")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Increased localized delivery of piroxicam by cationic nanoparticles after intra-articular injection
Authors Kim SR, Ho MJ, Kim SH, Cho HR, Kim HS, Choi YS, Choi YW , Kang MJ
Received 26 July 2016
Accepted for publication 21 September 2016
Published 16 November 2016 Volume 2016:10 Pages 3779—3787
DOI https://doi.org/10.2147/DDDT.S118145
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Frank Boeckler
Sung Rae Kim,1 Myoung Jin Ho,2 Sang Hyun Kim,1 Ha Ra Cho,2 Han Sol Kim,2 Yong Seok Choi,2 Young Wook Choi,1 Myung Joo Kang2
1Division of Pharmaceutical Sciences, College of Pharmacy, Chung-Ang University, Seoul, 2College of Pharmacy, Dankook University, Cheonan, Chungnam, South Korea
Abstract: Piroxicam (PRX), a potent nonsteroidal anti-inflammatory drug, is prescribed to relieve postoperative and/or chronic joint pain. However, its oral administration often results in serious gastrointestinal adverse effects including duodenal ulceration. Thus, a novel cationic nanoparticle (NP) was explored to minimize the systemic exposure and increase the retention time of PRX in the joint after intra-articular (IA) injection, by forming micrometer-sized electrostatic clusters with endogenous hyaluronic acid (HA) in the synovial cavity. PRX-loaded NPs consisting of poly(lactic-co-glycolic acid), Eudragit RL, and polyvinyl alcohol were constructed with the following characteristics: particle size of 220 nm, zeta potential of 11.5 mV in phosphate-buffered saline, and loading amount of 4.0% (w/w) of PRX. In optical and hyperspectral observations, the cationic NPs formed more than 50 µm-sized aggregates with HA, which was larger than the intercellular gaps between synoviocytes. In an in vivo pharmacokinetic study in rats, area under the plasma concentration–time curve (AUC0–24 h) and maximum plasma concentration (Cmax) of PRX after IA injection of the cationic NPs were <70% (P<0.05) and 60% (P<0.05), respectively, compared to those obtained from drug solution. Moreover, the drug concentration in joint tissue 24 h after dosing with the cationic NPs was 3.2-fold (P<0.05) and 1.8-fold (P<0.05) higher than that from drug solution and neutrally charged NPs, respectively. Therefore, we recommend the IA cationic NP therapy as an effective alternative to traditional oral therapy with PRX, as it increases drug retention selectively in the joint.
Keywords: piroxicam, polymeric nanoparticles, electrostatic interaction, intra-articular injection, local delivery, hyaluronic acid
Introduction
Intra-articular (IA) administration of therapeutic agents has been clinically explored to selectively deliver active compounds to their site of action in the treatment of osteoarthritis, rheumatoid arthritis, and joint pain.1 Direct IA delivery of active compounds to affected tissues offers the chance to boost therapeutic outcomes with lower dose, while reducing systemic exposure and undesirable adverse effects.1,2 Currently, corticosteroids, methotrexate, and nonsteroidal anti-inflammatory drugs (NSAIDs) are the common IA therapeutics for arthritic joints.2,3 Unfortunately, their therapeutic actions are often transient, because these molecules readily leak out from the synovial cavity and redistribute into the bloodstream. Knight and Levick4 revealed that the outer synovial membrane consisted of a discontinuous layer of synoviocytes with loose intercellular gaps (0.1–5.0 μm). It was reported that IA indomethacin showed a short elimination half-life of 2.8 h in patients.5 Moreover, several low molecular weight analgesics such as paracetamol, salicylate, and diclofenac exhibited only 1.1, 2.4, and 5.2 h elimination half-lives, respectively, in preclinical studies.6 There is an emerging interest in the development of polymeric nano- and micro-particulate systems as drug carriers to reduce the rapid efflux of therapeutic agents from the synovial cavity and extend drug retention time in joints, after IA injection.7,8 Poly(lactic-co-glycolic acid) (PLGA) has been widely employed to fabricate parenteral particulate systems, because of its long clinical use, favorable degradation characteristics, and sustained drug release profile.9,10 Horisawa et al11 revealed that PLGA-based nanoparticles (NPs) could prolong anti-inflammatory action of a corticosteroid, betamethasone sodium phosphate, by delaying the clearance rate of the active compound from the synovial cavity. Nevertheless, generally, NPs smaller than 250 nm in size quickly escape from the joint cavity, and thus longer retention times are still needed to permit less frequent IA injection.12 In our previous study, we reported on cationic NPs as a platform to acquire prolonged drug retention in the synovial joint.9 The positively surface-charged NPs composed of PLGA and a positively charged polymer, Eudragit RL, formed micrometer-sized clusters in synovial fluid with endogenous hyaluronic acid (HA) due to electrostatic interaction. This aggregate-forming behavior physically restricted the efflux of the cationic NPs through the interstitium of the synovial cavity after IA injection, thus increasing the retention time in the knees.
Piroxicam (PRX), a member of the oxicam group of NSAIDs, is often administered to reduce postoperative and/or chronic pain in joints, due to its strong and long-lasting analgesic and anti-inflammatory effects. However, this nonselective cyclooxygenase inhibitor, after repeated oral administration, occasionally causes serious adverse effects including gastroduodenal ulceration, bleeding, and renal disorder.13–15 To reduce the gastrointestinal damages of oral therapy, IA administration has been attempted.16 However, systemic exposure of PRX after IA injection was comparable to intramuscular injection of PRX because of its rapid clearance from the synovial cavity.16 Nevertheless, to the best of our knowledge, there are no investigations on the construction of IA-administered sustained-release PRX formulations.
Therefore, the objective of this study was to formulate a cationic NP delivery system for PRX to reduce the systemic exposure and simultaneously prolong the drug retention in joint tissue. PRX-loaded NPs were prepared by emulsification and solvent evaporation technique and were characterized in terms of particle size, surface charge, loading amount of drug, and in vitro release profile. Aggregate-forming behavior of cationic NPs with HA, which depended on the surface charge of the NPs, was visualized by a hyperspectral imaging system (Cytoviva, Auburn, AL, USA). In vivo systemic exposure and retention profile of PRX in the joint after IA injection of the optimized cationic NPs were evaluated in rats and compared to those after IA injection of drug solution or neutrally charged NPs.
Materials and methods
Materials
PRX powder (purity >99.0% w/w) was purchased from Nantong Jinghua Pharmaceutical Co. Ltd. (Jiangsu, China). PLGA (a lactide/glycolide ratio of 50:50 with a molecular weight of 38,000–54,000 kDa, Resomer® 504), polyvinyl alcohol (PVA with a molecular weight of 30,000–70,000 kDa), and phosphate-buffered saline (PBS) tablets were obtained from Sigma-Aldrich (St Louis, MO, USA). Sodium hyaluronate of microbial origin (molecular weight: 1,500–2,500 kDa) was purchased from Humedix (Sungnam, Korea). Eudragit RL (a copolymer of ethyl acrylate, methyl methacrylate, and a low content of methacrylic acid ester with quaternary ammonium groups) was kindly provided by Evonik (Darmstadt, Germany). Meloxicam (~98%), an internal standard (IS) for liquid chromatography tandem mass chromatography (LC–MS/MS) analyses, was obtained from Aladdin Industrial Corporation (Shanghai, China). Dichloromethane was purchased from Samchun Pure Chemical (Gyeonggi-do, Korea). Glacial acetic acid was purchased from Merck KgaA (Darmstadt, Germany). Water was prepared using a Millipore Milli-Q System (Millipore, Bedford, MA, USA). The grade of all other chemicals used for LC-MS/MS analyses was at least analytical grade.
Preparation of PRX-loaded NPs
PRX-loaded NPs were prepared by oil-in-water emulsification and solvent evaporation method with a slight modification.17 A total of 125 mg of the polymers (PLGA and Eudragit RL) and 25 mg of PRX were dissolved in 8 mL of dichloromethane. NPs prepared with PLGA and Eudragit RL with the weight ratios of 10:0, 9:1, 8:2, and 7:3 were named conventional NPs, E10-NPs, E20-NPs, and E30-NPs, respectively. The organic solution was then poured into 20 mL (0.1%; w/v) PVA solution and sonicated using a probe sonicator (Sonoplus, HD 2070; Bandelin Electronics, Berlin, Germany) operating at 55% power for 7 min. The oil-in-water emulsion was stirred at 400 rpm for 4 h to evaporate the organic phase, and the NP suspension was centrifuged at 20,000× g for 30 min. Finally, the supernatant was removed, and the NPs were re-dispersed in distilled water.
Physicochemical characterization of PRX-loaded NPs
Field-emission scanning electron microscopy (FE-SEM)
The morphological features of PRX-loaded NPs were observed using FE-SEM (Sigma, Carl Zeiss, Germany). NPs dispersed in water were dropped onto a cover glass and air dried under reduced pressure. The dried, sample-loaded cover glass was placed onto a copper grid using double-sided tape and coated with platinum for 2 min under vacuum. Samples were viewed at an acceleration voltage of 3 kV.
Measurement of particle size and zeta potential
The NP samples were diluted ~100-fold in 10 mM PBS solution and were examined for size distribution and polydispersity index (PDI) by a dynamic light scattering (DLS) method using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK) equipped with a 50 mV laser at a scattering angle of 90°. Samples were loaded into the disposable capillary cells, and the surface charge of the NPs was determined using the M3-PALS measurement technology equipped in the Zetasizer system. All measurements were performed in triplicate under ambient conditions.
Determination of the loading amount of drug in NPs
Approximately 10 mg of NP powder was added to 1 mL of acetonitrile and vortexed for 30 min. Then, the concentration of PRX in the organic solvent was analyzed by high-performance LC (HPLC).18 The HPLC system consisted of a pump (L-2130), ultraviolet (UV) detector (L-2400), a data station (LaChrom Elite, Hitachi, Japan), and a Kromasil C18 column (4.6 mm ×15 cm, 5 μm; Akzo Nobel, Bohus, Sweden). Acetonitrile–acetic acid (8%; v/v) in water (45:55) was used as mobile phase at a flow rate of 1 mL/min. The eluent was monitored at UV wavelength of 365 nm, and the PRX peak was observed at 4.3 min. The loading amount of PRX in NPs was calculated by the following equation:9
|
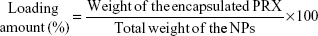
Optical and hyperspectral observations of PRX-loaded NP/HA aggregates
Optical and hyperspectral imaging systems (CytoViva, Auburn, AL, USA) were utilized to scrutinize the appearance of NP/HA aggregates and to track NPs in the electrostatic aggregates with HA. Approximately 250 μL of PRX-loaded NP suspension was admixed with 0.5 mL of HA solution (10 mM PBS, 2.2 mg/mL) and was gently shaken for 1 min. The sample was then spread on a glass slide and observed by optical or hyperspectral observation systems. The hyperspectral observation system included an Olympus microscope equipped with a visible-near infrared (VNIR) hyperspectral camera system, high-resolution adaptor, and the halogen light source illuminated by a fiber optic illuminator (Fiber-Lite DC-950; Dolan-Jenner Industries, Boxborough, MA, USA). Hyperspectral images were obtained from HyperVisual Software ENVI 4.8 (Exelis Visual Information Solutions, Boulder, CO, USA), by mapping spectral signals of PRX in the scanned images.
In vitro drug release test
In vitro release profile of PRX from drug powder, conventional and cationic NPs, and cationic NP/HA aggregates was assessed using the dialysis method.19 In brief, each formulation containing the equivalent amount of PRX (2 mg) was sealed in a dialysis bag (cellulose ester tubing with 50 kDa molecular weight cutoff; Spectrum Labs, Rancho Dominguez, CA, USA). Drug-containing bags were then immersed into 200 mL of 10 mM PBS solution (pH 7.4) and rotated at 50 rpm. At predetermined intervals, 500 μL of the release medium was collected and diluted twofold with acetonitrile. The aliquots were analyzed by HPLC to determine the drug concentration in the medium. The same volume of fresh PBS solution was supplemented to maintain a constant medium volume.
In vivo systemic exposure and joint retention of PRX after IA injection in rats
Animals
This animal experiment was approved by the Institutional Animal Care and Use Committee of Chung-Ang University in Seoul, Korea, and was carried out according to the National Institute of Health guidelines. Healthy Sprague Dawley rats (8-week-old, male) supplied by Orient Bio (Gyeonggi-do, Korea) were housed under controlled light cycle (12 h/12 h) and constant temperature (23°C) in the animal care laboratory. Standardized chow and water were available ad libitum. Prior to drug dosing, rats were fasted for ~12 h with free access to water.
Drug administration and plasma and joint tissue collections
Before drug administration, rats were assigned to three groups (n=6 in each group) by a stratified randomization scheme to get similar group mean body weights. One group received 50 μL of drug solution (2 mg/mL) intra-articularly using an insulin syringe (31 G) in the right and left knee joints. Rats in the second and third group received the equivalent amount of PRX in both knees, in the form of conventional NPs and E30-NPs, respectively. Then, blood samples of ~0.5 mL were collected from the retro-orbital plexus at predetermined time points of 0, 0.5, 1, 2, 4, 6, 8, 12, and 24 h using heparinized tubes. Blood samples were centrifuged at 16,000× g for 15 min. Obtained plasma samples were then stored at −80°C before being analyzed by LC-MS/MS. During the animal experiment, knee tissues were collected to estimate the level of PRX in joints. After 12 and 24 h of the IA injection, half of each group was sacrificed, and both knees were amputated using a bone cutter. The collected tissues were stored at −80°C until analysis, after removing any adhering tissue and washing out any residual substances.
Determination of PRX in plasma and joint tissue
Preparation of samples and their LC-MS/MS analyses were carried out following Kim et al’s20 methods. Briefly, in the case of a plasma sample, 100 μL of rat plasma was mixed with 10 μL of a 1,000 ng/mL IS in 50% (v/v) aqueous acetonitrile solution and 750 μL of acetonitrile for protein precipitation. A portion (5 μL) of supernatant was analyzed through an LC-MS/MS system (an LC-20 Prominence HPLC system from Shimadzu, Tokyo, Japan, and an API 2000 triple quadrupole mass spectrometer from AB/SCIEX, Foster City, CA, USA). For the preparation of an articular sample, the whole knee tissue was immersed into 2 mL of acetonitrile and incubated at 4°C by continuous shaking overnight to extract PRX from the tissue. After centrifugation of the extracted solution, 80 μL of the supernatant was mixed with 10 μL of the IS solution and 10 μL of acetonitrile. A portion (5 μL) of the resulting mixture was analyzed through the LC-MS/MS system. For separation, a Luna C18 column (2.0×150 mm, 5 μm; Phenomenex, Torrance, CA, USA) and the isocratic mobile phase condition of a 70% (v/v) aqueous acetonitrile solution in 0.1% formic acid were used with a flow rate of 0.25 mL/min and total analysis time of 5 min. PRX and IS eluted from the column were analyzed by multiple reaction monitoring in positive ion mode. The transitions of 331.9/95.0/31, precursor ion (m/z)/product ion (m/z)/collision energy (V), and 351.9/115.0/29 were monitored for PRX and IS, respectively. For the purpose of PRX and IS confirmation, additional transitions (331.9/77.9/71 for PRX and 351.9/141.0/31 for IS) were employed in the methods. Data acquisition/analyses were carried out using Analyst software (version 1.5.2; AB/SCIEX), and the screening transition peak area ratios of PRX to IS were used for the quantitation of PRX.
Calculation of pharmacokinetic parameters
Plasma data analysis was performed using BA Calc 2007 pharmacokinetic analysis program (Korea Food and Drug Administration, Korea). Area under the curve (AUC) from 0 to 24 h and elimination half-life (t1/2) of PRX was calculated using the linear trapezoidal rule of the analysis program. Maximum plasma concentration (Cmax) and the time taken to reach the maximum plasma concentration (Tmax) were measured directly from plasma concentration–time profiles.
Statistical analysis
All data are expressed as mean ± standard deviation, and results were considered to be significant at P<0.05.
Results and discussion
Morphological and physicochemical characteristics of PRX-loaded NPs
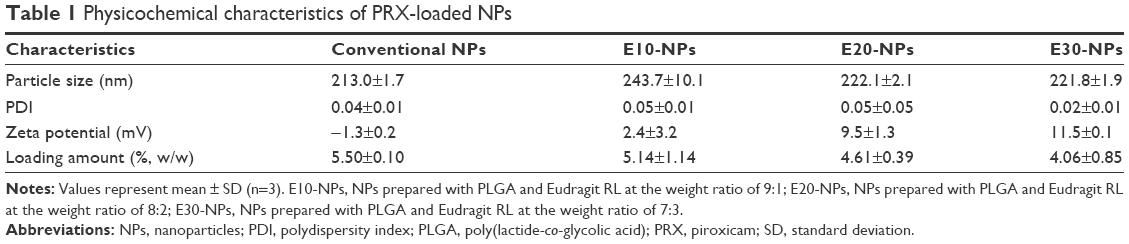
PRX-loaded cationic NPs consisting of PRX, PLGA, Eudragit RL, and PVA were fabricated by an emulsification and solvent evaporation method. In previous studies, PLGA and Eudragit RL formed homogeneous nanostructures when examined by differential scanning calorimeter, X-ray diffractometer, and Fourier transform infrared spectroscopy.21–23 PLGA polymer and hydrophobic group of Eudragit RL (methacrylic acid derivatives) preferentially formed the inner core structure of the NPs, while the positively charged moiety of Eudragit RL (quaternary ammonium groups) was on the surface of the NPs. In our study, three cationic NPs (E10-NPs, E20-NPs, and E30-NPs) containing different proportions of Eudragit RL were designed to investigate the influence of surface charge on the electrostatic interaction and aggregate-forming behavior with the anionic HA polymer. Hydrophobic PRX was predominantly entrapped inside the core compartment of the cationic NPs. A small quantity of PVA adsorbed on the surface helped the dispersion of the NPs in aqueous medium, with no aggregation and/or precipitation.24 The morphology of conventional and cationic NPs was scrutinized by FE-SEM. Both PRX-loaded conventional NPs (Figure 1A) and E30-NPs (Figure 1B) were uniform and spherical, and the surface of both was quite smooth. The appearance of the E10-NPs and E20-NPs was not noticeably different from E30-NPs (data not shown). The particle sizes of NPs as observed by SEM ranged from 200 to 300 nm, which was quite analogous to the mean hydrodynamic diameter between 200 and 250 nm as determined by DLS (Table 1). There was no obvious change in particle size, regardless of the ratio of PLGA polymer to cationic polymer. The low PDI values (<0.05) in all NP suspensions signify homogeneous size distribution in the aqueous medium.
 | Figure 1 Representative FE-SEM images of (A) conventional NPs and (B) cationic E30-NPs. |
 | Table 1 Physicochemical characteristics of PRX-loaded NPs |
On the other hand, the surface charge of PRX-loaded NPs increased as the proportion of cationic polymer was increased. The surface charge of conventional NPs composed of neutral PLGA and PVA polymers was neutral (−1.3 mV) in 10 mM PBS solution (pH 7.4). Conversely, the zeta potential values of E10-NPs, E20-NPs, and E30-NPs were 2.4, 9.5, and 11.5 mV, respectively, in the physiological environment. When the amount of Eudragit RL in the NPs was increased, the proportion of cationic quaternary ammonium groups residing on their surface might be expected to increase. We expected the positively charged groups on NPs to not only trigger the formation of micrometer-sized aggregates by electrostatic interaction with HA after IA injection but also improve the colloidal storage stability in aqueous medium by electrostatic repulsion mechanism.
Optical and hyperspectral imaging of PRX-loaded NP/HA aggregates
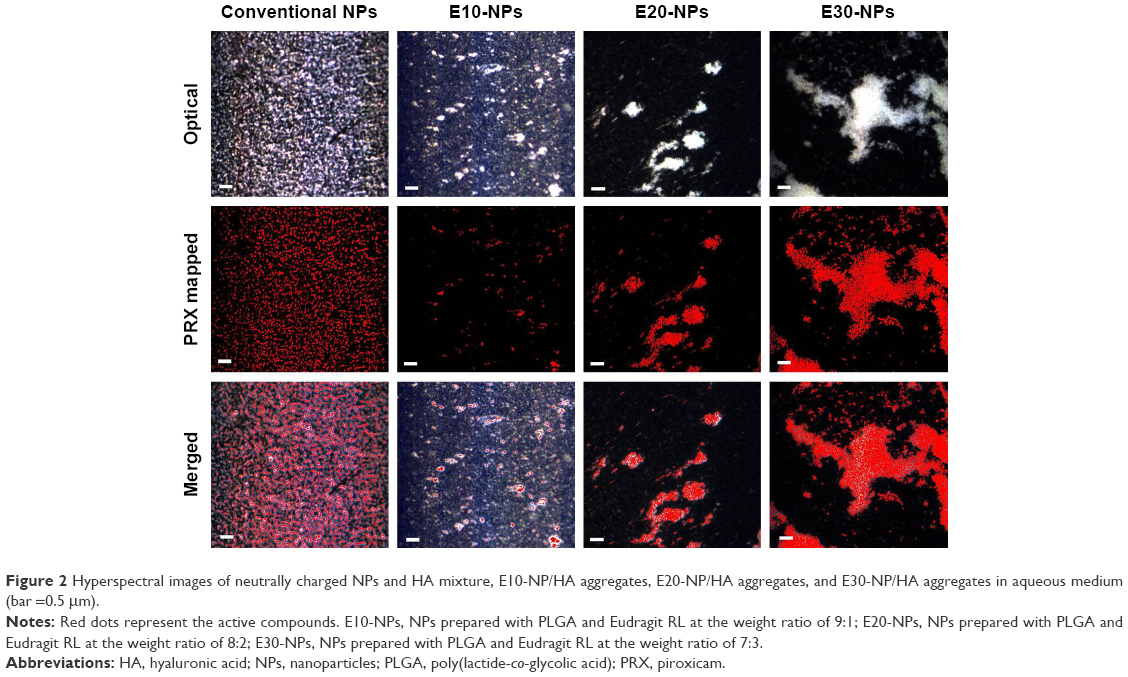
Optical and hyperspectral imaging tools were employed to scrutinize the appearance of NP/HA aggregates and to find the distribution of NPs in the aggregates. In our study, HA concentration in aqueous medium was set to 2.2 mg/mL, because HA concentration in synovial fluid was reported to be below 2.2 mg/mL in osteoarthritic joints.2 Figure 2 shows representative images of each NP system after admixing with HA solution, where the images are listed as optical (top), PRX mapped (center), and merged images (bottom). The spectrum derived from PRX was expressed in red color in the hyperspectral imaging. Conventional NPs with neutral surface charge did not form any aggregates and/or clusters with HA, as observed by optical microscope. PRX-loaded conventional NPs were evenly dispersed in the solution.
 | Figure 2 Hyperspectral images of neutrally charged NPs and HA mixture, E10-NP/HA aggregates, E20-NP/HA aggregates, and E30-NP/HA aggregates in aqueous medium (bar =0.5 μm). |
In contrast, every cationic NP (E10-NPs, E20-NPs, and E30-NPs) formed filamentous aggregates when in contact with the anionic polymer under gentle agitation. The diameter of the cluster became larger, when the surface charge of the NPs increased. In particular, E30-NPs constructed micrometer-sized, ionically cross-linked clusters with HA, which were larger than 50 μm. As observed in the merged images, the cationic NPs were predominantly gathered within the NP/HA aggregates, suggesting that charge interaction between the cationic PLGA NP and the anionic polymer principally could be attributed to the formation of insoluble aggregates. E30-NPs, which produced aggregates larger than the loose intercellular gaps (0.1–5.0 μm) in the synovial membrane, were employed for further in vitro and in vivo studies.
In vitro drug release profile from cationic NPs
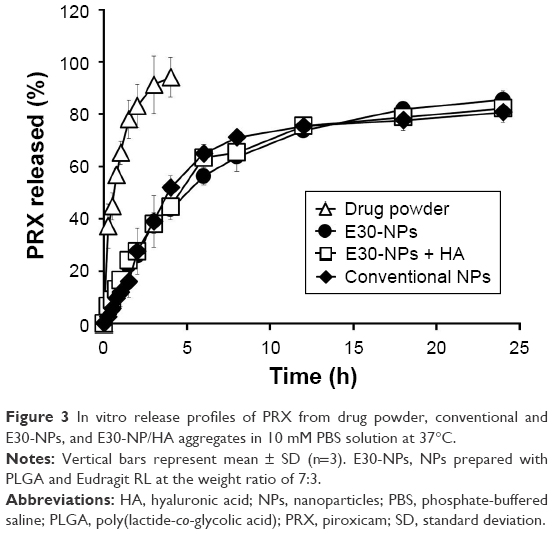
In vitro release profiles of PRX from drug powder, conventional and E30-NPs, and E30-NP/HA aggregates were assessed in 200 mL of PBS solution at 50 rpm stirring rate. This dissolution condition is assumed to accelerate drug release from the formulations, compared to the in vivo joint environment, because the actual volume of synovial fluid in normal human joints is only 0.5–2.0 mL.4,5 Moreover, the movement of synovial fluid in the knee is not fast, with a flow rate of 0.002 mL/cm2 per hour.25 Nevertheless, the drug release profile was assessed under these accelerated conditions for quicker comparison between formulations. The solubility of PRX in the medium was measured as 200 μg/mL, which assures appropriate sink condition to study the drug release from the formulations.
Under sink condition, the drug powder was readily dissolved in the aqueous medium and diffused out of the dialysis membrane, achieving 78% drug release after 90 min (Figure 3). On the other hand, drug release from both conventional and cationic E30-NPs became significantly delayed, exhibiting accumulative release of 16% and 22% after 90 min, respectively. There were no differences in the release profile between conventional and E30-NPs, regardless of the presence of Eudragit RL. The accumulated amounts of PRX which diffused out from E30-NPs were linear for 8 h, and increased by 26%, 44%, to 64% after 2, 4 and 8 h, respectively. Finally, a sustained release pattern was observed for 24 h in both conventional and E30-NP systems, showing 80% and 85% drug release after 24 h, respectively. In the preclinical and clinical setting, this sustained release profile of the cationic NPs would increase the drug retention period in the joint and, thus, extend the duration of pharmacological action of the compound.
 | Figure 3 In vitro release profiles of PRX from drug powder, conventional and E30-NPs, and E30-NP/HA aggregates in 10 mM PBS solution at 37°C. |
In vivo systemic exposure and joint retention of PRX after IA injection in rats
Although PRX exerts potent analgesic and anti-inflammatory effects in treating osteoarthritis, rheumatoid arthritis, and joint pain, its nonselective cyclooxygenase inhibition is strongly associated with serious gastrointestinal adverse events such as bleeding, ulceration, and perforation of the stomach or intestines, especially in elderly patients. It has been reported that the repeated oral intake of NSAIDs provokes drug-induced gastrointestinal effects, accounting for 10%–30% of gastric ulcers, 20%–35% of ulcer bleeding, ~20,000 hospitalizations, and 2,600 deaths annually in the US.26 Thus, in the current study, a novel NP-based IA delivery of PRX was explored as an alternative to minimize systemic drug exposure.
Plasma concentration–time profiles of PRX following IA administration of drug solution, conventional NPs, or E30-NPs are depicted in Figure 4. The pharmacokinetic parameters such as AUC, Cmax, Tmax, and t1/2 values are summarized in Table 2. After IA injection of PRX solution, the drug concentration in plasma rapidly rose, showing a Cmax value of 3.7 μg/mL at 0.8 h (Tmax) after dosing. This observation is in line with our earlier report that PRX rapidly redistributed from the joint to the blood stream within 1 h, following IA or intramuscular injection, denoting the rapid efflux of the small molecule from the joint cavity.17,27–29 In contrast, the plasma concentration of PRX after IA administration of conventional and E30-NPs gradually increased and peaked between 2.8 and 3.8 h after IA injection, as expected from in vitro release data. The Cmax values of PRX following conventional and E30-NP injection were 2.3 and 2.2 μg/mL, respectively, which are 62% (P<0.05) and 59% (P<0.05) that of the drug solution. Moreover, AUC0–24 h values obtained from conventional NP- and E30-NP-treated groups decreased to <78% (P<0.05) and 70% (P<0.05) of that obtained from the drug solution, respectively. These pharmacokinetic data show that these particulates reduced efflux from the synovial cavity into the blood, prolonging retention period of the drug in the joint. On the other hand, the cationic NPs exhibited slightly lower systemic exposure in terms of AUC0–24 h and Cmax values of PRX compared to the neutral NPs, but these differences were not statistically significant.
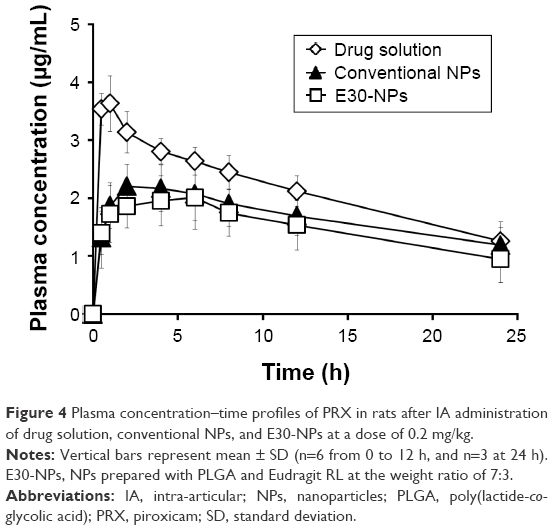 | Figure 4 Plasma concentration–time profiles of PRX in rats after IA administration of drug solution, conventional NPs, and E30-NPs at a dose of 0.2 mg/kg. |
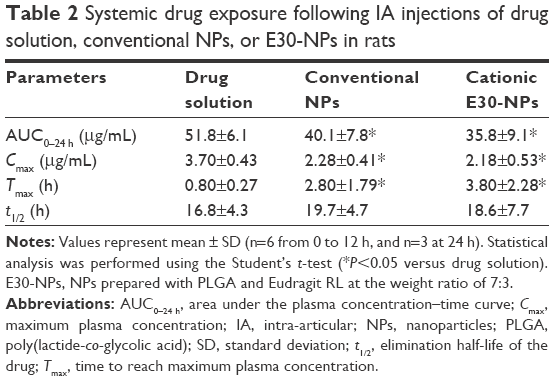 | Table 2 Systemic drug exposure following IA injections of drug solution, conventional NPs, or E30-NPs in rats |
The remains of PRX in the joint, 12 and 24 h after dosing, were determined following IA administration of drug solution, neutral NPs, and cationic NPs in rat knees (Figure 5). Following IA administration of drug solution, the concentration of PRX in the injected tissue decreased quickly. Only 0.3% and 0.2% of the drug administered was detected 12 and 24 h after dosing, respectively. However, the amount of drug remaining in the joint, after the administration of E30-NPs, was ~3.7-fold (P<0.05) and 3.2-fold (P<0.05) higher than its solution 12 and 24 h after dosing, respectively. The formation of micrometer-sized aggregates by electrical interaction between NPs and anionic HA might prevent rapid efflux of drug and/or drug-loaded NPs from the synovial cavity. Interestingly, the cationic NPs showed higher drug accumulation in joint tissue compared with neutral NPs, showing 55% and 83% (P<0.05) higher drug remaining 12 and 24 h after dosing, respectively. These results are in line with earlier reports that cationic NPs functionalized with chitosan or Eudragit RL have more prolonged retention in the joint after IA injection, as revealed by the higher fluorescence intensity in joints compared to solution or conventional NPs.9,25,30 Aggregate-forming behavior of the cationic NPs physically prevents the diffusion of the drug and/or drug-loaded carrier from the synovial cavity, whose boundary consists of a discontinuous layer of synoviocytes. Moreover, it is postulated that the positive surface charge of the NPs motivates their interaction with the immune system, which can lead to preferential uptake of NPs by phagocytic and/or nonphagocytic cells in synovial linings of the joint, compared to neutrally charged NPs.31
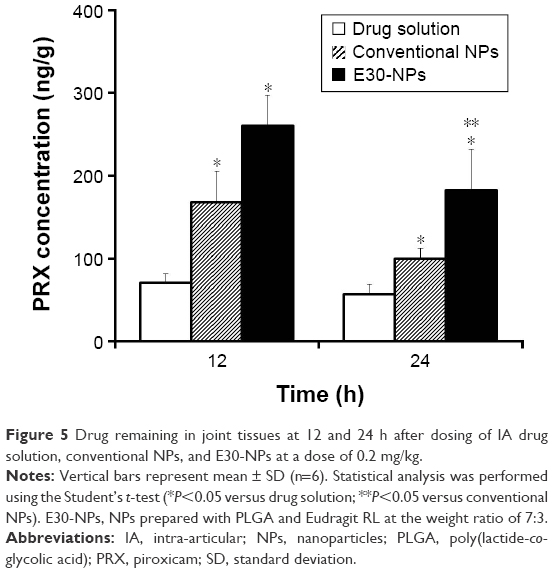 | Figure 5 Drug remaining in joint tissues at 12 and 24 h after dosing of IA drug solution, conventional NPs, and E30-NPs at a dose of 0.2 mg/kg. |
Conclusion
A cationic NP consisting of PLGA, Eudragit RL, and PVA was built to diminish systemic exposure and extend retention time of PRX in the joint after IA administration. The optimized NP, with a surface potential of >10 mV, promptly formed filamentous aggregates larger than 50 μm, by electrostatic interaction with anionic HA polymer. In vivo pharmacokinetic study in rats revealed that IA injection of the cationic NPs remarkably decreased systemic drug exposure compared to drug solution. Moreover, the amount of PRX in joint tissue after IA injection of the novel NPs was significantly more than by drug solution or neutrally charged NPs. Further investigations to estimate the local distribution of NPs in joint tissue, their local tolerability and/or toxicity, and a preclinical efficacy test are scheduled to develop this formulation for targeted delivery of PRX in osteoarthritic joints.
Acknowledgment
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (NRF-2013R1A1A1058935).
Disclosure
The authors report no conflicts of interest in this work.
References
Edwards SH. Intra-articular drug delivery: the challenge to extend drug residence time within the joint. Vet J. 2011;190(1):15–21. | ||
Gerwin N, Hops C, Lucke A. Intraarticular drug delivery in osteoarthritis. Adv Drug Deliv Rev. 2006;58(2):226–242. | ||
Uthman I, Raynauld JP, Haraoui B. Intra-articular therapy in osteoarthritis. Postgrad Med J. 2003;79(934):449–453. | ||
Knight AD, Levick JR. Morphometry of the ultrastructure of the blood-joint barrier in the rabbit knee. Q J Exp Physiol. 1984;69(2):271–288. | ||
Neander G, Eriksson LO, Wållin-Boll E, Ersmark H, Grahnen A. Pharmacokinetics of intraarticular indomethacin in patients with osteoarthritis. Eur J Clin Pharmacol. 1992;42(3):301–305. | ||
Owen SG, Francis HW, Roberts MS. Disappearance kinetics of solutes from synovial fluid after intra-articular injection. Br J Clin Pharmacol. 1994;38(4):349–355. | ||
Mitragotri S, Yoo JW. Designing micro- and nano-particles for treating rheumatoid arthritis. Arch Pharm Res. 2011;34(11):1887–1897. | ||
Butoescu N, Jordan O, Doelker E. Intra-articular drug delivery systems for the treatment of rheumatic diseases: a review of the factors influencing their performance. Eur J Pharm Biopharm. 2009;73(2):205–218. | ||
Kim SR, Ho MJ, Lee E, Lee JW, Choi YW, Kang MJ. Cationic PLGA/Eudragit RL nanoparticles for increasing retention time in synovial cavity after intra-articular injection in knee joint. Int J Nanomedicine. 2015;10:5263–5271. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Horisawa E, Hirota T, Kawazoe S, et al. Prolonged anti-inflammatory action of DL-lactide/glycolide copolymer nanospheres containing betamethasone sodium phosphate for an intra-articular delivery system in antigen-induced arthritic rabbit. Pharm Res. 2002;19(4):403–410. | ||
Levick JR. A method for estimating macromolecular reflection by human synovium, using measurements of intra-articular half lives. Ann Rheum Dis. 1998;57(6):339–344. | ||
Hart FD, Huskisson EC. Non-steroidal anti-inflammatory drugs: current status and rational therapeutic use. Drugs. 1984;27(3):232–255. | ||
Laake K, Kjeldaas L, Borchgrevink CF. Side-effects of piroxicam (Feldenee). A one-year material of 103 reports from Norway. Acta Med Scand. 1984;215(1):81–83. | ||
Deeks JJ, Smith LA, Bradley MD. Efficacy, tolerability, and upper gastrointestinal safety of celecoxib for treatment of osteoarthritis and rheumatoid arthritis: systematic review of randomized controlled trials. BMJ. 2002;325(7365):619. | ||
Park CW, Ma KW, Jang SW, Son M, Kang MJ. Comparison of piroxicam pharmacokinetics and anti-inflammatory effect in rats after intra-articular and intramuscular administration. Biomol Ther(Seoul). 2014;22(3):260–266. | ||
Budhian A, Siegel SJ, Winey KI. Haloperidol-loaded PLGA nanoparticles: systematic study of particle size and drug content. Int J Pharm. 2007;336(2):367–375. | ||
Belletti D, Tosi G, Forni F, et al. Chemico-physical investigation of tenofovir loaded polymeric nanoparticles. Int J Pharm. 2012;436(1):753–763. | ||
Park ES, Cui Y, Yun BJ, Ko IJ, Chi SC. Transdermal delivery of piroxicam using microemulsions. Arch Pharm Res. 2005;28(2):243–248. | ||
Kim HS, Cho HR, Ho MJ, Kang MJ, Choi YS. Determination of piroxicam from rat articular tissue and plasma based on LC-MS/MS. Arch Pharm Res. 2016. doi:10.1007/s12272-016-0845-2. | ||
Park MH, Baek JS, Lee CA, Kim DC, Cho CW. The effect of Eudragit type on BSA-loaded PLGA nanoparticles. J Pharm Invest. 2014;44(5):339–349. | ||
Dillen K, Vandervoort J, Van den Mooter G, Ludwig A. Evaluation of ciprofloxacin-loaded Eudragit RS100 or RL100/PLGA nanoparticles. Int J Pharm. 2006;314(1):72–82. | ||
Lalla JK, Sapna K. Biodegradable microspheres of poly(DL-lactic acid) containing piroxicam as a model drug for controlled release via the parenteral route. J Microencapsul. 1993;10(4):449–460. | ||
Feng S, Huang G. Effects of emulsifiers on the controlled release of paclitaxel (Taxol) from nanospheres of biodegradable polymers. J Control Release. 2001;71(1):53–69. | ||
Morgen M, Tung D, Boras B, Miller W, Malfait AM, Tortorella M. Nanoparticles for improved local retention after intra-articular injection into the knee joint. Pharm Res. 2013;30(1):257–268. | ||
Fosslien E. Adverse effects of nonsteroidal anti-inflammatory drugs on the gastrointestinal system. Ann Clin Lab Sci. 1998;28(2):67–81. | ||
Zhang Z, Bi X, Li H, Huang G. Enhanced targeting efficiency of PLGA microspheres loaded with Lornoxicam for intra-articular administration. Drug Deliv. 2011;18(7):536–544. | ||
Liang LS, Wong W, Burt HM. Pharmacokinetic study of methotrexate following intra-articular injection of methotrexate loaded poly (L-lactic acid) microspheres in rabbits. J Pharm Sci. 2005;94(6):1204–1215. | ||
Kawadkar J, Chauhan MK. Intra-articular delivery of genipin cross-linked chitosan microspheres of flurbiprofen: preparation, characterization, in vitro and in vivo studies. Eur J Pharm Biopharm. 2012;81(3):563–572. | ||
Kang ML, Ko JY, Kim JE, Im GI. Intra-articular delivery of kartogenin-conjugated chitosan nano/microparticles for cartilage regeneration. Biomaterials. 2014;35(37):9984–9994. | ||
Schwendener RA, Lagocki PA, Rahman YE. The effects of charge and size on the interaction of unilamellar liposomes with macrophages. Biochim Biophys Acta. 1984;772(1):93–101. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.