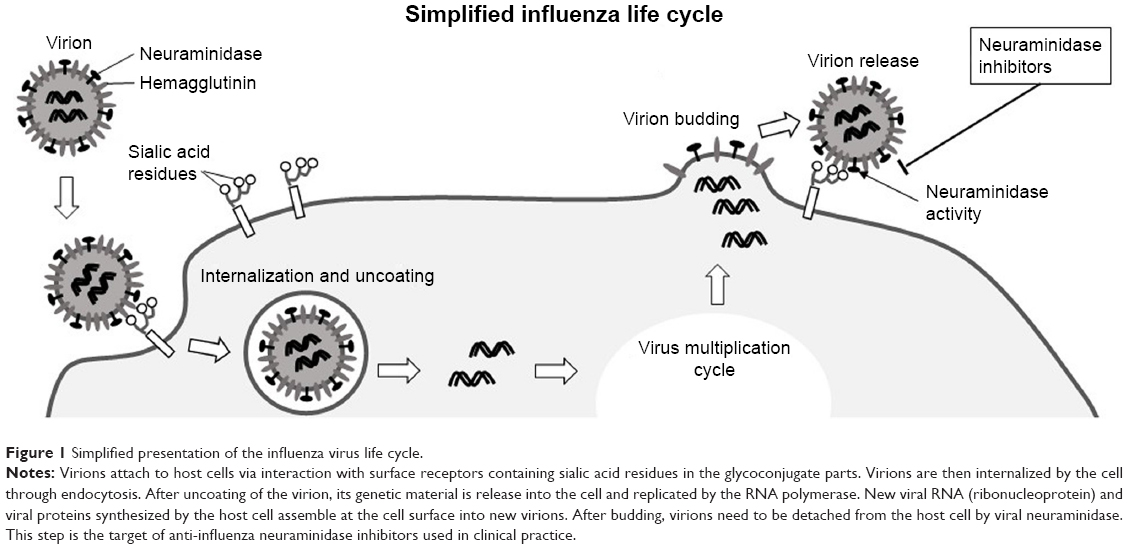 | Figure 1 Simplified presentation of the influenza virus life cycle. |
Vaccination remains the primary tool to prevent influenza infection and to manage the emerging epidemics and pandemics, while antiviral drugs provide a rescue option.26 The available antiviral drugs for treatment of influenza A infection include two main classes: adamantanes, such as amantadine and rimantadine, and neuraminidase inhibitors, such as oseltamivir, zanamivir, laninamivir, and peramivir.27
Structurally, influenza neuraminidases can be divided into two phylogenetic subtypes: group 1 (N1, N4, N5, and N8) and group 2 (N2, N3, N6, N7, and N9), with group 1 possessing the 150-cavity, which can have open and closed conformations and demonstrates a high sequence similarity between group 1 and group 2.28
Both viral neuraminidase and hemagglutinin bind to the terminal sialic acid residues of the host cell surface glycocalyx. Binding of hemagglutinin to sialic acid is necessary for viral internalization by the host cell. Neuraminidase also binds sialic acid and cleaves the α-(2,3) and α-(2,6) glycosidic bonds of the terminal residues, allowing the newly formed virion to detach from the host cell and preventing self-aggregation of the viral particles. Therefore, neuraminidase plays a key role in virus spread and infection propagation and represents an attractive therapeutic target. Three inhibitors have been developed based on sialic acid: zanamivir; its ethyl ester derivative oseltamivir; characterized by improved bioavailability; and peramivir, which was first used for treatment of hospitalized and pediatric patients.7 All neuraminidase inhibitors were shown to be effective only if administered no later than 36–48 hours after first manifestation of the symptoms.7 According to a recent estimation, ~99% of seasonal influenza viruses are sensitive to all licensed inhibitors, thus making these drugs an appropriate choice for influenza treatment.29 Current basic knowledge of common anti-influenza neuraminidase inhibitors is summarized in Table 1 and discussed in more detail in recent publications.30
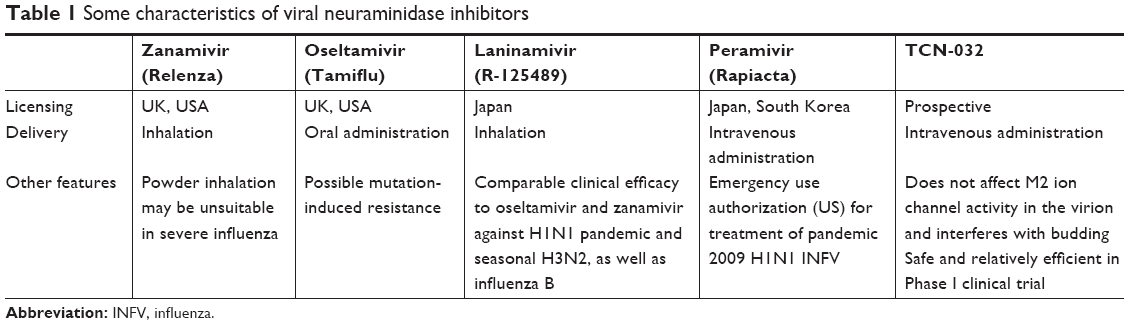 | Table 1 Some characteristics of viral neuraminidase inhibitors |
Alternative use of sialidase inhibitors
Since neuraminidases are produced by both viral and bacterial pathogens, beneficial effects of neuraminidase inhibitors for managing bacterial infections can be expected. This was the case with characterized substrate specificity of pneumococcal sialidases NanA, NanB, and NanC.31 Zanamivir and oseltamivir display either weak, as in case of zanamivir, or medium, as in case of oseltamivir, inhibitory effect on pneumococcal neuraminidase. The first report of efficient neuraminidase-inhibiting activity reducing bacterial adherence to pulmonary epithelial cells and hampering biofilm formation and bacterial growth and viability was published in 2015. In this work, artocarpin was described as both antipneumococcal and neuraminidase inhibitory compound.32 Potential therapeutic applications of the selective human NEU1 sialidase inhibitor C9-butyl-amide-2-deoxy-2,3-dehydro-N-acetylneuraminic acid (C9-BA-DANA) were explored in relation to treating sepsis, atherosclerotic arterial disease, idiopathic pulmonary fibrosis, and other human pathologies in a recent study.33 However, the authors highlighted the importance of selective approach, being aware of possible unpredictable effects of antibacterial and antiviral neuraminidase inhibitors’ administration on host–pathogen and pathogen–pathogen interactions.
Neuraminidase inhibitors might also be useful beyond the area of infectious diseases. Accumulating evidence highlights their potential beneficial effects in treatment of some cancers and in other human pathologies. For instance, study of a novel GPCR–MMP9–NEU1 signaling pathway, supposedly crucial for tumor progression, indicates that the therapeutic efficacy of oseltamivir phosphate targeting NEU1 is able to limit the tumor ability to form metastases.34 Therefore, NEU1 appears to be a novel potential therapeutic target for preventing tumor neovascularization, growth, metastases, and macrophage-mediated tumorigenesis. Inhibition of neuraminidases with N-acetyl-2,3-dehydro-2-deoxyneuraminic acid and oseltamivir could alleviate pulmonary fibrosis in a mouse model.35 Interestingly, the proposed pharmacological intervention may theoretically be beneficial for wound healing, since fibrosis has common features with the process of scar tissue formation. Importantly, neuraminidase inhibitors appear to be safe enough to be used during pregnancy, since there are no known associated increased risks of adverse fetal or neonatal outcomes.36
Challenges and future directions
There is an ongoing debate regarding the value of using neuraminidase inhibitors for reducing clinical outcomes of influenza, since the strongest evidence of their efficiency comes from large observational cohort-based studies of pandemic influenza A treatment and not from placebo-controlled randomized controlled trials.37 There is evidence of oseltamivir phosphate inhibiting mammalian neuraminidases, thus altering tumor sialylation state and leading to in vitro and in vivo increased canine mammary tumor aggressiveness.38 Furthermore, the available data on licensed inhibitors activity were reassessed. As a result, the position of oseltamivir in the list of essential medicines was changed from “core” to “complementary” by the WHO in 2017.39,40 Neuraminidase inhibitors are not free from side effects, such as nausea, vomiting, dizziness, sinusitis, runny or stuffy nose, cough, diarrhea, and headache.5
The interpretation of results of culture-based antiviral assays is complicated by several factors specific to neuraminidase inhibitors:
- balanced function of hemagglutinin and neuraminidase determines the results’ validity;
- possible negative effect of the receptor expression pattern of test cells; and
- changes in the efficiency of binding of hemagglutinin and sialic acid.7
Therefore, successful application of cell culture-based assays with approved inhibitors in antiviral research requires a careful evaluation.
Another current challenge is the possibility of acquisition of drug resistance by influenza viruses and the appearance of strains with reduced sensitivity to clinically used neuraminidase inhibitors. Constant monitoring is needed to identify the appearance of resistant strains and assess the risks.41 The potential risk of oseltamivir-induced resistance for influenza H1N1 and H3N2 viruses was assessed in a recent work.40 The authors highlighted two key factors affecting the epidemiology of drug resistance: the rate of drug resistance development in treated individuals and the fitness cost of resistance associated with mutation rate. The obtained results can be applied to detecting an epidemic outbreak at an early stage and controlling it, as well as to preventing the emergence and spread of drug resistance. Overall resistance in H1N1 and H5N1 virus subtypes is considered to be a result of loss of hydrophobicity within the binding pocket of neuraminidase active site, which leads to structural collapse of the available active site.42 Moreover, a substantial number of patients may develop resistance to oseltamivir as a result of its use, but resistance to zanamivir occurs only rarely.43
Current challenges in design and clinical implementation of neuraminidase inhibitors derive not only from the nature of inhibiting compounds but also from the properties of the target enzyme itself. This becomes particularly evident in case of human parainfluenza viruses, a leading cause of lower respiratory tract disease in children, with no available approved drug or vaccine.44 Study of recently designed and synthesized 4-deoxy-4-triazolo-Neu2en-based inhibitors revealed an interesting relationship between the size of the inhibitory molecule and its capacity to inhibit neuraminidase without altering the conformation of its active site. Slight structural changes in one part of the protein have a significant influence on other regions. There is still a need for optimal anti-parainfluenza virus drug design that would account for hPIV-3 neuraminidase protein flexibility and inhibitor-induced structural rearrangements in the protein. Moreover, this effect should be considered when targeting multiple individual structural features, since the expected synergistic effect may not be achieved.44
A simple mathematical model used to study the inhibition of virus release by sialidase inhibitors was modified to overcome the challenge of including the virus release in the model.45 To cope with overparameterization resulting from the addition of an explicit release rate to the model, the study considered a range of possible values of the release rate of influenza A virus. The simple model was compared against its variation that included an explicit term for virus release. The developed variant of the simple model showed that neglecting virus release with an explicit release term affects parameter and sialidase inhibitor efficacy estimation. It is therefore necessary to estimate the rate of influenza A virus release to interpret the results obtained from the simple mathematical model correctly. However, the drawback of the proposed strategy is that direct measurement of the virus release rate is difficult.45 Finally, the rate of viral resistance emergence in influenza patients remains to be determined.46
Conclusion
Identification of novel sialidase inhibitors is an urgent task of modern medicine accentuated by the need for treatment of constantly evolving and adapting influenza infection. However, viral drug resistance is not the only challenge impeding the progress in this field. Several factors, such as drug effective dose, synergistic or antagonistic relations with other drugs, safety, and patient’s resistance must be considered. Currently, zanamivir, oseltamivir, laninamivir, and peramivir are licensed in different countries as medications targeting viral sialidase. Some of these molecules have effects on mammalian cells as well, thus requiring further investigation of their metabolic involvement. With the rapid development of computational methods and implementing machine-learning algorithms, it is now possible to search for novel neuraminidase-inhibiting compounds at a larger scale. The pool of novel and promising synthetic compounds is being complemented with naturally occurring sialidase inhibitors, further emphasizing the importance of studying the structure, molecular basis of catalysis, and disease-associated changes in activity of sialidase.
Acknowledgment
This work was supported by Russian Science Foundation (Grant No 18-15-00254).
Disclosure
The authors report no conflicts of interest in this work.
References
Samraj AN, Läubli H, Varki N, Varki A. Involvement of a non-human sialic Acid in human cancer. Front Oncol. 2014;4:33. | ||
Miyagi T, Yamaguchi K. Mammalian sialidases: physiological and pathological roles in cellular functions. Glycobiology. 2012;22(7):880–896. | ||
Zhang Z, Wuhrer M, Holst S. Serum sialylation changes in cancer. Glycoconj J. 2018;35(2):139–160. | ||
Cline TD, Beck D, Bianchini E. Influenza virus replication in macrophages: balancing protection and pathogenesis. J Gen Virol. 2017; 98(10):2401–2412. | ||
von Itzstein M, Wu WY, Kok GB, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363(6428):418–423. | ||
Mallipeddi PL, Kumar G, White SW, Webb TR. Recent advances in computer-aided drug design as applied to anti-influenza drug discovery. Curr Top Med Chem. 2014;14(16):1875–1889. | ||
Grienke U, Schmidtke M, von Grafenstein S, Kirchmair J, Liedl KR, Rollinger JM. Influenza neuraminidase: a druggable target for natural products. Nat Prod Rep. 2012;29(1):11–36. | ||
Hsu KC, Hung HC, HuangFu WC, et al. Identification of neuraminidase inhibitors against dual H274Y/I222R mutant strains. Sci Rep. 2017;7(1):12336. | ||
Vavricka CJ, Muto C, Hasunuma T, et al. Synthesis of Sulfo-Sialic Acid Analogues: Potent Neuraminidase Inhibitors in Regards to Anomeric Functionality. Sci Rep. 2017;7(1):8239. | ||
Colombo C, Podlipnik Č, Lo Presti L, Niikura M, Bennet AJ, Bernardi A. Design and synthesis of constrained bicyclic molecules as candidate inhibitors of influenza A neuraminidase. PLoS One. 2018;13(2):e0193623. | ||
Chamni S, De-Eknamkul W. Recent progress and challenges in the discovery of new neuraminidase inhibitors. Expert Opin Ther Pat. 2013;23(4):409–423. | ||
Belardo G, Cenciarelli O, La Frazia S, Rossignol JF, Santoro MG. Synergistic effect of nitazoxanide with neuraminidase inhibitors against influenza A viruses in vitro. Antimicrob Agents Chemother. 2015;59(2):1061–1069. | ||
Zhang L, Ai HX, Li SM, et al. Virtual screening approach to identifying influenza virus neuraminidase inhibitors using molecular docking combined with machine-learning-based scoring function. Oncotarget. 2017;8(47):83142–83154. | ||
Ikram NK, Durrant JD, Muchtaridi M, et al. A virtual screening approach for identifying plants with anti H5N1 neuraminidase activity. J Chem Inf Model. 2015;55(2):308–316. | ||
Zhu Q, Bang TH, Ohnuki K, Sawai T, Sawai K, Shimizu K. Inhibition of neuraminidase by Ganoderma triterpenoids and implications for neuraminidase inhibitor design. Sci Rep. 2015;5:13194. | ||
Zhao YM, Wang LH, Luo SF, et al. Magnetic beads-based neuraminidase enzyme microreactor as a drug discovery tool for screening inhibitors from compound libraries and fishing ligands from natural products. J Chromatogr A. 2018;1568:123–130. | ||
Liu J, Zu M, Chen K, et al. Screening of neuraminidase inhibitory activities of some medicinal plants traditionally used in Lingnan Chinese medicines. BMC Complement Altern Med. 2018;18(1):102. | ||
Liu F, Cao W, Deng C, Wu Z, Zeng G, Zhou Y. Polyphenolic glycosides isolated from Pogostemon cablin (Blanco) Benth as novel influenza neuraminidase inhibitors. Chem Cent J. 2016;10:51. | ||
Enkhtaivan G, Muthuraman P, Kim DH, Mistry B. Discovery of berberine based derivatives as anti-influenza agent through blocking of neuraminidase. Bioorg Med Chem. 2017;25(20):5185–5193. | ||
Ide K, Kawasaki Y, Kawakami K, Yamada H. Anti-influenza virus effects of catechins: A molecular and clinical review. Curr Med Chem. 2016;23(42):4773–4783. | ||
Gilbertson B, Ng WC, Crawford S, McKimm-Breschkin JL, Brown LE. Mouse Saliva Inhibits Transit of Influenza Virus to the Lower Respiratory Tract by Efficiently Blocking Influenza Virus Neuraminidase Activity. J Virol. 2017;91(14):e00145-17. | ||
Cheng CK, Tsai CH, Shie JJ, Fang JM. From neuraminidase inhibitors to conjugates: a step towards better anti-influenza drugs? Future Med Chem. 2014;6(7):757–774. | ||
World Health Organization. Influenza at the human-animal interface. Summary and assessment, 26 January to 2 March 2018;2018:1–5. Available from: http://www.who.int/entity/influenza/human_animal_interface/Influenza_Summary_IRA_HA_interface_09_27_2017.pdf?ua=1. Accessed April 1, 2018. | ||
Nikolaidis NM, Noel JG, Pitstick LB, et al. Mitogenic stimulation accelerates influenza-induced mortality by increasing susceptibility of alveolar type II cells to infection. Proc Natl Acad Sci U S A. 2017;114(32):E6613–E6622. | ||
Zanin M, Duan S, Wong SS, et al. An Amino Acid in the Stalk Domain of N1 Neuraminidase Is Critical for Enzymatic Activity. J Virol. 2017;91(2):e00868-16. | ||
Burnham AJ, Baranovich T, Govorkova EA. Neuraminidase inhibitors for influenza B virus infection: efficacy and resistance. Antiviral Res. 2013;100(2):520–534. | ||
McKimm-Breschkin JL. Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance. Influenza Other Respir Viruses. 2013;7(Suppl 1):25–36. | ||
Kerry PS, Mohan S, Russell RJ, Bance N, Niikura M, Pinto BM. Structural basis for a class of nanomolar influenza A neuraminidase inhibitors. Sci Rep. 2013;3:2871. | ||
Gubareva LV, Besselaar TG, Daniels RS, et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2015–2016. Antiviral Res. 2017;146:12–20. | ||
Tyrrell BE, Sayce AC, Warfield KL, Miller JL, Zitzmann N. Iminosugars: Promising therapeutics for influenza infection. Crit Rev Microbiol. 2017;43(5):521–545. | ||
McCombs JE, Diaz JP, Luebke KJ, Kohler JJ. Glycan specificity of neuraminidases determined in microarray format. Carbohydr Res. 2016;428:31–40. | ||
Walther E, Richter M, Xu Z, et al. Antipneumococcal activity of neuraminidase inhibiting artocarpin. Int J Med Microbiol. 2015;305(3):289–297. | ||
Hyun SW, Liu A, Liu Z, et al. The NEU1-selective sialidase inhibitor, C9-butyl-amide-DANA, blocks sialidase activity and NEU1-mediated bioactivities in human lung in vitro and murine lung in vivo. Glycobiology. 2016;26(8):834–849. | ||
Haxho F, Neufeld RJ, Szewczuk MR. Neuraminidase-1: a novel therapeutic target in multistage tumorigenesis. Oncotarget. 2016;7(26):40860–40881. | ||
Karhadkar TR, Pilling D, Cox N, Gomer RH. Sialidase inhibitors attenuate pulmonary fibrosis in a mouse model. Sci Rep. 2017;7(1):15069. | ||
Graner S, Svensson T, Beau AB, et al. Neuraminidase inhibitors during pregnancy and risk of adverse neonatal outcomes and congenital malformations: population based European register study. BMJ. 2017;356:j629. | ||
Bradbury N, Nguyen-Van-Tam J, Lim WS. Clinicians’ attitude towards a placebo-controlled randomised clinical trial investigating the effect of neuraminidase inhibitors in adults hospitalised with influenza. BMC Health Serv Res. 2018;18(1):311. | ||
de Oliveira JT, Santos AL, Gomes C, et al. Anti-influenza neuraminidase inhibitor oseltamivir phosphate induces canine mammary cancer cell aggressiveness. PLoS One. 2015;10(4):e0121590. | ||
Ebell MH. WHO downgrades status of oseltamivir. BMJ. 2017;358:j3266. | ||
Hsieh NH, Lin YJ, Yang YF, Liao CM. Assessing the oseltamivir-induced resistance risk and implications for influenza infection control strategies. Infect Drug Resist. 2017;10:215–226. | ||
Lackenby A, Besselaar TG, Daniels RS, et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors and status of novel antivirals, 2016–2017. Antiviral Res. 2018;157:38–46. | ||
Singh A, Soliman ME. Understanding the cross-resistance of oseltamivir to H1N1 and H5N1 influenza A neuraminidase mutations using multidimensional computational analyses. Drug Des Devel Ther. 2015;9:4137–4154. | ||
Thorlund K, Awad T, Boivin G, Thabane L. Systematic review of influenza resistance to the neuraminidase inhibitors. BMC Infect Dis. 2011;11:134. | ||
Dirr L, El-Deeb IM, Chavas LMG, Guillon P, Itzstein MV. The impact of the butterfly effect on human parainfluenza virus haemagglutinin-neuraminidase inhibitor design. Sci Rep. 2017;7(1):4507. | ||
Liao LE, Kowal S, Cardenas DA, Beauchemin CAA. Exploring virus release as a bottleneck for the spread of influenza A virus infection in vitro and the implications for antiviral therapy with neuraminidase inhibitors. PLoS One. 2017;12(8):e0183621. | ||
Itoh Y, Shichinohe S, Nakayama M, et al. Emergence of H7N9 influenza A virus resistant to neuraminidase inhibitors in nonhuman primates. Antimicrob Agents Chemother. 2015;59(8):4962–4973. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.