")
Back to Journals » Journal of Blood Medicine » Volume 10
Impact Of TP53 Gene Promoter Methylation On Chronic Lymphocytic Leukemia Pathogenesis And Progression
Authors Saeed WH, Eissa AA , Al-Doski AA
Received 2 July 2019
Accepted for publication 5 November 2019
Published 25 November 2019 Volume 2019:10 Pages 399—404
DOI https://doi.org/10.2147/JBM.S221707
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Waleed Haji Saeed,1 Adil Abozaid Eissa,2 Adnan Anwar Al-Doski2
1Department of Nursing, Akre Technical Institute, Duhok Polytechnic University, Duhok, Iraq; 2Department of Pathology, College of Medicine, University of Duhok, Duhok, Iraq
Correspondence: Adil Abozaid Eissa
Department of Pathology, College of Medicine, University of Duhok, 9 Azadi Hospital Road, Duhok, Iraq
Tel +9647504576051
Email [email protected]
Background: Chronic lymphocytic leukemia (CLL) is a malignant lymphoid disorder that results from the overgrowth of mature-looking lymphoid cells in the blood and lymphatic tissue. Various clinical presentations have been attributed to the disease as a result of the different underlying genetic and epigenetic alterations. The current study has been initiated to study the role of an epigenetic alteration affecting the promoter of the TP53gene on CLL pathogenesis and progression.
Methods: The current study involved 54 newly diagnosed patients presenting with CLL as well as 30 normal individuals as controls. After obtaining verbal consent, data collection was done and the blood collected from all enrolled individuals for hematological investigations as well as for molecular categorization of TP53 methylation status. Methylation-specific polymerase chain reaction (MS-PCR) technique was used to define the methylation status of the TP53 gene promoter that encompasses DNA extraction, bisulfite conversion, conventional PCR amplification, running on agarose gel and documentation. Finally, statistical analysis was done to assess any correlation of the TP53 epigenetic alteration to the disease etiology and the progression.
Results: In the current study, all controls and 42 of 54 patients show unmethylated TP53 gene promoter; on the other hand, the methylated promoter was detected among 12 patients with a p-value of 0.001. TP53 gene promoter methylation significantly linked to reduced platelet count (p-value of 0.047) and advanced stage at presentation (p-value of 0.076). No significant differences were seen among both methylated and unmethylated TP53 promoters in relation to the age of the affected individuals, total white blood cell counts and hemoglobin level of the affected individuals.
Conclusion: The current study revealed a significant correlation of TP53 gene promoter methylation to chronic lymphocytic leukemia pathogenesis and lower platelet counts.
Keywords: chronic lymphocytic leukemia, epigenetic alteration, TP53 gene promoter
Background
Chronic lymphocytic leukemia (CLL) is a clonal disorder that results in the accumulation of morphologically mature-looking and immunologically incompetent lymphoid cells in the bone marrow, peripheral blood, and lymphatic tissues.1,2 Until recently, it was thought that CLL developed mainly from impaired apoptosis and evasion of cell death; however, it has been acknowledged that CLL-affected tissue comprising the bone marrow contains proliferation centers that support the top-up theory of the CLL cell population.3–5 CLL is a heterogeneous disease as some patients may progress rapidly, even though they may fail to respond to therapy, and others may remain stable for years without any intervention, and this mirrors the genetic configuration and the epigenetic modification of different genes that lead to disease development, stability, progression, and response to different chemotherapeutic agents.6,7
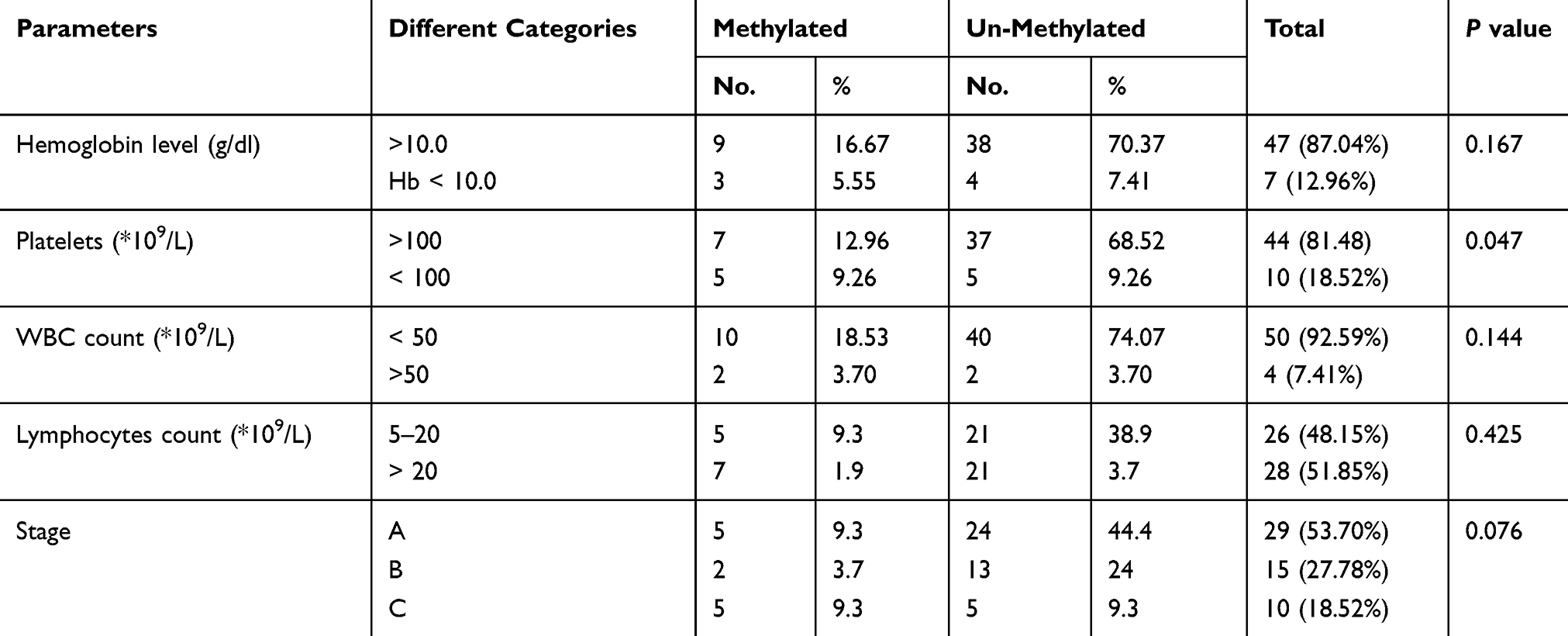 |
Table 1 Correlation Of TP53 Gene Methylation Status To Hematological Data And Staging Categories |
Epigenetic modification may result from different strategies including DNA methylation/hypomethylation, histone alterations, and chromatin conversion. The epigenetic alteration has significant roles in controlling different genes' activities, genomic imprint, reproduction, embryonic development, inactivation of the X chromosome, tissue remodeling, as well as in disease pathogenesis, progression, and chemo-refractoriness.8–12 DNA methylation arises as a consequence of the addition of a methyl group to cytosine in the CpG dinucleotides that are characteristically seen as collections in small stretches of DNA termed “CpG islands” in the promoter regions of different housekeeping, the tumor suppressor, and some tissue-specific genes.13,14 Hypermethylation in the promoter regions through the action of DNA methyltransferases enzyme results in gene suppression and inactivation, while hypomethylation results in gene activation.8,15
Protein 53 (p53), an end product for the TP53 gene, plays a dynamic role in sustaining genetic stability and regulating the cellular response to DNA damage and other replicative stresses. Activation of p53 can ultimately lead to either DNA repair or programmed cell death, thus preventing cancer formation, i.e., it functions as a tumor suppressor and TP53 is classified as a tumor suppressor gene.16 Inactivation of the TP53gene through different mutations like in Li-Fraumeni syndrome or hypo-activation through epigenetic alteration by methylation impairs apoptosis and may lead to many types of cancers, including CLL.17
The current study was initiated to estimate the effect of methylation of different genes functions and to correlate the TP53 non-functioning (through methylation) with the pathogenesis and progression of CLL.
Methods
The current study represents a case-control study. A total of 54 newly diagnosed patients with CLL attending Nanakali hospital for blood diseases in Erbil, Iraq from January 1, 2017 to July 30, 2018 were enrolled. These patients were diagnosed as described by WHO criteria, which include absolute lymphocytosis (absolute lymphocytosis >5*109/L) with typical immunological expression by flowcytometry.5 Also, 30 age- and sex-matched individuals with a negative history of previous “hematological and non-hematological malignancies“ were recruited for the purpose of the current study and used as controls. At first, verbal informed consent was obtained from all enrolled patients and the study was approved by the appropriate ethical committee at the University of Duhok College of Medicine and Duhok Directorate of Health, Duhok, Iraq and followed the Declaration of Helsinki ethical values for medical research concerning human beings.
Supportive information was collected from all enrolled individuals and then blood (also bone marrow sample was taken from patients) were collected in K2-EDTA tubes from each enrollee and all had their DNA extracted by a modified salting-out extraction method adopted by Iranpur-Mubarakeh and his colleague that yield high quantity with high purity DNA.18,19
The methylation-specific polymerase chain reaction (MS-PCR) technique was used in the current study, which requires an initial conversion of DNA structure and then amplification using specific primer sets for methylated and unmethylated DNA targets.20
At first, DNA bisulfite conversion is done with the aid of the EpiTect fast bisulfite conversion kit from Qiagen (Germany) and all steps were done according to the manufacturer's instructions. Then the converted DNA samples were amplified using M-P53-F 5ʹ-TTCGGTAGGCGGATTATTTG-3ʹ and M-P53-R 5ʹ-AAATATCCCCGAAACCCAAC-3ʹ primers for the methylated TP53gene and U-P53-F 5ʹ-TTGGTAGGTGGATTATTTGTTT-3ʹ and U-P53-R 5ʹ-CCAATCCAAAAAAACATATCAC-3ʹ primers for the un-methylated TP53gene.21 Initially a master mix was prepared using 2X Hot Start Master Mix from GeNet Bio (South Korea) and then amplification was performed using a PCR thermal cycler (2720) from Applied Biosystems, Hitachi (Japan). PCR conditions were as follows: for methylated TP53gene (94°C for 5 min, 35 cycles at 94°C for 30 s, 60°C for 40 s, 72°C for 60 s, 72°C for 7 min) and for unmethylated gene (94°C for 5 min, 35 cycles at 94°C for 30 s, 53°C for 40 s, 72°C for 60 s, 72°C for 7 min). Finally, the amplified products were subjected to 2% agarose gel electrophoresis. As shown in Figure 1, the methylated DNA sample produced a 193-bp DNA product with the M-P53 primers set, while the unmethylated DNA samples produced no band. On the other hand, the unmethylated DNA samples produced a 247-bp DNA product with the U-P53 primers set, while the methylated DNA samples produced no band. Appropriate methylated and unmethylated DNA controls were obtained from Qiagen [Epitec®Control DNA (Human), unmethylated and bisulfite converted (100)] and ran in parallel to patients’ samples in all runs.
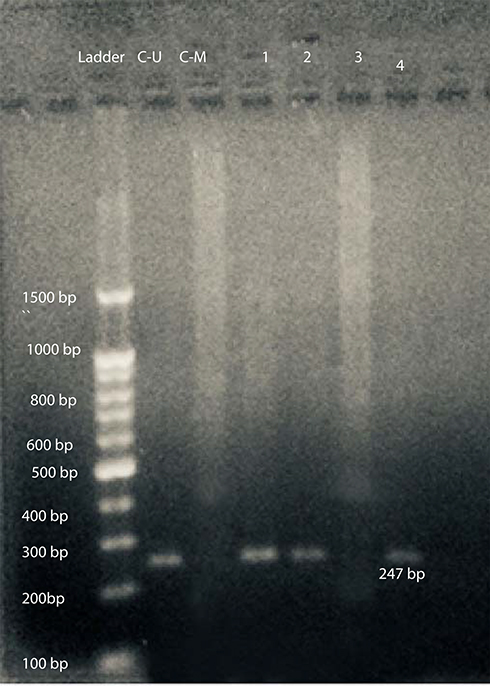 |
Figure 1 Gel electrophoresis of amplified products with U-P53 primers for determination of P53 methylation status. L: represents the 100-bp ladder; CU: unmethylated commercial P53 control DNA (247 bp); C-M: methylated commercial P53 control DNA; 1, 2, & 4 reveals unmethylated P53 DNA patient samples; and 3: represents methylated P53 DNA patient samples. |
After data collection, data were analyzed using SPSS 24. Frequency and frequency percentages were used to describe the study sample. Student’s t-test was used for continuous variables, and chi-square or Fisher exact test were used for categorical variables. P-value was considered to be significant if less than 0.05.
Results
A total of 54 patients with CLL were enrolled in the current study. Their ages ranged from 44 to 80 years with a median age of 62.2 ± 8.2 years. Of these, 37 (68.5%) were male and 17 (31.5%) were female, with a male to female ratio of 2.18.
After amplification with specific primers designed for determination of TP53gene methylation status, samples from 42 patients (77.8%) and all 30 controls (100%) show a 247-bp pattern with U-P53 (F&R) primers that is consistent with unmethylation status (Figure 1). On the other hand, samples from 12 patients (22.2%) revealed no band with U-P53 (F&R) primers, but show a 193-bp band with M-P53 (F&R) primers, which is consistent with methylation status (Figure 2). The methylated status was seen among 17.6% of female patients (3 females) and only among 23.1% of male patients (9 males) with a p-value of 0.539.
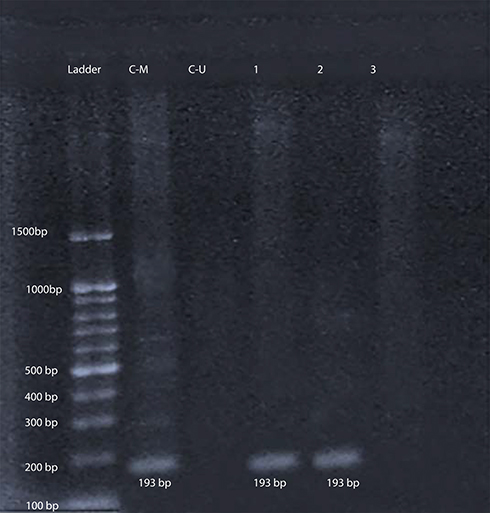 |
Figure 2 Gel electrophoresis of amplified products with M-P53 for determination of P53 methylation status. L: represents the 100-bp ladder; CM: methylated commercial P53 control DNA (193 bp); CU: unmethylated commercial P53 control DNA; 1 & 2 reveal methylated P53 DNA patient samples; and 3: represents unmethylated P53 DNA patient samples. |
Methylated P53 had been associated significantly with CLL groups in comparison to controls with a p-value of <0.001. Using a t-test as shown in Table 1, the methylated status shows a significant correlation with low platelet count (p-value 0.047) as well as advanced stage (p-value 0.076); despite this, it does not show a significant correlation with hemoglobin level (p-value 0.167), total white blood cell count (p-value 0.144), and absolute lymphocytes count (p-value 0.425).
Discussion
The exact mechanism underlying CLL is still unclear; nevertheless, various molecular changes (genetic and epigenetic) have been implicated in CLL tumorigenesis and disease progression.1,22
From these, TP53 gene modification disturbs the process of apoptosis and results in prolonged survival of the malignant B cells in tissue and peripheral blood.23 Also, TP53 inactivation had been criticized for fludarabine resistance in 30–40% of patients with CLL, the backbone drug for this leukemia. Only a quarter of cases had contributed to TP53 gene deletion and the remaining three-fourths had contributed to gene inactivation through methylation of the promoter region of the gene.24,25 As the methylation results in gene inactivation and in accordance with the previous review written by Wang and Wang, the current study revealed a significant association of TP53 gene promoter methylation with the CLL development.26 Not only the pathogenesis but also the advanced stage (low platelets) had been associated significantly with TP53 gene suppression as through promoter methylation and this poor prognosis was in accordance with the previous study conducted by Zenz and his coworkers.23,25 On the other hand, methylation of the TP53 gene promoter was not significantly associated with low hemoglobin level or high total white cells counts, and this may explain the heterogeneous nature of CLL and its correlation with other gene malfunctions. The various effect of TP53 methylation on platelet count, hemoglobin level, and total white cell count may represent the different effects of TP53 dysfunction on the micro-environment and interaction of different genes with the hemopoietic proliferation and maturation.27,28
Epigenetic modification of promoters of different genes contributes to gene activation or suppression as a result of hypo- or hypermethylation, respectively, and targeting this mechanism, e.g., with 5-aza-2ʹ-deoxycytidine (5-aza-dC) that upregulates expression of TP53 mRNA and protein, may be valuable in fighting CLL in the future as such drugs have been used extensively in myelodysplastic syndrome with excess blasts that show a characteristically high ratio of hypermethylation and different gene inactivation.29
Follow-up of the patients involved in the current study has been initiated to determine the effect of the methylation on the progression of the disease and the response to various chemotherapeutic agents.
Conclusion
The current study revealed that TP53 methylation contributes significantly to CLL development and progression. Further workups are recommended to study their relation with other genetic changes as malignancies are multifactorial and heterogeneous that arise from the interaction of different genetic changes.
Abbreviations
CLL, chronic lymphocytic leukemia; TP53, tumor protein 53; WBC, white blood cells; K2-EDTA, dipotassium-ethylenediaminetetraacetic acid; PCR, polymerase chain reaction; CPG, cytosine–guanine dinucleotide.
Ethics Approval And Consent To Participate
The current study was approved by the ethical committee at the University of Duhok College of Medicine and Duhok Directorate of Health and followed the Declaration of Helsinki ethical values for medical research concerning human beings. As the current study does not include any therapeutic modality and has a minor threat of injury to enrolled subjects, only verbal informed consent was taken from all enrolled individuals according to the general guidelines of Duhok Directorate of Health for obtaining consent.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Gaidano G, Foà R, Dalla-Favera R. Molecular pathogenesis of chronic lymphocytic leukemia. J Clin Invest. 2012;122(10):3432–3438. doi:10.1172/JCI64101
2. Ghia P, Ferreri AM, Caligaris-Cappio F. Chronic lymphocytic leukemia. Crit Rev Oncol Hematol. 2007;64(3):234–246. PMID: 17544290. doi:10.1016/j.critrevonc.2007.04.008.
3. Ponzoni M, Doglioni C, Caligaris-Cappio F. Chronic lymphocytic leukemia: the pathologist’s view of lymph node microenvironment. Semin Diagn Pathol. 2011;28(2):161–166. doi:10.1146/annurev-pathol-020712-163955
4. Damle RN, Calissano C, Chiorazzi N. Chronic lymphocytic leukaemia: a disease of activated monoclonal B cells. Best Pract Res Clin Haematol. 2010;23(1):33–45. doi:10.1016/j.beha.2010.02.001
5. Campo E, Ghia P, Montserrat E, et al. Chronic lymphocytic leukaemia/small lymphocytic lymphoma. In: Swerdlow SH, Campo E, Harris N, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.
6. Zenz T, Mertens D, Kuppers R, Dohner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010;10:37–50. doi:10.1038/nrc2764
7. Zenz T, Gribben JG, Hallek M, Ohner H, Keating MJ, Stilgenbauer S. Risk categories and refractory CLL in the era of chemoimmunotherapy. Blood. 2012;119:4101–4107. doi:10.1182/blood-2011-11-312421
8. Lee CJ, Evans J, Kim K, Chae H, Kim S. Determining the effect of DNA methylation on gene expression in cancer cells. Methods Mol Biol. 2014;1101:161–178. PMID: 24233782. doi:10.1007/978-1-62703-721-1_9
9. Cheedipudi S, Genolet O, Dobreva G. Epigenetic inheritance of cell fates during embryonic development. Front Genet. 2014;5:19. doi:10.3389/fgene.2014.00019
10. Tang W, Ho S-M. Epigenetic reprogramming and imprinting in origins of disease. Rev Endocr Metab Disord. 2007;8(2):173–182. doi:10.1007/s11154-007-9042-4
11. Hagood J. Beyond the genome: epigenetic mechanisms in lung remodeling. Physiology (Bethesda). 2014;29(3):177–185. doi:10.1152/physiol.00048.2013
12. Ross SA, Milner J. Epigenetic modulation and cancer: effect of metabolic syndrome? Am J Clin Nutr. 2007;86(3):872S–877S. doi:10.1093/ajcn/86.3.872S
13. Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1:11–19. doi:10.1038/35049533
14. Hube F, Reverdiau P, Iochmann S, Gruel Y. Improved PCR method for amplification of GC-rich DNA sequences. Mol Biotechnol. 2005;31:81–84. doi:10.1385/MB:31:1:081
15. Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21:5427–5440. doi:10.1038/sj.onc.1205600
16. Wang L, Kwong Y, Wong K, et al. Epigenetic inactivation of mir-34b/c in addition to mir-34a and DAPK1 in chronic lymphocytic leukemia. J Transl Med. 2014;12:52. PMCID: PMC3941938; PMID: 24559316. doi:10.1186/1479-5876-12-52.
17. Hollstein M, Sidransky D, Vogelstein B, Harris C. p53 mutations in human cancers. Science. 1991;253(5015):49–53. PMID 1905840. doi:10.1126/science.1905840.
18. Iranpur-Mubarakeh V, Esmailizadeh AK. Rapid extraction of high quality DNA from whole blood stored at 4 _C for long period; 2010. Available from: http://www.protocol-online.org/prot/Protocols/Rapid-Extraction-of-High-Quality-DNA-from-Whole-Blood-Stored-at-4-C-for-Long-Period-4175.html.
19. Kashmoola M, Eissa A, Al-Takay D, Al-Allawi N. Molecular characterization of G6PD deficient variants in Nineveh province, Northwestern Iraq. Indian J Hematol Blood Transfus. 2015;3:133–136. doi:10.1007/s12288-014-0368-2
20. Galm O, Herman J. Methylation-specific polymerase chain reaction. Methods Mol Med. 2005;113:279–291. PMID:15968111. doi:10.1385/1-59259-916-8:279
21. Jarzynski A, Papiernik K, Polz-Dacewicz M. Analysis of mutation and promoter methylation of TP53 gene in tumors of the head and neck. Curr Issues Pharm Med Sci. 2016;29(2):53–56. doi:10.1515/cipms-2016-0011
22. Fernàndez V, Jares P, Salaverria I, et al. Gene expression profile and genomic changes in disease progression of early-stage chronic lymphocytic leukemia. Haematologica. 2008;93(1):132–136. doi:10.3324/haematol.11694
23. Zenz T, Eichhorst B, Busch R, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010;28(29):4473–4479. doi:10.1200/JCO.2009.27.8762
24. Zenz T, Kröber A, Scherer K, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112:3322–3329. doi:10.1182/blood-2008-04-154070
25. Stilgenbauer S, Zenz T. Understanding and managing ultra high-risk chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2010;2010:481–488. doi:10.1182/asheducation-2010.1.481
26. Wang C, Wang X.The role of TP53 network in the pathogenesis of chronic lymphocytic leukemia. Int J Clin Exp Pathol. 2013;6(7):1223–1229. ISSN:1936-2625/IJCEP1304037.
27. Gaiti F, Chaligne R, Gu H, et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature. 2019;569:576–580. doi:10.1038/s41586-019-1198-z
28. Hudson J, Shoaibi M, Maestro R, Carnero A, Hannon G, Beach D. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190(10):1375–1382. doi:10.1084/jem.190.10.1375
29. Schroeder T, Rautenberg C, Haas R, Germing U, Kobbe G. Hypomethylating agents for treatment and prevention of relapse after allogeneic blood stem cell transplantation. Int J Hematol. 2018;107(2):138–150. doi:10.1007/s12185-017-2364-4
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.