")
Back to Journals » Drug Design, Development and Therapy » Volume 14
In silico Design and Synthesis of Tetrahydropyrimidinones and Tetrahydropyrimidinethiones as Potential Thymidylate Kinase Inhibitors Exerting Anti-TB Activity Against Mycobacterium tuberculosis
Authors Venugopala KN , Tratrat C , Pillay M , Chandrashekharappa S , Al-Attraqchi OHA, Aldhubiab BE , Attimarad M , Alwassil OI , Nair AB , Sreeharsha N , Venugopala R , Morsy MA , Haroun M , Kumalo HM , Odhav B, Mlisana K
Received 23 August 2019
Accepted for publication 20 February 2020
Published 9 March 2020 Volume 2020:14 Pages 1027—1039
DOI https://doi.org/10.2147/DDDT.S228381
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Tin Wui Wong
Katharigatta N Venugopala, 1, 2 Christophe Tratrat, 1 Melendhran Pillay, 3 Sandeep Chandrashekharappa, 4 Omar Husham Ahmed Al-Attraqchi, 5 Bandar E Aldhubiab, 1 Mahesh Attimarad, 1 Osama I Alwassil, 6 Anroop B Nair, 1 Nagaraja Sreeharsha, 1 Rashmi Venugopala, 7 Mohamed A Morsy, 1, 8 Michelyne Haroun, 1 Hezekiel M Kumalo, 9 Bharti Odhav, 2 Koleka Mlisana 3
1Department of Pharmaceutical Sciences, College of Clinical Pharmacy, King Faisal University, Al-Ahsa 31982, Kingdom of Saudi Arabia; 2Department of Biotechnology and Food Technology, Durban University of Technology, Durban 4001, South Africa; 3Department of Microbiology, National Health Laboratory Services, KZN Academic Complex, Inkosi Albert Luthuli Central Hospital, Durban 4001, South Africa; 4Institute for Stem Cell Biology and Regenerative Medicine, NCBS, TIFR, Bangalore 560 065, India; 5Department of Pharmaceutical Sciences, Faculty of Pharmacy, Philadelphia University, Amman 19392, Jordan; 6Department of Pharmaceutical Sciences, College of Pharmacy, King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia; 7Department of Public Health Medicine, University of KwaZulu-Natal, Howard College Campus, Durban 4001, South Africa; 8Department of Pharmacology, Faculty of Medicine, Minia University, El-Minia 61511, Egypt; 9Department of Medical Biochemistry, School of Laboratory Medicine and Medical Sciences, University of KwaZulu-Natal, Medical School, Durban 4001, South Africa
Correspondence: Katharigatta N Venugopala
Department of Pharmaceutical Sciences, College of Clinical Pharmacy, King Faisal University, P.o. Box-400, Al-Ahsa 31982, Kingdom of Saudi Arabia
Tel +96613 589 8842
Email [email protected]
Sandeep Chandrashekharappa
Institute for Stem Cell Biology and Regenerative Medicine, National Center for Biological Sciences, Tata Institute of Fundamental Research, GKVK, Bellary Road, Bangalore 560 065, India
Tel +9194486 39413
Email [email protected]
Background and Purpose: Tuberculosis has been reported to be the worldwide leading cause of death resulting from a sole infectious agent. The emergence of multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis has made the battle against the infection more difficult since most currently available therapeutic options are ineffective against these resistant strains. Therefore, novel molecules need to be developed to effectively treat tuberculosis disease. Preliminary docking studies revealed that tetrahydropyrimidinone derivatives have favorable interactions with the thymidylate kinase receptor. In the present investigation, we report the synthesis and the mycobacterial activity of several pyrimidinones and pyrimidinethiones as potential thymidylate kinase inhibitors.
Methods: The title compounds ( 1a–d) and ( 2a–b) were synthesized by a one-pot three-component Biginelli reaction. They were subsequently characterized and used for whole-cell anti-TB screening against H37Rv and multidrug-resistant (MDR) strains of Mycobacterium tuberculosis (MTB) by the resazurin microplate assay (REMA) plate method. Molecular modeling was conducted using the Accelry’s Discovery Studio 4.0 client program to explain the observed bioactivity of the compounds. The pharmacokinetic properties of the synthesized compounds were predicted and analyzed.
Results: Of the compounds tested for anti-TB activity, pyrimidinone 1a and pyrimidinethione 2a displayed moderate activity against susceptible MTB H37Rv strains at 16 and 32 μg/mL, respectively. Only compound 2a was observed to exert modest activity at 128 μg/mL against MTB strains with cross-resistance to rifampicin and isoniazid. The presence of the trifluoromethyl group was essential to retain the inhibitory activity of compounds 1a and 2a. Molecular modeling studies of these compounds against thymidylate kinase targets demonstrated a positive correlation between the bioactivity and structure of the compounds. The in-silico ADME (absorption, distribution, metabolism, and excretion) prediction indicated favorable pharmacokinetic and drug-like properties for most compounds.
Conclusion: Pyrimidinone 1a and pyrimidinethione 2a were identified as the leading compounds and can serve as a starting point to develop novel anti-TB therapeutic agents.
Keywords: thymidylate kinase inhibitors, tetrahydropyrimidinones, tetrahydropyrimidinethiones, multidrug resistance, Mycobacterium tuberculosis, molecular modeling
Introduction
Tuberculosis (TB) is a prevalent infectious disease that affects the broader population. This disease is caused by Mycobacterium tuberculosis (MTB). According to the World Health Organization’s (WHO) Global Tuberculosis Report 2018, approximately 1.3 million deaths were caused by TB in human immunodeficiency virus (HIV)-negative people. In addition, about 250,000 deaths were caused by TB in HIV-positive people.1 TB was also reported to be the leading cause of death resulting from a sole infectious agent worldwide.2 The emergence of multidrug-resistant TB (MDR-TB)3 and extensively drug-resistant tuberculosis (XDR-TB)4 has made the battle against TB more difficult because most currently available therapeutic options are ineffective against these resistant strains.5 Furthermore, the appearance of totally drug-resistant (TDR) strains has heightened this challenge because these strains are unaffected by the currently available anti-TB agents.6 Thus, developing novel therapeutic agents that are capable of combating resistant strains of MTB is necessary. After decades of academic and pharmaceutical industry investigation, in December 2012, bedaquiline became one of the first novel anti-TB drugs to be approved by the United States Food and Drug Administration (US FDA) for the treatment of MDR-TB.7 Following this, delamanid was approved by the European Medicines Agency in 2013.8
MTB thymidylate kinase (MTB-TMK) is involved in DNA synthesis and is an essential target in the discovery of anti-TB compounds. Thymidine monophosphate kinase (TMK) catalyzes the phosphorylation of thymidine monophosphate (dTMP) to thymidine diphosphate (dTDP) using adenosine triphosphate (ATP) as the source of the phosphoryl group. The action of TMK is essential for maintaining the required levels of thymidine triphosphate (dTTP), which is a DNA building block required for DNA replication. Thus, inhibiting this enzyme is fatal to M. tuberculosis cells by rendering them unable to replicate and survive. The position of this enzyme in the dTTP synthesis pathway is crucial: it lies at the intersection of the de novo and salvage pathways. Further, this enzyme is the last specific enzyme involved in the synthesis of dTTP.9 Another important property of the MTB-TMK enzyme that makes it attractive for developing therapeutic agents is that it only shares a 22% sequence identity with human TMK. This allows for the development of selective inhibitors that target MTB-TMK without affecting healthy human cells.10
Although most MTB-TMK inhibitors are thymidine monophosphate analogues that contain a nucleoside core,11–17 some thymine non-nucleoside derivatives have also been reported to be potent MTB-TMK inhibitors.18–21 Two novel classes, 3-cyanopyridones and 1.6-naphthyridin-2-ones, were recently identified as MTB-TMK inhibitors via high-throughput screening and structural optimization to improve potency (Figure 1).22 These two scaffolds share a common substructure with thymidine, namely, the unsubstituted lactam functional group, which is important for the formation of hydrogen bonds with thymidylate kinase receptors.
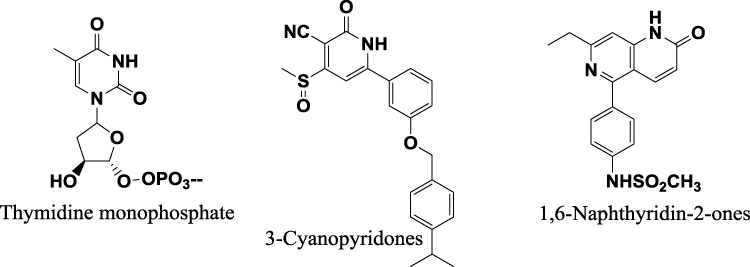 |
Figure 1 The representative scaffold of Mycobacterium tuberculosis thymidylate kinase inhibitors. |
The tetrahydropyrimidine (THP) pharmacophore is found in various heterocyclic structures, which are known for their various pharmacological actions since they serve as antiviral,23 anticancer,24 antihypertensive,25 calcium-channel-blocking,26 antitubercular,27,28 antimicrobial,29,30 anti–inflammatory,31,32 antidiabetic,33 and larvicidal and insecticide agents.34–36 Herein, we report the anti-TB activity of THP derivatives against H37Rv and multidrug-resistant (MDR) strains of MTB. On the basis of our previous findings on the promising anti-TB activity of THP scaffolds27,28 on whole-cell anti-TB properties and in a continuous effort to identify novel heterocyclic scaffolds that demonstrate potential anti-TB activity,37–40 we screened six 1,2,3,4-tetrahydropyrimidinone (1a–d) and 1,2,3,4-tetrahydropyrimidinethione (2a–b) analogues (Scheme 1) for whole-cell anti-TB activity against H37Rv and MDR strains of MTB by the resazurin microplate assay (REMA) plate method.
Materials and Methods
Chemistry
The chemicals reported here were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA), while the solvents were obtained from Millipore Sigma (Burlington, MA, USA). Thin-layer chromatography (TLC) using silica gel (Sigma-Aldrich Co.) on aluminum foil was employed to observe the chemical reactions; n-hexane and ethyl acetate (4:6) were used as the solvent. The reactions were visualized under an ultraviolet (UV)-light/iodine chamber. B-545 was used to measure the melting points (Büchi, Labortechnik, Flawil, Switzerland). Infrared (IR) spectra were recorded on a Nicolet 6700 Fourier-transform infrared (FT-IR) spectrometer. Further, 1H and 13C-NMR spectra were recorded using Bruker AVANCE III 600 MHz (Bruker Corporation, Billerica, MA, USA) with CDCl3 (solvent). Chemical shifts (δ) were indicated in ppm, with tetramethylsilane (TMS) as a reference; coupling constants (J) were recorded (Hz). The splitting pattern was documented as follows: s, singlet; d, doublet; q, quartet; and m, multiplet. Liquid chromatography/mass spectrometry (LC–MS; Agilent 1100 series) was used to measure the mass spectra in conjunction with MSD and 0.1% aqueous trifluoroacetic acid in an acetonitrile system on a C18-BDS column. Then, an elemental analysis was carried out using the analyzer FLASH EA 1112 CHN (Thermo Finnigan LLC, New York, NY, USA).
The test compounds 1,2,3,4-tetrahydropyrimidinone (1a–c) and 1,2,3,4-tetrahydropyrimidinethione (2b) (Scheme 1) were synthesized using our previously reported procedures,41 and novel compounds 1d and 2a were synthesized by following a similar procedure. Table 1 details the physicochemical constants of the test compounds.
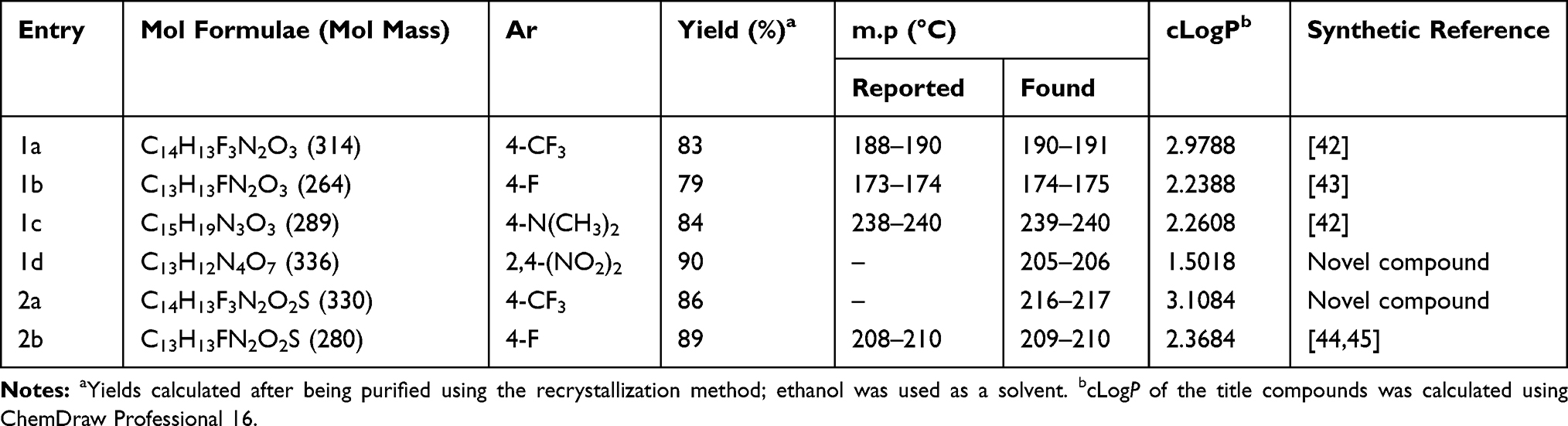 |
Table 1 Physicochemical Characteristics of Methyl 6-Methyl-2-Oxo-4-Substituedphenyl-1,2,3,4-Tetrahydropyrimidine-5-Carboxylate (1a–d) and Methyl 6-Methyl-4-Substituedphenyl-2-Thioxo-1,2,3,4-Tetrahydropyrimidine-5-Carboxylate (2a–b) |
The Synthesis of Methyl 4-(2,4-Dinitrophenyl)-6-Methyl-2-Oxo-1,2,3,4-Tetrahydropyrimidine-5-Carboxylate (1d)
A mixture of 2.4-dinitrophenyl benzaldehyde (1.2 mmol), urea (1.2 mmol), and methyl acetoacetate (1.5 mmol) in methanol was added to a 50 mL round-bottomed flask and refluxed for 15 hrs using concentrated hydrochloric acid as a catalyst. The reaction was examined on a thin-layer chromatography plate. Once the reaction was achieved, the reaction medium was poured into ice-cold water with stirring. The obtained precipitate was filtered and washed with aqueous methanol and then dried. The resulting product was recrystallized using ethanol to obtain the pure compound. The remaining compounds were prepared using the aforementioned procedure; the physicochemical characteristics are tabulated in Table 1.
Methyl 4-(2,4-Dinitrophenyl)-6-Methyl-2-Oxo-1,2,3,4-Tetrahydropyrimidine-5-Carboxylate (1d)
Appearance: brown amorphous compound; FT-IR (KBr) cm–1 = 3220, 3087, 1728, 1677, 1596, 1421. 1H-NMR (600 MHz CDCl3) δ = 9.54 (1H, bs), 8.65–8.64 (1H, d, J = 1.8 Hz), 8.53–8.52 (1H, m), 8.04 (1H, bs), 7.86–7.85 (1H, d, J = 7.2 Hz), 5.86 (1H, s), 3.39 (3H, s), 2.27 (3H, s); 13C-NMR (100 MHz CDCl3) δ = 165.41, 151.44, 150.89, 147.67, 147.04, 145.96, 131.52, 128.82, 119.93, 97.73, 51.36, 49.96, 18.40. LC–MS (ESI Positive): m/z = (M+H)+: 337. Anal. calculated for C13H12N4O7; C, 46.44, H, 3.60, N, 16.66; Found; C, 46.45, H, 3.65, N, 16.61.
The Synthesis of Methyl 6-Methyl-2-Thioxo-4-(4-(Trifluoromethyl)Phenyl)-1,2,3,4-Tetrahydropyrimidine-5-Carboxylate (2a)
A mixture of thiourea (1.2 mmol), 4-trifluoromethyl benzaldehyde (1.2 mmol), and methyl acetoacetate (1.5 mmol) in methanol was added to a 50 mL round-bottomed flask and refluxed for 15 hrs using concentrated hydrochloric acid as a catalyst. The reaction was examined on a thin-layer chromatography plate. Once the reaction was achieved, the cooled reaction medium was poured into ice-cold water with constant stirring. The obtained precipitate was filtered and washed with aqueous methanol and then dried over sodium sulfate. The resulting product was recrystallized using ethanol to obtain the pure compound. The remaining compound 2b was prepared using the aforementioned procedure; the physicochemical characteristics are tabulated in Table 1.
Methyl 6-Methyl-2-Thioxo-4-(4-(Trifluoromethyl)Phenyl)-1,2,3,4-Tetrahydropyrimidine-5-Carboxylate (2a)
Appearance: yellow amorphous powder; FT-IR (KBr) cm–1 = 3390, 3124, 1716, 1666, 1517, 1456, 1236. 1H-NMR (600 MHz CDCl3) δ = 8.01 (1H, bs), 7.62–7.59 (2H, m), 7.51 (1H, bs), 7.44–7.43 (2H, m), 5.48 (1H, s), 3.68 (3H, s), 2.39 (3H, s); 13C-NMR (150 MHz CDCl3) δ = 175.8, 165.46, 145.9, 143.45, 129.05, 127.07, 126.00, 125.98, 125.79, 101.2, 55.60, 51.6, 18.58; LC–MS (ESI Positive): m/z = (M+H)+: 331. Anal. calculated for C14H13F3N2O2S; C, 50.91, H, 3.97, N, 8.48; Found; C, 50.95, H, 3.95, N, 8.49.
Anti-Tubercular Activity
Resazurin Microplate Assay (REMA)
Anti-TB screening of test compounds 1a–d and 2a–b was carried out against H37Rv and MDR strains of Mycobacterium tuberculosis via the REMA plate method, as described in our previous communication.28,46 Testing was conducted on stored MDR-MTB cultures isolated from patient sputum specimens; pure colonies from a single strain were retrieved from storage and subcultured for further testing. Clinical isolates of a well-characterized MDR strain of Mycobacterium tuberculosis with resistance to rifampicin (Rif) and isoniazid (Inh) were selected for testing. Mutations within the rpoB gene and katG or inhA genes conferred resistance to Rif and Inh, respectively.
Determining the Minimum Inhibitory Concentration (MIC)
All six test compounds 1a–d and 2a–b were assessed using the agar incorporation method, which was performed three times against the H37Rv strain and the MDR-TB strain, which demonstrates resistance to rifampicin (1 µg/mL) and isoniazid (0.2 µg/mL). MIC was determined.47 The MTB reference strain H37Rv (American Type Culture Collection [ATCC]: 25177), as well as MDR-TB were cultured for a total of 3 weeks in Middlebrook 7H11 medium,48 and were then supplemented with OADC (0.005%, v/v, oleic acid; 0.2%, w/v, glucose; 0.085%, w/v, NaCl; 0.02%, v/v, catalase; and 0.5%, 171 w/v, bovine serum albumin [BSA]). Incubation was set at 37°C. The obtained cultures were utilized to prepare an inoculum in a sterile tube with 0.05% Tween 80 and 4.5 mL of phosphate buffer with glass beads (5 mm in diameter) by vortexing. After this, the cultures settled for a total of 45 mins; the clear bacterial supernatant was standardized to McFarland Number 1 using sterile water. The resulting bacterial concentration was approximately 1×107 CFU/mL, which was then diluted with sterile water. Overall, 100 µL of the dilution was added onto Middlebrook 7H10 agar plates containing 8–0.125 µg/mL of the agent. The drug (8 µg/mL) was dissolved in distilled water and diluted to the required concentration before being added to the agar medium. The drug MICs were read three weeks following 37°C incubation, which were regarded as the minimum drug concentration that could inhibit >99% growth of the bacterial culture when compared to controls. The results of this evaluation are presented in Table 2.
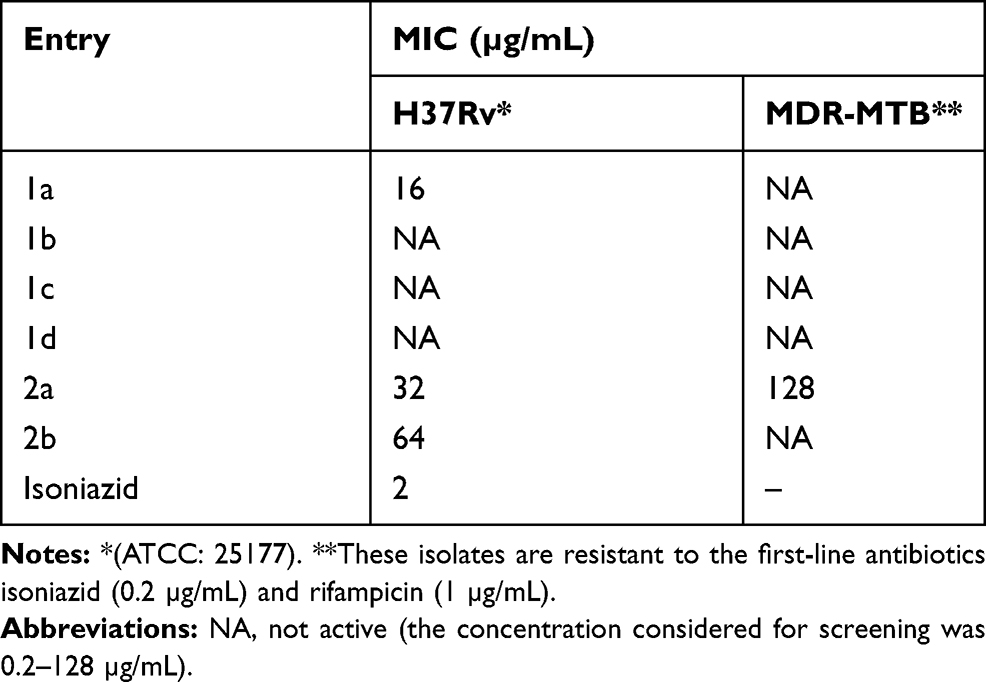 |
Table 2 The Anti-Tubercular Activity of Methyl 6-Methyl-2-Oxo-4-Substituedphenyl-1,2,3,4-Tetrahydropyrimidine-5-Carboxylate (1a–d) and Methyl 6-Methyl-4-Substituedphenyl-2-Thioxo-1,2,3,4-Tetrahydropyrimidine-5-Carboxylate (2a–b) Analogues Against H37Rv and MDR-MTB Strains of Mycobacterium tuberculosis |
Computational Chemistry
Accelry’s Discovery Studio 4.0 was used for molecular modeling by following the reported docking protocol.49 The CHARMm force-fields algorithm was employed. In the docking study, the X-ray co-crystal structures of the ligand and enzyme (PDB code: 5NQ5) were used to evaluate the binding affinity of the new THP. In prior docking studies, the protein was retrieved from the Protein Data Bank (PDB) and was subjected to the following sequences: Ligands and water exclusion, protein preparation and minimization energy, and the concerned ligand into the minimized protein to select the cavity site responsible for the binding domain. Molecular docking was conducted using the CDocker protocol. Docking scores obtained from the 10 best conformations were recorded as CDocker energy and CDocker interaction energy. The scores were then ranked according to CDocker energy. Stronger negative CDocker scores indicate more favorable binding interactions. The highest negative scores for PLP1, PLP2, Jain, and PMF demonstrated the strongest binding affinities between receptors and ligands; this was used to refine the binding pose. The docking protocol was validated by re-docking the original ligand to the receptor to ensure the correct binding pose, as reported in X-ray co-crystal PDB 5NQ5. The root mean square deviation difference between the docked and the actual crystal bound conformation of the native ligand was found to be 0.175 Å (Figure S7).
ADME Prediction
The SwissADME Web server50 was used to predict the pharmacokinetic properties, including absorption, distribution, metabolism, and excretion (ADME), of all compounds. The Lipinski rule of five conditions, the number of rotatable bonds, and total polar surface area (TPSA) were calculated to gain insights into the oral bioavailability of the compounds since these factors are known to influence it.51,52 The probability of a compound being a substrate for p-glycoprotein (p-gp) was also calculated because this protein is responsible for the efflux of xenobiotics outside the cells, which serves as a drug-resistance mechanism.53,54 Blood–brain barrier (BBB) permeability was also predicted for all compounds to consider the possibility of the agent entering the central nervous system (CNS), which may be undesirable and would require structural modification of the compounds. Additionally, the potential inhibition of some important cytochrome P450 (CYP 450) isoforms (namely, CYP1A2, CYP2C9, and CYP2D6) was predicted to investigate possible drug–drug interactions that can result from inhibiting one or more of these CYP 450 isoforms.55
Results and Discussion
Rational Design
In the current investigation, the title compounds, incorporating a pyrimidine core, have been designed as potential MTB-TMK inhibitors (Figure 2A). MTB-TMK inhibitors must be able to form critical hydrophilic interactions with amino acid residues ASN 100, ARG 74, and ARG 95 to ensure binding. The docking pose of thymidine monophosphate in the active site, depicted in Figure 2B, reveals the carbonyl and NH group of the thymine core-forming hydrogen bonds with the amino-acid backbone comprising ASN 100 and ARG 74, respectively; an additional hydrogen bond is formed between the oxygen atom of phosphate and the residue ARG 95.
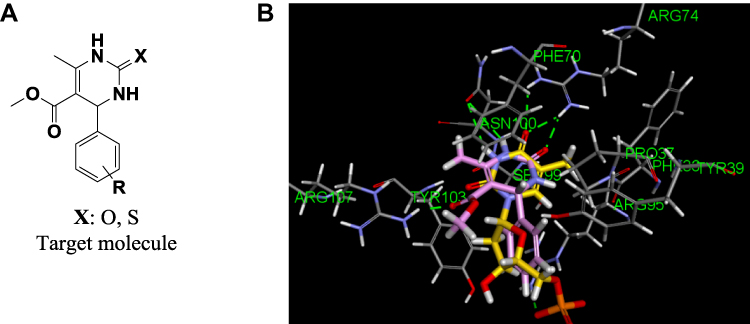 |
Figure 2 (A) Design of tetrahydro-pyrimidines as potential MTB-TMK inhibitors. (B) Superposition of thymidine monophosphate (yellow) and the molecular target pyrimidinone (violet) in the active site of MTB-TMK (PDB: 5NQ5). |
When exploiting the general binding feature of the TMK receptor, the application of a fragment-based design reveals a 1,2,3,4-tetrahydropyrimidinone (THP)-based scaffold for potential MTB-TMK inhibitors. We sought to investigate the MTB activity of six substituted phenyl THP derivatives (Scheme 1). THP was chosen as a molecular target for many reasons, including the structural similarities that it shares with MTB-TMK inhibitors, the variety of analogues that can be generated via a simple synthetic operation, and its excellent pharmacological and safety profile. The starting point of our study was to recognize that the unsubstituted lactam functional group present in the THP core would be essential for interacting with the thymidylate kinase receptor. Indeed, a preliminary docking study of the unsubstituted aryl THP into the binding site suggested that THP interacts favorably with the receptor by hydrogen bonding with the key residues ASN 100 and ARG 74 (Figure 2B). In addition, the aryl group of THP occupies the same pocket as the ribose moiety of thymidine monophosphate. The presence of an appropriate substituent on the phenyl ring would, therefore, be a determinant when generating a bioactive molecule. Therefore, we hypothesized that the target molecule might be an effective anti-TB agent.
Chemistry
The synthetic routes used to construct test compounds 1,2,3,4-tetrahydropyrimidinones (1a–d) and 1,2,3,4-tetrahydropyrimidinethiones (2a–b) are depicted in Scheme 1. The compounds were synthesized by the Biginelli reaction, and the yields were found to be satisfactory.
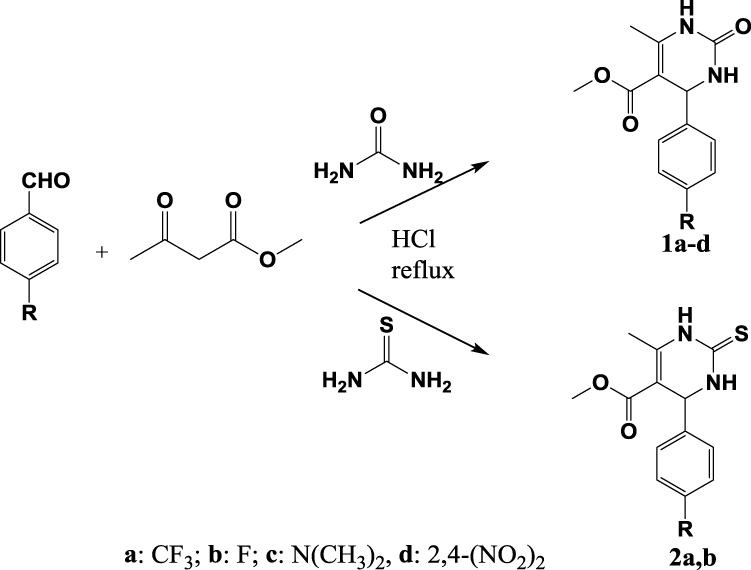 |
Scheme 1 Synthetic route to pyrimidinones 1a–d and pyrimidinethiones 2a, b. |
The test compounds were purified via recrystallization; the purity of the test compounds was ascertained by HPLC and determined to be >99%. The identities of the reported compounds were confirmed by comparing their melting points and molecular masses from LC–MS with the reported and calculated values, respectively. Fourier-transform infrared (FT-IR) spectra of compounds 1d (Figure S1) and 2a (Figure S4) revealed carbonyl stretching of the ester functional group in the range of 1716–1728 cm–1. In proton nuclear magnetic resonance (NMR), the methyl group on 1,2,3,4-tetrahydropyrimidine and ester group were observed at δ 3.39–3.68 and δ 2.27–2.39, respectively (Figures S2 and S5). In carbon-13 NMR, the carbonyl of the ester functional group appeared in the range of 165.41–175.8 ppm (Figures S3 and S6). The molecular ion peaks in LC–MS corresponded with the proposed molecular weight. The elemental analysis results fell within ±0.4% of the obtained values, and the cLogP of the title compound was calculated with ChemDraw Professional 16 (range: 1.5018–3.1084).
Anti-Tubercular Activity
Test compounds 1,2,3,4-tetrahydropyrimidinones (1a–d) and 1,2,3,4-tetrahydropyrimidinethiones (2a–b) were evaluated for their whole-cell anti-TB activity against the H37Rv and MDR strains of MTB. Well-characterized clinical isolates with resistance to rifampicin (Rif) and isoniazid (Inh) were selected for testing. Mutations within the rpoB gene and katG or inhA genes conferred resistance to Rif and Inh, respectively. MDR-MTB was found to be resistant to isoniazid and rifampicin at 0.2 and 1 µg/mL, respectively. The growth inhibition of the tested compounds was given in Table 2. From our studies, compound 2a demonstrated potential as a promising anti-TB agent against the H37Rv and MDR strains of MTB at 32 and 128 µg/mL respectively. Although compounds 1a and 2b demonstrated anti-TB activity against the H37Rv strain of MTB, they failed to exhibit activity against the MDR strain of MTB, which was resistant to the first-line antituberculosis drugs rifampicin and isoniazid. In addition, compound 1a, substituted by a trifluoromethyl group at fourth position of the benzene ring, was the most potent compound in the series and found to be 8 times less active than the reference isoniazid drug. Compound 1a was observed to exhibit two-fold higher activity than compound 2a, demonstrating the importance of having carbonyl instead of thiocarbonyl in the pyrimidine ring. Lastly, compounds 1b, 1c, and 1d did not show any anti-TB activity up to a concentration of 128 µg/mL against both MTB strains. It is interesting to note that the type and the position of the substituent on the aromatic ring play an important factor to obtain a bioactive THP compound. The presence of the trifluoromethyl group at the fourth position was highly favorable to the MTB growth inhibition while replacing by N(CH3)2 substituent leads to the inactive compound. The molecule 1d bearing two nitro groups at ortho and para position on the aryl ring was detrimental to the activity of the compounds. Replacement of the trifluoromethyl group by fluorine atom at the fourth position of the benzene ring reduces the potency of the compound. Molecular modeling investigation will provide insight into the structural requirement contributing to the potency of THP and may provide additionally an explanation for the inactivity of compounds 1b, 1c, and 1d.
Computational Chemistry
The thymidylate kinase receptor was chosen for this study on the basis of the results of our preliminary docking study conducted with unsubstituted 4-phenyltetrahydopyrimidinone (Scheme 1). It is interesting to note that the lack of activity observed for compounds 1a and 2a against cross-drug resistance to first-line anti-TB drugs may provide evidence for their mechanism of action. Our compounds may potentially act as either RNA polymerase inhibitor (rifampicin molecular target) or InhA inhibitor (isoniazid molecular target). However, the computational study of our compounds against both receptors did not convincingly explain the inactivity of derivatives 1c and 1d, which demonstrated favorable docking interactions against rifampicin and isoniazid molecular receptors. This is the reason why our computational chemistry was conducted against the thymidylate kinase receptor. Another aspect of this computational study that should be addressed is that THP exists as R and S stereoisomers as a result of a stereogenic center at position 4 of the structure. According to the docking results reported in Table 3, the stereoisomer (R) 1a displayed the highest docking score with a CDocker energy interaction of −36.75 kcal/mol. The interaction pattern of (R) 1a with the active site revealed four H-bonds with residues ASN 100, ARG 74, ARG 95, and ARG 107 (Figure 3). As expected, the THP ring interacted favorably through a hydrophilic interaction of ASN 100 and ARG 74 with carbonyl and NH, respectively. The trifluoromethyl group formed a moderate H-bond with the residue ARG 95, and a strong H-bond interaction was observed between the carbonyl ester and the amino acid ARG 107. These four key interactions may strongly contribute to the anti-mycobacterial activity of THP (R) 1a. In addition, two π interactions were observed between the phenyl ring and TYR 103 (π–π) and ARG 95 (π–cation). However, the stereoisomer (S) 1a displayed only two H-bond interactions between the THP ring and residues ARG 74 and ASN 100. The trifluoromethyl group pointed away from ARG 95, preventing any possible hydrophilic interaction. Further, stereoisomer (S) 1a did not show any π interactions. We also noted that the docking score of the stereoisomer (S) 1a is similar to that of (R) 1a, most likely because of the stronger interaction observed between (S) 1a and the residue ASN 100, with an H-bond distance of 2.61 Å, compared with 3.01 Å for (R) 1a. In general, the activity of THP derivatives against mycobacteria was attributed to the R form. The loss of potency for compound 1b and the weak potency for thiooxopyrimidines 2a and 2b can be explained by the type and strength of the interaction. Indeed, derivatives (R) 1a and (R) 2a and 2b did not show H-bonding with ASN 100 or a π–π interaction with TYR 103. As for the inactivity of compounds 1c and 1d, it could be attributed to their binding modes, which differ from those of the active compounds 1a, 2a, and 2b. Indeed, the binding modes of 1c and 1d were found opposite to those of active derivatives in which their pending aromatic ring of THP 1c and 1d were located to the pyrimidine core of active compounds and their pyrimidine core of inactive compounds 1c and 1d were found to interact with the ribose pocket. The presence of a moderate group at the ortho position of phenyl such as NO2 and a large group at the para position of phenyl such as -N(CH3)2 was not favorable and prevents them to interact strongly with ribose pocket. The lack of interaction is due to the steric hindrance effect between these groups and the amino acid residues present in the ribose site that shift the aromatic ring to another binding site. This proves that the THP may act as potential TMK inhibitors. On the basis of the docking study, the substituent at position 4 of the phenyl group in the (R) form was more favorable for interacting with the residue ARG 95 because of its close proximity to the ribose interacting pocket compared with the corresponding S stereoisomer. However, further study is needed to determine which enantiomer form, R or S, would be responsible for MTB activity, hence confirming their mode of action.
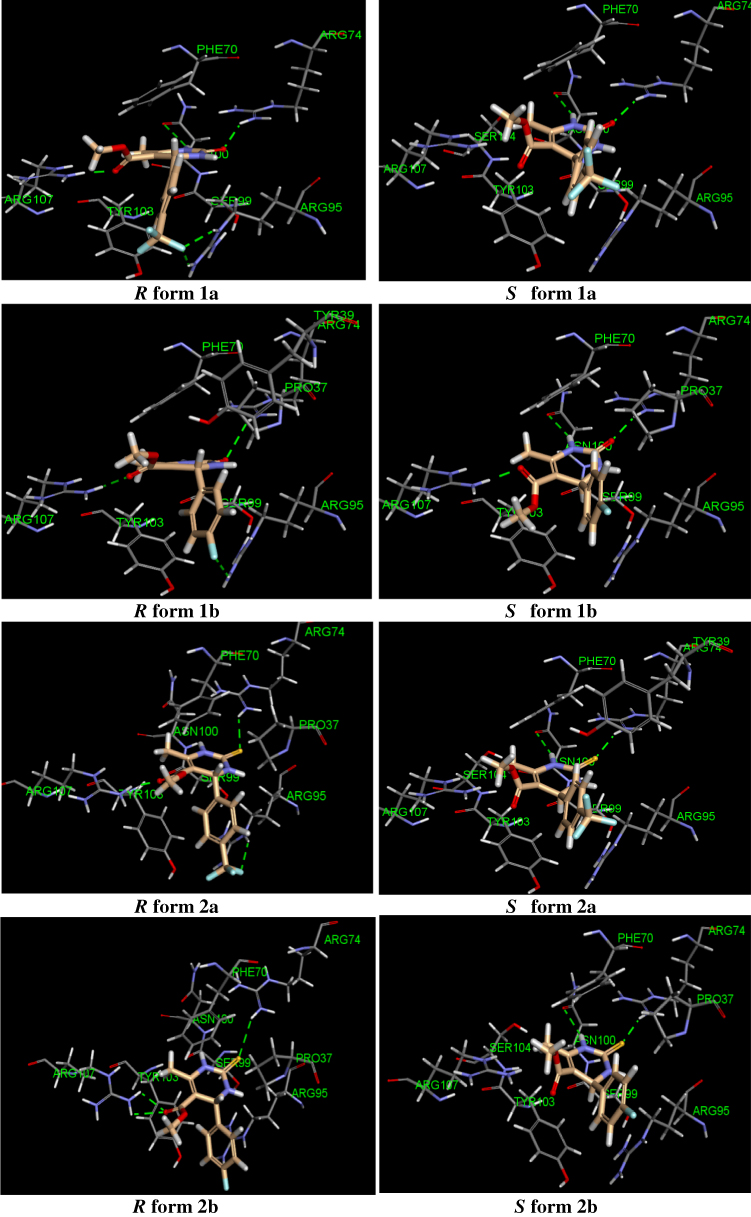 |
Figure 3 The predicted docking poses of tetrahydropyrimidinone derivatives in the active site of MTB thymidylate kinase (PDB: 5NQ5). H-bond interactions are depicted as dotted green lines. |
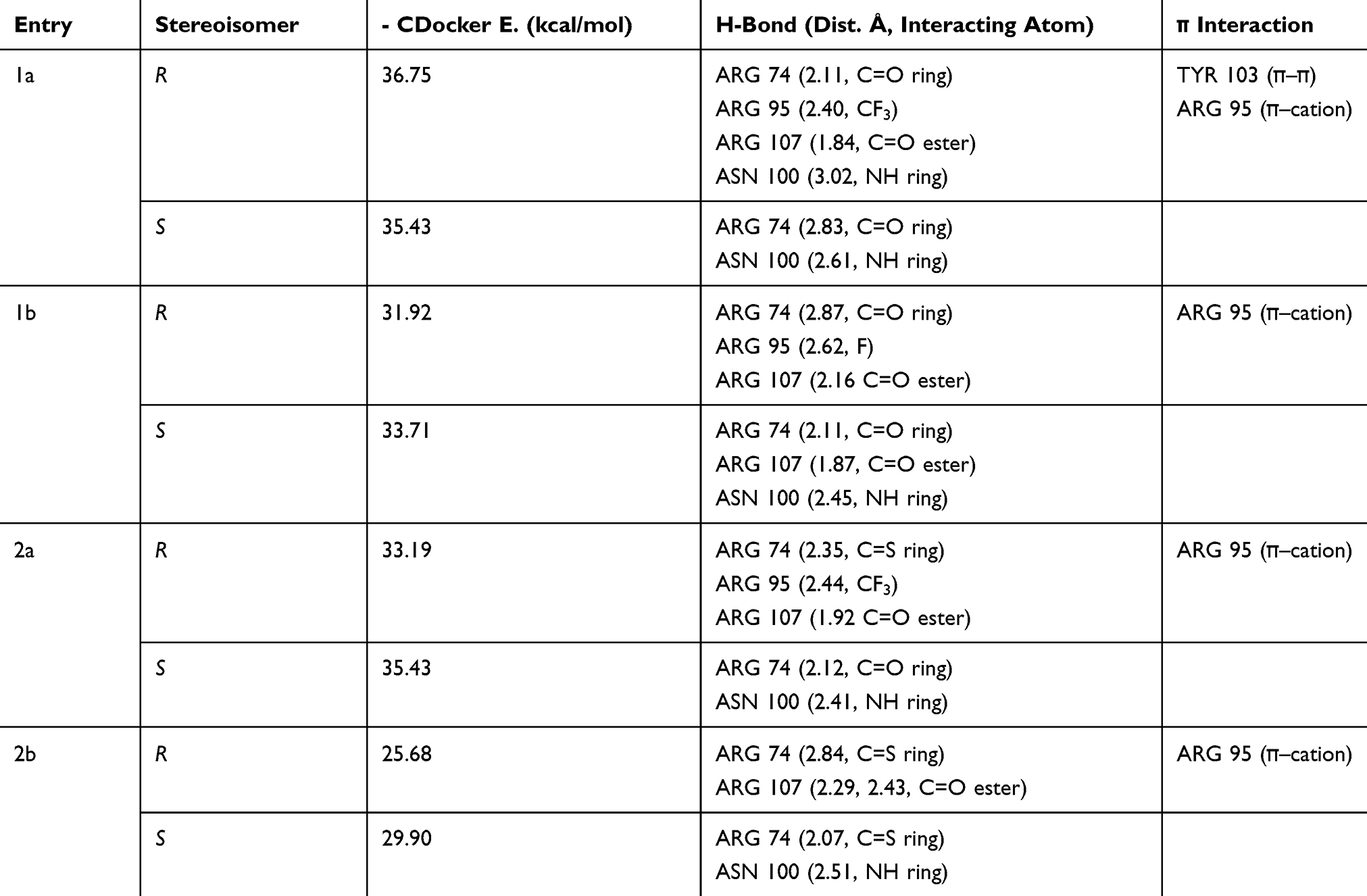 |
Table 3 Docking Results for Compounds 1a, b and 2a, b with the MTB Thymidylate Kinase Receptor (PDB: 5NQ5) |
On the basis of the bioactivity, the leading compound 1a was identified as a potential anti-TB agent. Further direction will be pursued to design more potent inhibitors through a structure-based drug design approach. Figure 4 illustrates the design of a novel possible inhibitor. In terms of receptor specificity, the preliminary docking study revealed favorable interactions with substituted tetrahydropyrimidinone in R1 by the methyl and phenyl group and in R2 by –CF3, –F, –OH, –NH2, and –CO2H in both the para and meta positions. This new research direction will also help us to confirm the mechanism of action by which the molecules inhibit MTB.
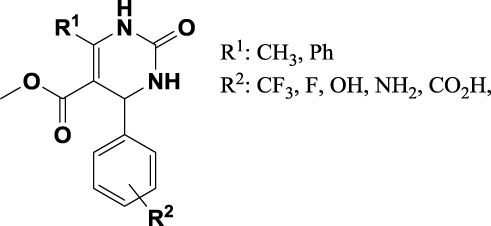 |
Figure 4 Future development of anti-TB drug discovery research on novel substituted pyrimidinones. |
ADME Prediction
The results of the ADME prediction by the SwissADME webserver are presented in Table 4. All compounds obey the Lipinski rule of five conditions, except for compound 1d, which has a single violation—specifically, the number of oxygen and nitrogen atoms is >10. The number of rotatable bonds in each compound is <7, which is preferred because it leads to a higher probability of being orally bioavailable. The cLogP and TPSA values (Tables 1 and 4, respectively) are also in the optimum range for most of the compounds. The predicted gastrointestinal absorption is high for all compounds, except for compound 1d, which has a low predicted gastrointestinal absorption, possibly because of the high TPSA resulting from a large number of oxygen and nitrogen atoms in its structure. It can be concluded that the compounds, in general, have favorable properties that indicate oral bioavailability, with the exception of compound 1d.
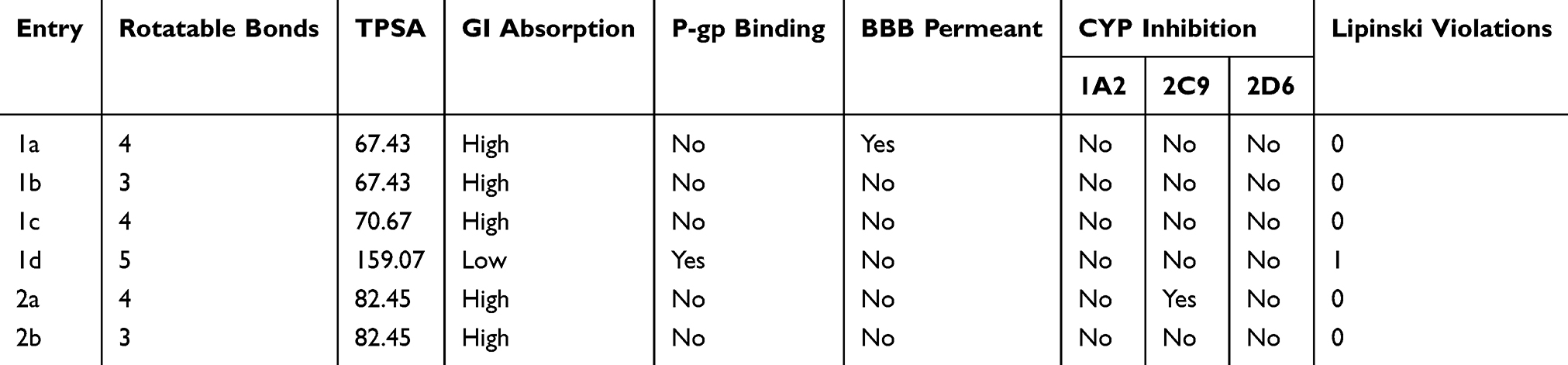 |
Table 4 The Predicted ADME Properties of 1,2,3,4-Tetrahydropyrimidinone (1a–d) and 1,2,3,4-Tetrahydropyrimidinethione (2a–b) Compounds |
The predicted p-gp binding probability shows that none of the compounds, except for compound 1d, are candidate substrates for p-gp; thus, the compounds are not expected to result in resistance via the efflux mechanism. The BBB permeability prediction indicates that the compounds are not permeable to the BBB; hence, they cannot enter the central nervous system. Only compound 1a is predicted to be permeable to the BBB. Finally, the results of the CYP inhibition study show that most compounds do not inhibit any of the evaluated CYP isoforms; only compound 2a is predicted to be a possible inhibitor of CYP2C9.
Conclusion
In the present study, we designed several pyrimidinones and pyrimidinethiones as potential thymidylate kinase inhibitors, and the compounds were synthesized and evaluated for their anti-MTB activity. Our findings show that only pyrimidinone 1a and pyrimidinethione 2a demonstrate moderate anti-TB activity against MTB H37Rv strains. All compounds were found to be inactive against MTB strains resistant to the first-line antituberculosis drugs rifampicin and isoniazid, with the exception of pyrimidinethione 2a, which showed weak inhibition. The presence of a trifluoromethyl group in the para position of the pendant aromatic ring is essential for retaining the activity of compounds 1a and 2a. Thymidylate kinase might be the molecular target responsible for the bioactivity of these compounds, although they did not exhibit any activity or demonstrated weak activity against rifampicin- and isoniazid-resistant MTB strains. The molecular docking studies reveal the importance of the size and the position of the substituent on the pendant aromatic ring of THP to interact favorably with the thymidylate kinase receptor. The trifluoromethyl group at 4-position of aryl of 1a and 2a was found to interact through hydrogen bonding with the ribose pocket receptor. The predicted binding modes of the inactive compounds 1c and 1d differ from those of 1a and 2a, explaining their lack of activity due to the steric hindrance effect of the group on the aromatic ring. The prediction and analysis of the ADME properties of the active compounds demonstrate favorable ADME properties; hence, compounds 1a and 2a can serve as potential lead compounds in the development of potential anti-TB drugs. However, further studies are needed to elucidate the mechanism of action by developing more potent inhibitors through an in silico structured-based design approach, as well as to identify which enantiomer form, R or S, is responsible for the bioactivity.
Acknowledgments
The authors wish to express their gratitude to the Deanship of Scientific Research, King Faisal University, Kingdom of Saudi Arabia, for providing support and encouragement. The funders had no role in the study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors declare no conflicts of interest.
References
1. WHO. Global tuberculosis report 2018, Geneva; 2018. Availabe from: https://www.who.int/tb/publications/global_report/en/.
2. Shruthi T, Eswaran S, Shivarudraiah P, Narayanan S, Subramanian S. Synthesis, antituberculosis studies and biological evaluation of new quinoline derivatives carrying 1, 2, 4-oxadiazole moiety. Bioorg Med Chem Lett. 2019;29(1):97–102. doi:10.1016/j.bmcl.2018.11.002
3. Marcos AE. The global situation of MDR-TB. Tuberculosis. 2003;83:44–51. doi:10.1016/S1472-9792(02)00058-6
4. Caminero JA, Sotgiu G, Zumla A, Migliori GB. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect Dis. 2010;10(9):621–629. doi:10.1016/S1473-3099(10)70139-0
5. Hu Y, Xu L, He YL, et al. Prevalence and molecular characterization of second-line drugs resistance among multidrug-resistant Mycobacterium tuberculosis isolates in Southwest of China. Biomed Res Int. 2017;2017:4563826. doi:10.1155/2017/4563826
6. Parida SK, Axelsson-Robertson R, Rao MV, et al. Totally drug-resistant tuberculosis and adjunct therapies. J Intern Med. 2015;277(4):388–405. doi:10.1111/joim.2015.277.issue-4
7. Cox E, Laessig K. FDA approval of bedaquiline — the benefit–risk balance for drug-resistant tuberculosis. N Engl J Med. 2014;371(8):689–691. doi:10.1056/NEJMp1314385
8. Barry Iii CE. Timing is everything for compassionate use of delamanid. Nat Med. 2015;21(3):211. doi:10.1038/nm.3823
9. Familiar O, Munier‐Lehmann H, Negri A, et al. Exploring acyclic nucleoside analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. ChemMedChem. 2008;3(7):1083–1093. doi:10.1002/cmdc.v3:7
10. Vanheusden V, Munier-Lehmann H, Pochet S, Herdewijn P, Van Calenbergh S. Synthesis and evaluation of thymidine-5′-O-monophosphate analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. Bioorg Med Chem Lett. 2002;12(19):2695–2698. doi:10.1016/S0960-894X(02)00551-6
11. Alexandrova LA, Chekhov VO, Shmalenyuk ER, Kochetkov SN, El-Asrar RA, Herdewijn P. Synthesis and evaluation of C-5 modified 2’-deoxyuridine monophosphates as inhibitors of M. tuberculosis thymidylate synthase. Bioorg Med Chem. 2015;23(22):7131–7137. doi:10.1016/j.bmc.2015.09.053
12. Shmalenyuk ER, Chernousova LN, Karpenko IL, et al. Inhibition of Mycobacterium tuberculosis strains H37Rv and MDR MS-115 by a new set of C5 modified pyrimidine nucleosides. Bioorg Med Chem. 2013;21(17):4874–4884. doi:10.1016/j.bmc.2013.07.003
13. Toti KS, Verbeke F, Risseeuw MD, Frecer V, Munier-Lehmann H, Van Calenbergh S. Synthesis and evaluation of 5′-modified thymidines and 5-hydroxymethyl-2′-deoxyuridines as Mycobacterium tuberculosis thymidylate kinase inhibitors. Bioorg Med Chem. 2013;21(1):257–268. doi:10.1016/j.bmc.2012.10.018
14. Van Poecke S, Munier-Lehmann H, Helynck O, Froeyen M, Van Calenbergh S. Synthesis and inhibitory activity of thymidine analogues targeting Mycobacterium tuberculosis thymidine monophosphate kinase. Bioorg Med Chem. 2011;19(24):7603–7611. doi:10.1016/j.bmc.2011.10.021
15. Familiar O, Munier-Lehmann H, Aínsa JA, Camarasa M-J, Pérez-Pérez M-J. Design, synthesis and inhibitory activity against Mycobacterium tuberculosis thymidine monophosphate kinase of acyclic nucleoside analogues with a distal imidazoquinolinone. Eur J Med Chem. 2010;45(12):5910–5918. doi:10.1016/j.ejmech.2010.09.056
16. Fioravanti E, Adam V, Munier-Lehmann H, Bourgeois D. The crystal structure of Mycobacterium tuberculosis thymidylate kinase in complex with 3‘-azidodeoxythymidine monophosphate suggests a mechanism for competitive inhibition. Biochemistry. 2005;44(1):130–137. doi:10.1021/bi0484163
17. Pochet S, Dugue L, Labesse G, Delepierre M, Munier-Lehmann H. Comparative study of purine and pyrimidine nucleoside analogues acting on the thymidylate kinases of Mycobacterium tuberculosis and of humans. Chembiochem. 2003;4(8):742–747. doi:10.1002/cbic.200300608
18. Song L, Merceron R, Gracia B, et al. Structure guided lead generation toward nonchiral M. tuberculosis thymidylate kinase inhibitors. J Med Chem. 2018;61(7):2753–2775. doi:10.1021/acs.jmedchem.7b01570
19. Kawatkar SP, Keating TA, Olivier NB, et al. Antibacterial inhibitors of gram-positive thymidylate kinase: structure–activity relationships and chiral preference of a new hydrophobic binding region. J Med Chem. 2014;57(11):4584–4597. doi:10.1021/jm500463c
20. Martinez-Botella G, Breen JN, Duffy JE, et al. Discovery of selective and potent inhibitors of gram-positive bacterial thymidylate kinase (TMK). J Med Chem. 2012;55(22):10010–10021. doi:10.1021/jm3011806
21. Gasse C, Douguet D, Huteau V, Marchal G, Munier-Lehmann H, Pochet S. Substituted benzyl-pyrimidines targeting thymidine monophosphate kinase of Mycobacterium tuberculosis: synthesis and in vitro anti-mycobacterial activity. Bioorg Med Chem. 2008;16(11):6075–6085. doi:10.1016/j.bmc.2008.04.045
22. Naik M, Raichurkar A, Bandodkar BS, et al. Structure guided lead generation for M. tuberculosis thymidylate kinase (Mtb TMK): discovery of 3-cyanopyridone and 1,6-naphthyridin-2-one as potent inhibitors. J Med Chem. 2015;58(2):753–766. doi:10.1021/jm5012947
23. Hoffmann -H-H, Kunz A, Simon VA, Palese P, Shaw ML. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc Natl Acad Sci U S A. 2011;108(14):
24. Prachayasittikul S, Pingaew R, Worachartcheewan A, et al. Roles of pyridine and pyrimidine derivatives as privileged scaffolds in anticancer agents. Mini Rev Med Chem. 2017;17(10):869–901. doi:10.2174/1389557516666160923125801
25. Karnail SA, Brian NS, Steven EU, et al. Dihydropyrimidine calcium channel blockers. 3. 3-Carbamoyl-4-aryl-1,2,3,4-tetrahydro-6-methyl-5-pyrimidinecarboxylic acid esters as orally effective antihypertensive agents. J Med Chem. 1991;34(2):806–811. doi:10.1021/jm00106a048
26. Jauk B, Pernat T, Kappe CO. Design and synthesis of a conformationally rigid mimic of the dihydropyrimidine calcium channel modulator SQ 32,926. Molecules. 2000;5:227–239. doi:10.3390/50300227
27. Venugopala KN, Nayak SK, Pillay M, Prasanna R, Coovadia YM, Odhav B. Synthesis and antitubercular activity of 2-(substituted phenyl/benzyl-amino)-6-(4-chlorophenyl)-5-(methoxycarbonyl)-4-methyl-3,6-dihydropyrimidin-1-ium chlorides. Chem Biol Drug Des. 2013;81(2):219–227. doi:10.1111/cbdd.12065
28. Venugopala KN, Dharma Rao GB, Bhandary S, et al. Design, synthesis, and characterization of (1-(4-aryl)-1H-1,2,3-triazol-4-yl)methyl, substituted phenyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates against Mycobacterium tuberculosis. Drug Des Dev Ther. 2016;10:2681–2690. doi:10.2147/DDDT
29. Wael AES, Ibrahim FN, Adel AH, Abdel R. C-Furyl glycosides, II: synthesis and antimicrobial evaluation of C-furyl glycosides bearing pyrazolines, isoxazolines, and 5,6-dihydropyrimidine-2(1H)-thiones. Monatsh Chem. 2009;140:365–370. doi:10.1007/s00706-008-0033-2
30. Shah TB, Gupte A, Patel MR, Chaudhari VS, Patel H, Patel VC. Synthesis and in vitro study of biological activity of heterocyclic N-Mannich bases of 3,4-dihydropyrimidine-2(1H)-thiones. Indian J Chem. 2010;49 B(05):578–586.
31. Sushilkumar SB, Devanand BS. Synthesis and anti-inflammatory activity of some 2-amino-6-(4-substituted aryl)-4-(4-substituted phenyl)-1,6-dihydropyrimidine-5-yl-acetic acid derivatives. Acta Pharm. 2003;53:223–229.
32. Nofal ZM, Fahmy HH, Zarea ES, El-Eraky W. Synthesis of new pyrimidine derivatives with evaluation of their anti-inflammatory and analgesic activities. Acta Pol Pharm. 2011;68(4):507–517.
33. Keshab MB, Nancy SY, Promise ME, et al. Anti-diabetic activity of dihydropyrimidine scaffolds and structural insight by single crystal X-ray studies. Med Chem. 2019.
34. Rajanarendar E, Reddy MN, Murthy KR, et al. Synthesis, antimicrobial, and mosquito larvicidal activity of 1-aryl-4-methyl-3,6-bis-(5-methylisoxazol-3-yl)-2-thioxo-2,3,6,10b-tetrahydro-1H-pyrimido[5,4-c]quinolin-5-ones. Bioorg Med Chem Lett. 2010;20(20):6052–6055. doi:10.1016/j.bmcl.2010.08.060
35. Venugopala KN, Gleiser RM, Chalannavar RK, Odhav B. Antimosquito properties of 2-substituted phenyl/benzylamino-6-(4-chlorophenyl)-5-methoxycarbonyl-4-methyl-3,6-dihydropyrimidinium chlorides against Anopheles arabiensis. Med Chem. 2014;10(2):211–219. doi:10.2174/157340641002140131164945
36. Bairagi KM, Venugopala KN, Mondal PK, et al. Larvicidal study of tetrahydropyrimidine scaffolds against Anopheles arabiensis and structural insight by single crystal X-ray studies. Chem Biol Drug Des. 2018;92(6):1924–1932. doi:10.1111/cbdd.2018.92.issue-6
37. Khedr MA, Pillay M, Chandrashekharappa S, et al. Molecular modeling studies and anti-TB activity of trisubstituted indolizine analogues; molecular docking and dynamic inputs. J Biomol Struct Dyn. 2018;36(8):2163–2178. doi:10.1080/07391102.2017.1345325
38. Venugopala KN, Chandrashekharappa S, Pillay M, et al. Synthesis and structural elucidation of novel benzothiazole derivatives as anti-tubercular agents: in-silico screening for possible target identification. Med Chem. 2019;15(3):311–326. doi:10.2174/1573406414666180703121815
39. Venugopala KN, Sandeep C, Pillay M, et al. Computational, crystallographic studies, cytotoxicity and anti-tubercular activity of substituted 7-methoxy-indolizine analogues. PLoS One. 2019;14(6):
40. Venugopala KN, Tratrat C, Pillay M, et al. Anti-tubercular activity of substituted 7-methyl and 7-formylindolizines and in silico study for prospective molecular target identification. Antibiotics. 2019;8(247):1–16. doi:10.3390/antibiotics8040247
41. Nayak SK, Venugopala KN, Chopra D, Row TNG. Insights into conformational and packing features in a series of aryl substituted ethyl-6-methyl-4-phenyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates. CrystEngComm. 2011;13(2):591–605. doi:10.1039/C0CE00045K
42. Ali F, Khan KM, Salar U, et al. Dihydropyrimidones: as novel class of beta-glucuronidase inhibitors. Bioorg Med Chem. 2016;24(16):3624–3635. doi:10.1016/j.bmc.2016.06.002
43. Dondoni A, Massi A. Parallel synthesis of dihydropyrimidinones using Yb(III)-resin and polymer-supported scavengers under solvent-free conditions. A green chemistry approach to the Biginelli reaction. Tetrahedron Lett. 2001;42(45):7975–7978. doi:10.1016/S0040-4039(01)01728-2
44. Mohamadpour F, Lashkari M. Three-component reaction of β-keto esters, aromatic aldehydes and urea/thiourea promoted by caffeine, a green and natural, biodegradable catalyst for eco-safe Biginelli synthesis of 3,4-dihydropyrimidin-2(1H)-ones/thiones derivatives under solvent-free conditions. J Serb Chem Soc. 2018;83(6):673–684.
45. Liu Q, Pan N, Xu J, Zhang W, Kong F. Microwave-assisted and iodine-catalyzed synthesis of dihydropyrimidin-2-thiones via biginelli reaction under solvent-free conditions. Synth Commun. 2013;43(1):139–146. doi:10.1080/00397911.2011.593289
46. Martin A, Morcillo N, Lemus D, et al. Multicenter study of MTT and resazurin assays for testing susceptibility to first-line anti-tuberculosis drugs. Int J Tuberc Lung Dis. 2005;9(8):901–906.
47. Yoshikuni O, Mayumi T, Kenichi S. Inhibitory activity of quinolones against DNA gyrase of Mycobacterium tuberculosis. J Antimicrob Chemother. 2001;47:447–450. doi:10.1093/jac/47.4.447
48. Middlebrook G, Reggiards Z, Tigertt WD. Automable radiometric detection of growth of Mycobacterium tuberculosis in selective media. Am Rev Respir Dis. 1977;115:1067–1069.
49. Chandrashekharappa S, Venugopala KN, Tratrat C, et al. Efficient synthesis and characterization of novel indolizines: exploration of in vitro COX-2 inhibitory activity and molecular modelling studies. New J Chem. 2018;42(7):4893–4901. doi:10.1039/C7NJ05010K
50. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi:10.1038/srep42717
51. Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623. doi:10.1021/jm020017n
52. Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1(4):337–341. doi:10.1016/j.ddtec.2004.11.007
53. Yu DK. The contribution of P‐glycoprotein to pharmacokinetic drug‐drug interactions. J Clin Pharmacol. 1999;39(12):1203–1211. doi:10.1177/00912709922012006
54. Fromm M. Importance of P‐glycoprotein for drug disposition in humans. E J Clin Invest. 2003;33:6–9. doi:10.1046/j.1365-2362.33.s2.4.x
55. Lin JH. CYP induction-mediated drug interactions: in vitro assessment and clinical implications. Pharm Res. 2006;23(6):1089–1116. doi:10.1007/s11095-006-0277-7
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.