")
Back to Journals » International Journal of Nanomedicine » Volume 13
Hypoxia-activated prodrugs and redox-responsive nanocarriers
Authors Zeng Y , Ma J, Zhan Y , Xu X, Zeng Q , Liang J , Chen X
Received 1 June 2018
Accepted for publication 22 August 2018
Published 18 October 2018 Volume 2018:13 Pages 6551—6574
DOI https://doi.org/10.2147/IJN.S173431
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Yun Zeng,1,* Jingwen Ma,2,* Yonghua Zhan,1 Xinyi Xu,1 Qi Zeng,1 Jimin Liang,1 Xueli Chen1
1Engineering Research Center of Molecular and Neuro Imaging of the Ministry of Education, School of Life Science and Technology, Xidian University, Xi’an 710071, Shaanxi Province, People’s Republic of China; 2Department of Pharmaceutical Analysis, China Pharmaceutical University, Nanjing 210009, Jiangsu Province, People’s Republic of China
*These authors contributed equally to this work
Abstract: Hypoxia is one of the marked features of malignant tumors, which is associated with several adaptation changes in the microenvironment of tumor cells. Therefore, targeting tumor hypoxia is a research hotspot for cancer therapy. In this review, we summarize the developing chemotherapeutic drugs for targeting hypoxia, including quinones, nitroaromatic/nitroimidazole, N-oxides, and transition metal complexes. In addition, redox-responsive bonds, such as nitroimidazole groups, azo-groups, and disulfide bonds, are frequently used in drug delivery systems for targeting the redox environment of tumors. Both hypoxia-activated prodrugs and redox-responsive drug delivery nanocarriers have significant effects on targeting tumor hypoxia for cancer therapy. Hypoxia-activated prodrugs are commonly used in clinical trials with favorable prospects, while redox-responsive nanocarriers are currently at the experimental stage.
Keywords: antitumor drugs, hypoxia, nanoparticles, redox-sensitive, tumor therapy
Introduction
Hypoxia is one of the key features of malignant tumors.1 It can significantly affect tumor treatment and prognosis because it can influence the expression of many genes.2 In recent years, being inspired by some antioxidants from plants3 and their antioxidant effects,4 several hypoxia-sensitive prodrugs have been designed for oxygen-selective chemotherapy on the basis of their ability to target the tumor hypoxia reductive microenvironment,5 and ultraviolet-visible spectrophotometer or high-performance liquid chromatography was performed to determine the quantitative assessment of drugs6 or dominant compounds in extracts.7 These prodrugs are nontoxic under normoxia but can be reduced to toxic chemotherapeutic drugs by specific hypoxia-responsive enzymes in cells. Therefore, prodrugs function through direct targeting of specific sites or indirectly by producing free radicals to kill tumor cells.
To overcome the low solubility of hydrophobic crude drugs,8 micro/nano-capsules or nano-micelles were introduced to control release.9 Liposome was the first commercial pharmaceutical formulation to encapsulate therapeutic agents.10 Naturally, apart from hypoxia-activated prodrugs, redox-responsive drug delivery systems have also been developed to target tumor cells.11 The concentration of reducing agents,12 such as glutathione (GSH), is much higher in the hypoxic tumor microenvironment than in normoxic cells. Thus, to target the tissue hypoxic microenvironment, redox-responsive bonds, such as disulfide bonds, are usually introduced to drug delivery nanocarriers assembled by artificial polymers or natural polysaccharides such as chitosan whose infrared spectra were determined.13
Both hypoxia-activated prodrugs and redox-responsive drug delivery nanocarriers are effective for targeting the hypoxic microenvironment (Scheme 1). Hypoxia-activated prodrugs are usually used in clinical trials with favorable prospect, and the redox-responsive nanocarriers are commonly investigated at the experimental research stage. Here, we summarize the hypoxia-activated prodrugs and redox-responsive nanocarriers and discuss the advantages and disadvantages of these hypoxia-targeting strategies.
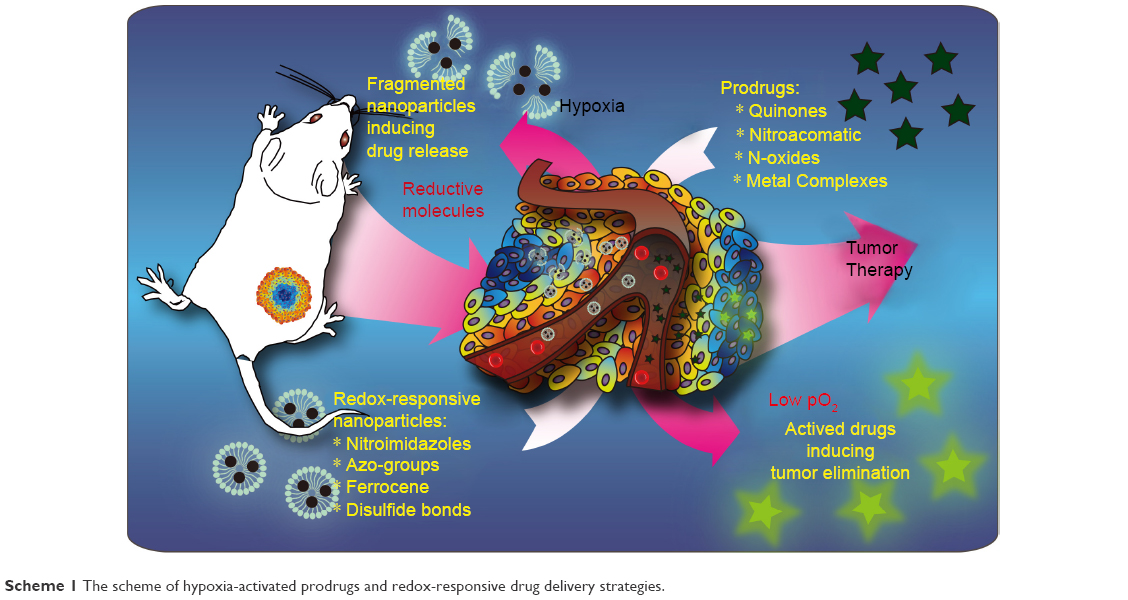 | Scheme 1 The scheme of hypoxia-activated prodrugs and redox-responsive drug delivery strategies. |
Hypoxia-activated prodrugs
There are four main types of hypoxia-activated prodrugs based on the skeleton structure, which are quinones, nitro-group, N-oxide analogs, and transition metal complexes.1,14 Quinone-based prodrugs, such as EO9 and RH1, are the earliest developed hypoxia-activated prodrugs, but with limited effects in clinical trials. Nitro-group chemicals include nitroaromatics and nitroimidazole. Nitroaromatics-based prodrugs include PR-104A, CB1954, SN29730, and KS119W, and nitroimidazole-based prodrugs include TH-302 and NQCQ-1. The most common N-oxide analog prodrug is the tertiary amine-oxides banoxantrone (AQ4N), whose reductive compound AQ4 can compete with topoisomerase II for binding to double-stranded DNA and subsequently leads to apoptosis and tumor cell death. Moreover, these prodrugs include particular aromatic di-N-oxides, such as trapazamin (TPZ) and SN30000. Lastly, prodrugs that are based on transition metal complexes include Co(III), Cu(II), V(II), and Fe(III), as well as other complexes.
Quinones
Quinones and their derivatives are the earliest developed hypoxia-targeting prodrugs. Quinones can be reduced to hydroquinones by two enzymatic steps, including CYP450-mediated one-electron transfer pathway and the DT-diaphorase-mediated two-electron transfer pathway (Figure 1).5,15 The functions of these drugs are dependent on structural modification. Mitomycin C (MMC) is the original quinone hypoxia-activated alkylating prodrug, which can be bio-reduced by the aromatization process. EO9, a derivative of MMC, is an indolequinone bioreductive prodrug that can be activated by DT-diaphorase under hypoxic and normoxic environments to induce DNA damage. To improve EO9’s pharmacokinetics and tissue penetration ability, the 2,3,5-position of indolequinone has been modified, and the derivatives were reported to maintain reasonable hypoxia sensitivity and low cytotoxicity. A series of indolequinone prodrugs have been designed to target quinone oxidoreductases. One of these compounds showed antiproliferative activities in the HT-29 and A549 cell lines with IC50 values of 0.386 and 0.263 mM, respectively.16 AZQ analogs, which are based on benzoquinone/benzimidazolequinone and naphthoquinone, have also been studied as potential bioreductive antitumor drugs.
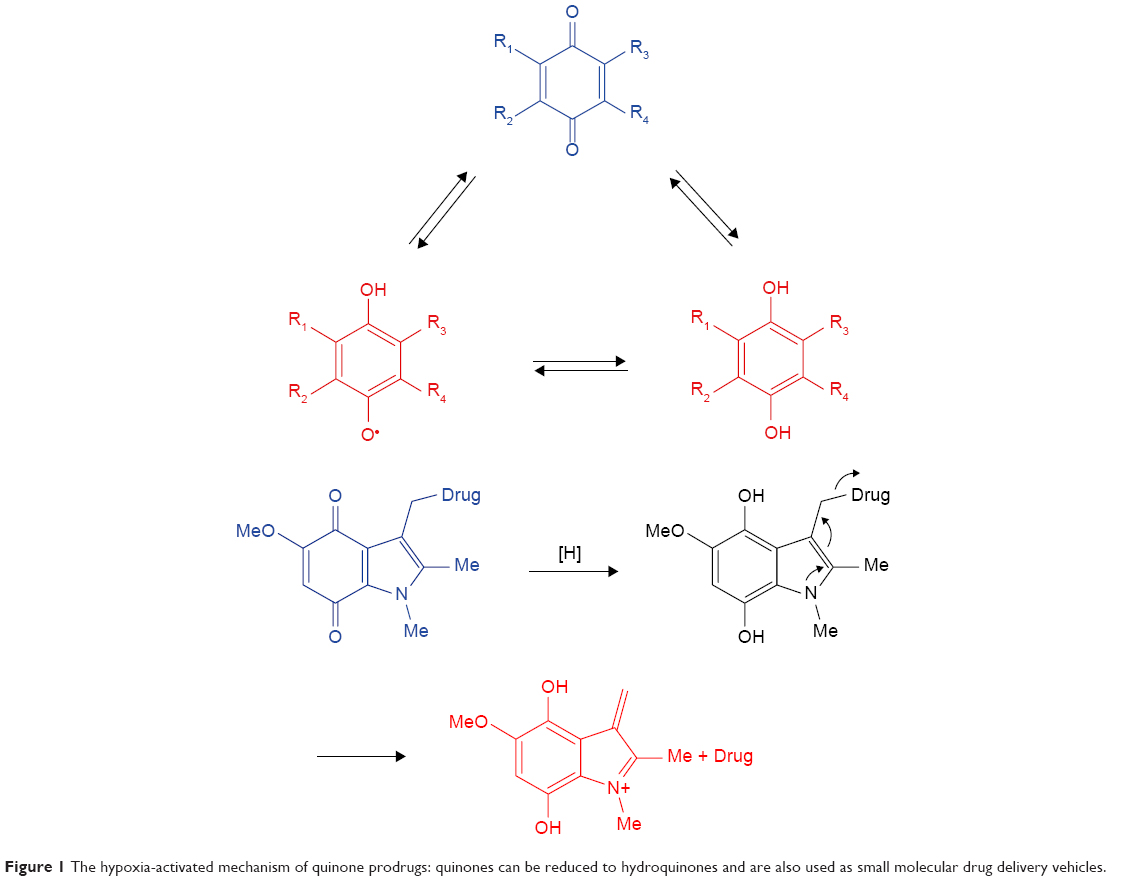 | Figure 1 The hypoxia-activated mechanism of quinone prodrugs: quinones can be reduced to hydroquinones and are also used as small molecular drug delivery vehicles. |
In addition, quinones are used as small molecular drug delivery vehicles. For instance, indolequinones, naphthoquinones, and benzoquinones could release antitumor agents by bioreductive- or radiochemical-mediated cleavage at the 3-position of the quinone ring.17,18 EO9 itself can be considered as a promoiety and is used to conjugate other antitumor drugs at the 3-position, in order to form a new dual-function prodrug. Moreover, the 2-position of the quinone ring can function as a substrate, as it is more sensitive than the 3-position during DT-diaphorase activation. Antitumor drugs can also be released from benzoquinones. The trimethyl lock, a trigger for molecular release, is activated by benzoquinones promoiety drugs, as well as naphthoquinones and benzoimidazolequinones. A series of seco-CBI-indole2 N-acyl O-amino derivatives have been investigated as promoiety of duocarmycin and CC-1065, which could be specifically cleaved from promoiety in hypoxic tumor regions.19 INDQ/NO system can generate NO through DT-diaphorase activation and induce DNA damage to inhibit tumor proliferation.20 Indolequinone SN38, coupled with target tumor ligand c(RGDyK) agents, has been designed to release SN38 under bioreductive conditions.21 Later, a new compound was synthesized by coupling SN38 and nitroaromatic groups, which could be specifically activated under hypoxia conditions in the A549 and HeLa cell lines. The released active drug could accumulate in tumor regions and inhibit tumor growth in a mouse tumor-bearing model.22
A series of 4-nitrobenzyloxycarbonyl O6-benzylguanine (O6-BG) prodrugs were synthesized, which were considered as O6-alkylguanine-DNA-alkyltransferase (AGT) inhibitors, which could be activated by reductase in the absence of O2.23 Three of these prodrugs could activate the reductase system under insufficient O2 levels, and among these, the most water-soluble exhibited the highest nitro-group reduction efficiency. The same compound had little effect in a normoxic environment but showed enhanced cytotoxicity in a low O2 environment, indicating that these AGT inhibitors can be more specifically released under hypoxic conditions. Comparable AGT inhibitors, 6-(benzylozy)-2-(aryldiazenyl)-9H-purines, are also O6-BG prodrugs.24 Researchers have improved water solubility and cytotoxicity by synthesizing 6-((3-((dimethylamino)methyl)benzyl)oxy)-9H-purin-2-amine, in which the 3-aminomethyl moiety was protected through methylation. This prodrug contained an α,α-dimethyl-4-nitrobenzyloxycarbonyl moiety, which successfully attained AGT hypoxia sensitivity with improvement in water solubility in EMT6 and DU145 cells.25
ChK1/Aurora A inhibitor redox-responsive prodrugs can be used under certain hypoxia conditions. When CH-01 was coupled with bioresponsive groups, the ChK1/Aurora A inhibition activity was decreased. When the nitro-groups were reduced, CH-01 was activated and subsequently induced chromosome instability and inhibited tumor cells survival.26 Two types of cyclic N-acyl O-amino phenol agents have been used as duocarmycin family prodrugs based on cleavage principles.27 Drugs could be released from these prodrugs by nucleophile reduction in the hypoxic tumor environment, which extraordinarily showed 40-fold higher cytotoxicity than the noncleaved prodrugs in vivo. A series of tetrazole structured pyranonaphthoquinone derivatives, such as nanaomycin and eleutherin, have also been synthesized to evaluate cytotoxicity in the presence of one- or two-electron quinone bioreductases (NADPH:CP450 or NAD(P)H:NQO1; DT-diaphorase).28 The cytotoxicity of these prodrugs increased along with the reduction of O2 levels, indicating that cytotoxicity was dependent on their reductive degree.
Quinone propionic acids (QPAs) can be attached to prodrugs or on the surface of liposomes, and their release can be induced by reductases, such as NQO1.29 The bioresponsive properties of these prodrugs can be adjusted by the structure of QPAs, which could be affected by the Flavin isoalloxazine ring located at the rhNQO1 active site. Nanoparticles (NPs) loading paclitaxel (PTX) are redox-responsive due to their attachment to QPAs. Furthermore, controlled release tests have shown that >80% of PTX was released in 24 hours in a reductive environment, while the control release was just 26.5%–41.2% in vitro. Therefore, these nanocarriers could rapidly release PTX in breast tumor cells under hypoxic conditions.30 Alternative biodegradable polymer can be used to synthesize bioreductive nanocarriers loading PTX by trimethyl-locked benzoquinone, and these nanocarriers could be activated by reductases to release PTX.31
Nitroaromatics/nitroimidazole
The earliest hypoxia-selective prodrugs were synthesized from nitroimidazole radioisotope. Figure 2 illustrates the hypoxia-selective mechanism of nitroaromatics/nitroimidazole. RSU1069 is the original dual nitroimidazole prodrug that is derived from NSC639862. NLA-1 or NLCQ-1, the prodrug of 2-nitroimidazole attached to acridine or quinoline, or other compounds with similar structure, such as nitroaromatic, also have comparable hypoxia-responsive effects. 1-Nitroacridine and nitroquinoline are considered as good hypoxia-selective prodrugs, which function through binding to DNA. Nitrobenzene is an alternative alkylating agent, and its nitro-groups can be reduced to hydroxylamine by bio-reduction. The oldest anticancer prodrug, CB1954, has shown little therapeutic effects but SN23862 showed potential therapeutic effects for targeting severe anoxia in tumor cells.32 PR-104, a phosphate ester-based pro-prodrug, was obtained through the modification of SN23862 by carboxamide moiety cleavage, and its release was promoted by phosphatases.33 Finally, 5-hydroxylamino and 5-amino metabolites could be metabolized to release mustard moiety through NADPH:CP450 activation under bioreductive conditions. Two tyrosine kinase inhibitors that were incorporated in 2-nitroimidazole-based hypoxia-activated prodrugs demonstrated antitumor and signaling inhibitory effects.34
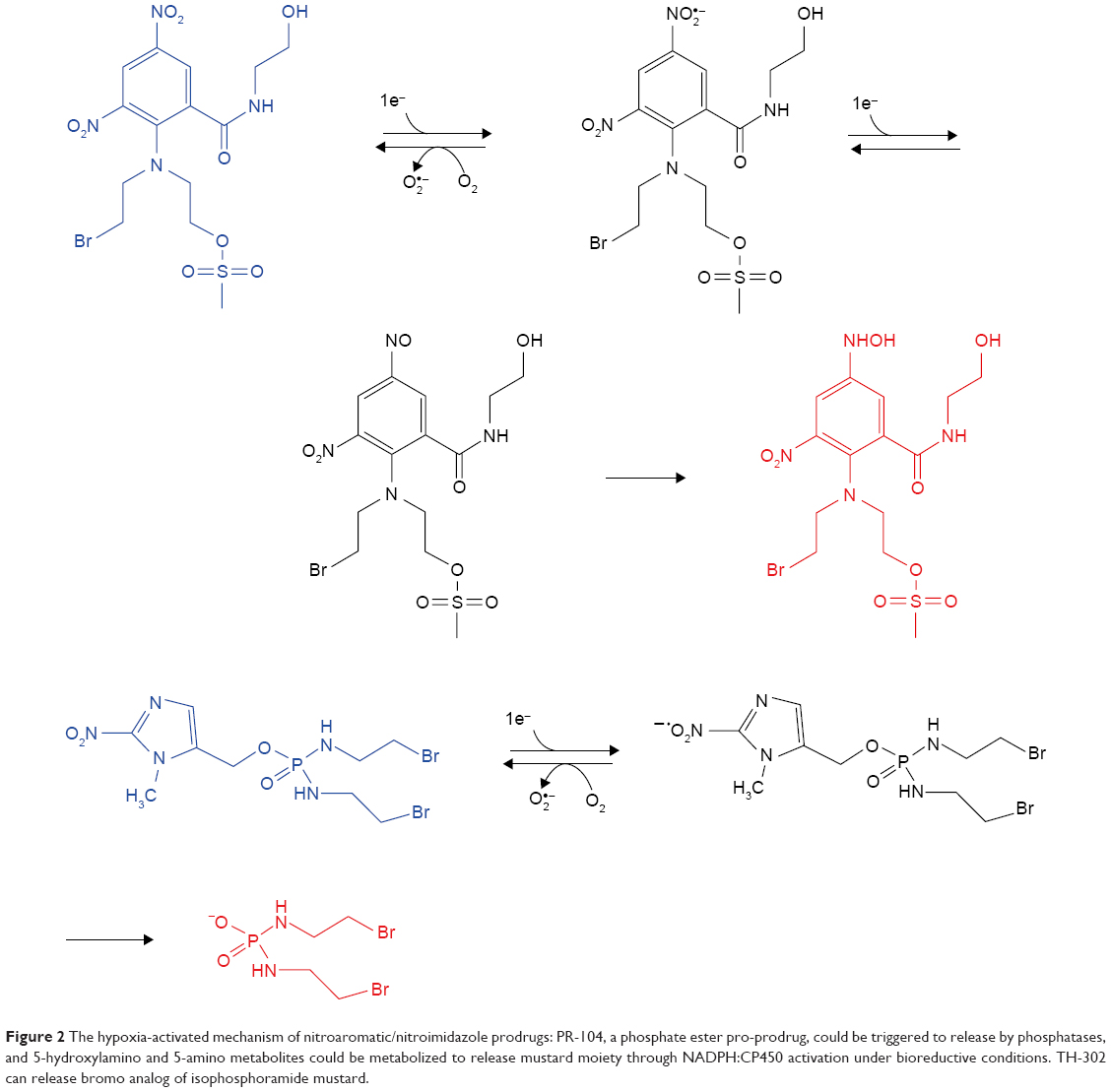 | Figure 2 The hypoxia-activated mechanism of nitroaromatic/nitroimidazole prodrugs: PR-104, a phosphate ester pro-prodrug, could be triggered to release by phosphatases, and 5-hydroxylamino and 5-amino metabolites could be metabolized to release mustard moiety through NADPH:CP450 activation under bioreductive conditions. TH-302 can release bromo analog of isophosphoramide mustard. |
Nitrobenzyl mustard quaternary ammonium salts are new water-soluble hypoxia-selective prodrugs, but their performance is more effective under in vitro than in vivo conditions. Nitroimidazole and nitropyrrole demonstrated quite high mechlorethamine formation yield, but weak in vivo cytotoxicity. Elimination process is another method to trigger hypoxia-activated prodrugs, in which the phosphoramide mustard at the β-position of nitroquinoline can be released by nitro reduction in a hypoxic environment, and β-elimination reaction can be promoted by nearby basic quinoline nitrogen.5 Numerous phosphoramide nitrogen mustards have been designed, and their nitro-groups can be reduced to hydroxylamine intermediates, while phosphorodiamidates can be spontaneously released by 1,6-elimination. The same principle has also been applied on nitrofuryl and nitrothienyl derivatives as hypoxia-selective alkylating agents. Cyclophosphamide derivatives can be activated by CP450 oxidoreductase and converted to 4-hydroxycyclophosphamide through β-elimination. 4-Nitrophenyl showed 100-fold higher cytotoxicity relative to cyclophosphamide and 1,000-fold increased cytotoxicity in the presence of bacterial nitroreductase. Nitroaryl methylheterocycle can be loaded and release combretastatin via bioreduction. Nitroacyl methylcarbamate is one of the typical hypoxia-activated prodrugs, among which 4-nitrobenzyl carbamate derivatives can act as alkylating, antibiotic, and DNA intercalating agents. TH-302 is well known among these derivatives, and it could release bromo analogs of isophosphoramide mustard.35–37 SN30000 showed equivalent cytotoxicity to TPZ and TH-302 prodrugs but higher cytotoxicity than PR-104A and SN29428.38,39
Pyrrolobenzodiazepine-containing TH-302 released similar hypoxia-activated compounds through the action of P450 reductase under both normoxic and hypoxic conditions. However, the cytotoxicity ratio of normoxic to hypoxic was still lower than TH-302.40 TH-302 could suppress canine lymphoma survival by downregulating HIF-1α and its downstream genes.41 Triggering Br-IPM release from TH-302 was accompanied by enhanced cytotoxicity when pO2 was <76 mmHg, according to electron paramagnetic resonance monitoring.42 Nonpharmacological methods could exacerbate tumor hypoxia and therefore increase the in vitro and in vivo TH-302 cytotoxicity in pancreatic tumors.43 Tumor growth was efficiently inhibited by the combined treatment of ionizing radiation with TH-302 in pancreatic xenografts, but this inhibitory effect was not obvious in tumor stem cells.44 It has also been reported that TH-302 could overcome hypoxia-induced tumor radio-resistance, while combinatorial therapy with TH-302 and radiation could inhibit pancreatic tumor growth.45 Osteosarcoma tumor growth could be suppressed by the synergistic function of TH-302 with doxorubicin (DOX), in which bone tumor burden, bone damage, and lung metastases could be simultaneously inhibited.46
KS119 is one of the 1,2-bis(sulfonyl)hydrazine prodrugs, which can be reduced to the active intermediate 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazine, and then alkylated at the O6-position of guanine. Compared with other AGT inhibitors, KS119 demonstrated the strongest cytotoxicity in cells expressing AGT and was able to overcome AGT-mediated drug resistance.47 BCCA621C and IC86621 can inhibit DNA double-strand repair, because their target is the DNA-dependent protein kinase (DNA-PK). BCCA621C was able to release DNA-PK inhibitor in hypoxia-selective cells, especially in mouse liver microsomes and NCI-H460 cells, and increased radiotherapy sensitivity, in contrast to normoxic cells.48 Both O2-(2,4-dinitrophenyl)-diazeniumdiolate and oleanolic acid could be triggered by GSTπ enzymes to release NO in HCC tumor cells but not in normal cells. The released NO could induce apoptosis in HepG2 cells by arresting the cell cycle at the G2/M phase and activate mitochondria and MAPK-mediated pathways to enhance reactive oxygen species (ROS) generation.49 A series of 4-anilinoquinazoline derivatives, which were synthesized by conjugating 2-nitroimidazole to 4-anilinoquinazoline, can function as epidermal growth factor receptor (EGFR) inhibitors. These derivatives could be reduced under hypoxic conditions and induce apoptosis in tumor cells.50 Sulfonate prodrug (5-nitro-2,3-dihydro-1H-benzo[e]indol-1-yl)methyl contains releasable sulfonate groups which can act as alkylating agents for binding to the DNA minor groove. They exhibited 4,090-fold increased cytotoxicity during in vitro hypoxic conditions and showed strong antitumor effects in vivo.51
N-oxides
N-oxides are considered as the most potential hypoxia-activated class of prodrugs because of their low cytotoxicity and can be selectively reduced, especially by reductases, to effective therapeutic compounds in a hypoxic environment (Figure 3). These features make them valuable for the synthesis of potent hypoxia-selective prodrugs. Two representative drugs, TPZ and AQ4N, in combination with radiotherapy and chemotherapy, have performed well in clinical trials.
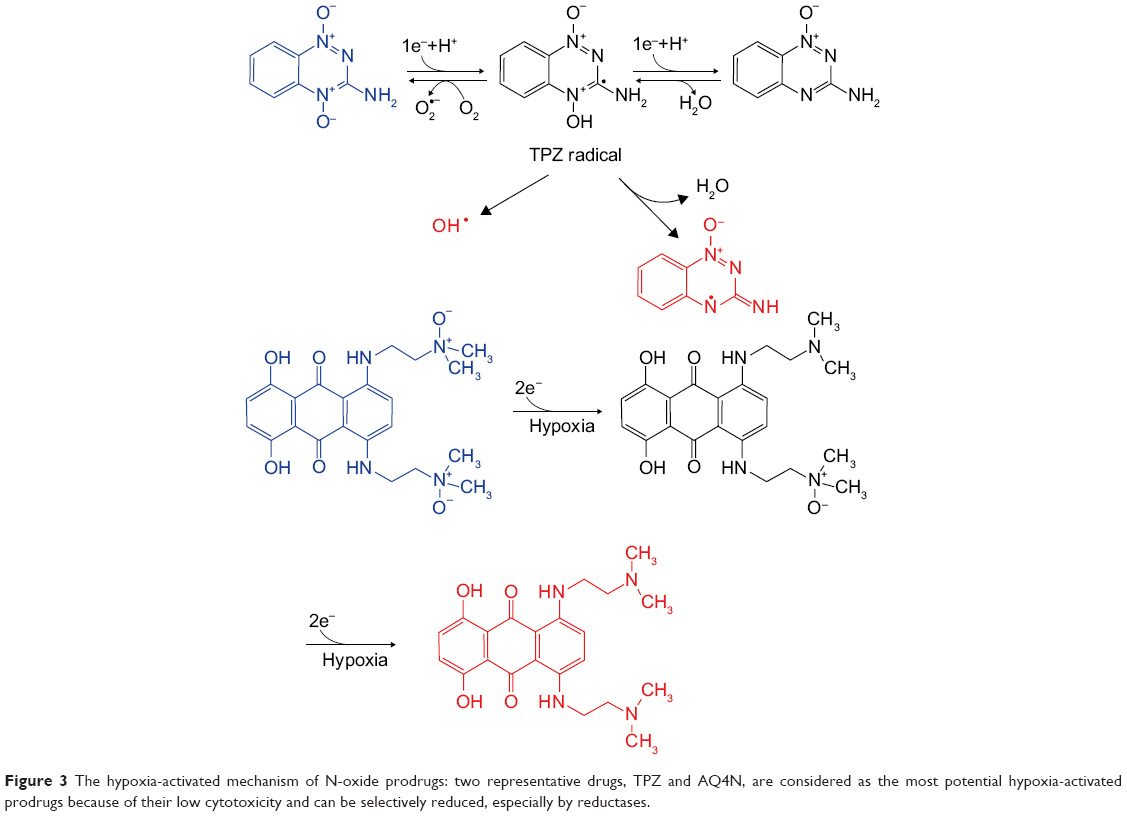 | Figure 3 The hypoxia-activated mechanism of N-oxide prodrugs: two representative drugs, TPZ and AQ4N, are considered as the most potential hypoxia-activated prodrugs because of their low cytotoxicity and can be selectively reduced, especially by reductases. |
TPZ is considered as an aromatic N-oxide and can be reduced by CP450 through the one-electron transport chain, which generates short-lived oxidation radicals that can cause DNA damage. Compared to quinones and nitroaromatics, the effective O2 concentration range is broader in hypoxic cells under TPZ treatment. Mono-N-oxide metabolism is also involved in TPZ treatment, which might be induced by changes in electron transport chain. In addition, TPZ, which contains a nitrogen mustard at the 6-position of its structure, demonstrated that 4-oxide elimination could increase nitrogen mustard activation, which proved that TPZ could have a differential hypoxic cytotoxicity as a prodrug without masking the group’s activity. A series of TPZ analogs illustrated better water solubility and therapeutic effects.52 3-Position modification could increase cytotoxicity and water solubility under hypoxic conditions,53 while 7-position modification could increase the in vitro therapeutic effects. In addition, benzo-1,2,4-triazine 1,4-dioxide derivatives54 and other heterocyclic aromatic N-oxides, such as quinoxaline 1,4-dioxide, imidazoquinoxaline N-oxides, imidazopyridopyrazine N-oxides, oxadizole N-oxides, trazine N-oxides, and phenazine 5,10-dioxides, have been synthesized.
Alkyl tertiary amine N-oxides AQ4N can be reduced to AQ4 by CP450 enzymes through two-electron reduction.55 AQ4, an inhibitor of topoisomerase II, showed considerable therapeutic effects in preclinical trials. However, similar drugs such as nitroacrine with a tertiary amine side chain showed minor activities. Both amonafide and mitonafide contain a naphthalimide structure and could inhibit topoisomerase activity but without considerable effects in vitro. OCT1002, a deuterated analog of AQ4N, was not only able to inhibit LNCaP, 22Rv1, and PC3 tumor growth and revascularization but could also enhance the antitumor synergistic effects with other chemotherapeutic agents.56,57 Synergistic liposomes containing both hypoxia-activated AQ4N and photosensitizer chlorin e6 have been introduced in hypoxic tumor therapy, in which hypoxia induced by photosensitizer chlorin e6 in PDT was used to subsequently trigger AQ4N activation.58 Shen et al investigated AQ4N and gossypol combination chemotherapy in HA@AQ4N-Cu(II)-gossypol NPs and demonstrated that AQ4N could function as self-carrier material and auto-fluorescence indicator.59
Phenazine 5,10-dioxides have also shown hypoxia-selective drug cytotoxicity. The mechanism is still unclear, and their cytotoxicity has just been demonstrated in vitro but with little effects in vivo. In a previous study, the function of 7-bromo-2-hydroxyphenzine-5,10-dioxide was studied in the cytosol of rat liver,60 using different enzyme inhibitors such as dicoumarol, ketoconazole, and menadione to determine which enzymatic pathways were involved. Activation of the natural product myxin, a bioreductive di-N-oxide, generates free radicals that cause DNA double-strand breaks.61 However, it does not show any obvious hypoxia-selective effects as the free radicals generated by phenazine 5,10-dioxides could not induce apoptosis in Caco-2 cells.62
Moreover, several novel derivatives of phenazine 5,10-dioxides have been synthesized to investigate the inhibition rate between DNA and topoisomerase II, showing cytotoxicity in V79 cells in both hypoxic and normoxic environments.63 Some of these derivatives illustrated cytotoxicity but did not exhibit hypoxia specificity. However, 7-fluoro-2-aminophenzine 5,10-dioxide showed hypoxia-selective cytotoxicity, and fluorescence tests indicated a stronger binding to DNA after hypoxia activation.64
Many N-oxides based on ROS generation have been investigated. 3-Trifluoromethyl-quinoxaline 1,4-dioxides could release·OH radicals through CP450 by one-electron reduction that could be suppressed by O2.65 Hence, its cytotoxicity enhancement depended on the concentration of O2. 1,2,4-Benzotriazine 1,4-dioxides were significant hypoxia-selective antitumor prodrugs that function through a one-electron reduction to generate free radicals and induce DNA double-strand breaks.54
Transition metal complexes
Transition metal complexes, especially Co(III) complexes,66 are usually considered as potential targeting hypoxia prodrugs.67 These complexes are coordinated with cytotoxic ligands of relatively low cytotoxicity for specific binding to their target sites. When the center metal ions are at a higher oxidation status, they are coordinated with ligands and cannot be readily metabolized in cells. However, when the center metal ions are reduced to a lower unstable reduced status, the corresponding ligands are triggered to be released (Figure 4). For instance, Co(III) can be reduced to Co(II) and release dissociated ligands as the active drugs. In fact, this procedure is reversible as Co(II) can be oxidized back to Co(III) in the present of O2.
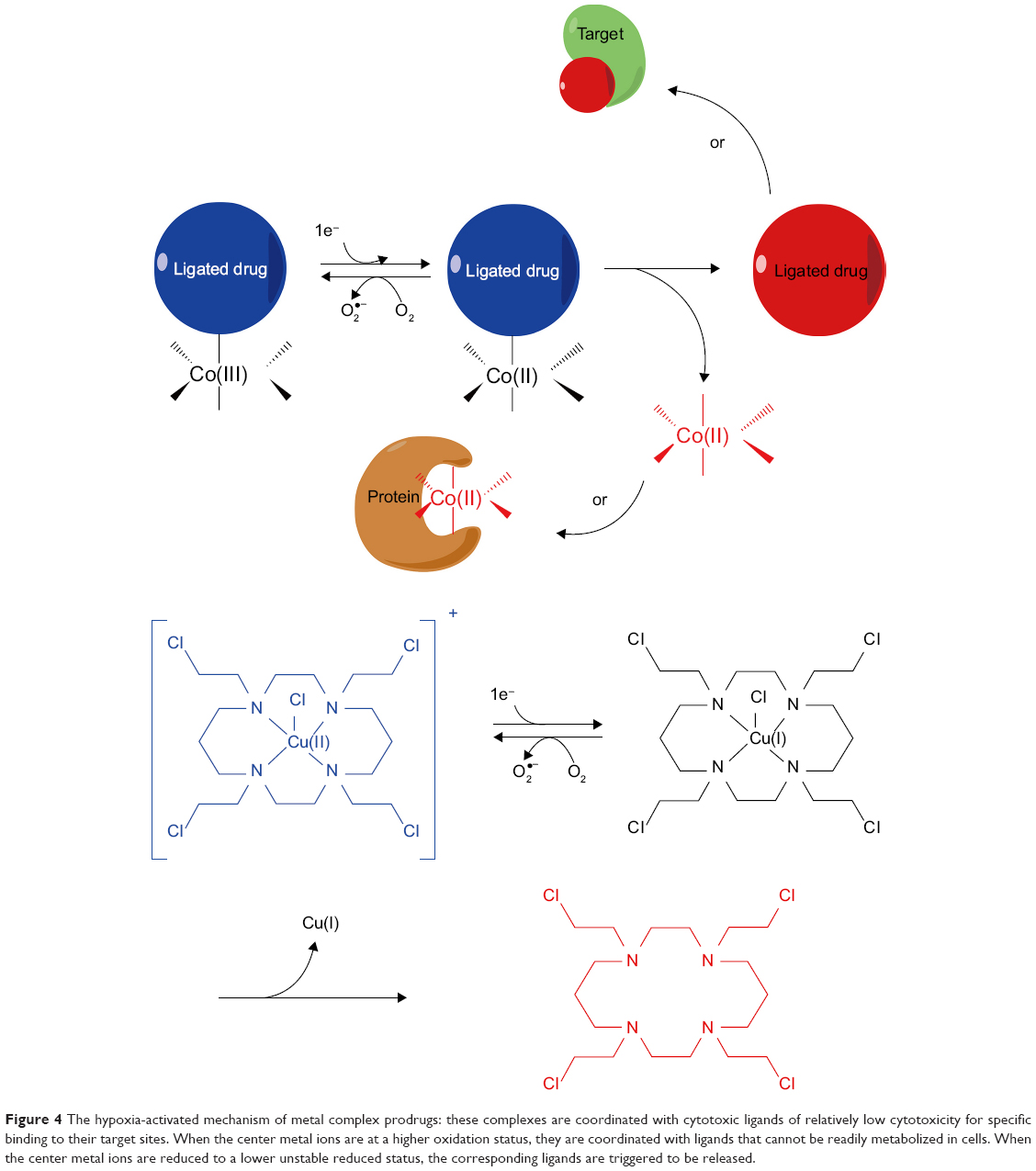 | Figure 4 The hypoxia-activated mechanism of metal complex prodrugs: these complexes are coordinated with cytotoxic ligands of relatively low cytotoxicity for specific binding to their target sites. When the center metal ions are at a higher oxidation status, they are coordinated with ligands that cannot be readily metabolized in cells. When the center metal ions are reduced to a lower unstable reduced status, the corresponding ligands are triggered to be released. |
In order to study the drug release process, fluorescence coumarin Co(III) chaperones were used to target hypoxic regions. These anionic complexes could specifically release drugs in acidic and hypoxic regions. The releasing properties of C343haH2 enable its application as a typical fluorophore tumor indicator instead of antitumor drug.68 Two series of Co(III) complexes with fluorescence auxiliary ligands have been shown to target hypoxia cells, according to assays on Eos photon-convertible green fluorescent protein (GFP) live hypoxic 3D cell models. After reduction of Co(III) to Co(II), the release of fluorescent ligands is induced, and fluorescence can be monitored to localize their targets.69
Cobalt-mustard complexes, such as [Co(Racac)2(DCE)]+, are significant in vitro hypoxia-selective delivery systems. In order to design novel EGFR inhibitors, Co(III) complexes with hypoxia-responsive properties were synthesized to selectively release EGFR inhibitors in the hypoxic microenvironment of solid tumors.70 The related DNA minor groove alkylating agent, (1-(chloromethyl)-5-hydroxy-1H-pyrrolo[3,2-f]quinolin-3(2H)-yl)(5,6,7-trimethoxy-1H-indol-2-yl)methanone, could be synthesized with cyclam or cyclen auxiliary ligands, which is derived from cross-linking to ethylene or N,N′-dimethyl analogs.71 These prodrugs demonstrated noticeable alkylating effects and marked hypoxia-selective cytotoxicity.
Co(III) complexes with H2bhnq ligands could be considered as bioreductive drugs. For instance, Co(III) complexes [Co(bhnq)(L1)]BF4·H2O and [Co(bhnq)(L2)]BF4·H2O [where L1: N,N′-bis(pyridine-2-ylmethyl) ethylenediamine and L2: N,N′-dimethyl-N,N′-bis(pyridine-2-ylmethyl) ethylenediamine] have been synthesized.72 Compared to nonmodified cyclen ligands, Co(III) complexes coordinated with N-substituted cyclen ligands and showed from 2- to 30-fold lower cytotoxicity and from 2- to 22-fold differences between hypoxic and normoxic cells.73
Other Cu(II) or Fe(II) complexes are usually coordinated with tetradendate ligands, such as tris(2-methylyridyl)-amine (tpa) or N,N-bis(salicylidene) ethane-1,2-diimine (salen). Novel Cu(II) complexes with 1,4,7-tetraazacyclododecane as ligands showed 24-fold higher cytotoxicity in hypoxic rather than in normoxic in vitro conditions. Similar metal complex redox pairs such as reduction of Pt(IV) to Pt(II), Ru(III) to Ru(II), Cu(II) to Cu(I), and Fe(III) to Fe(II) were also commonly applied as hypoxia-triggered release strategies. The release mechanism was based on altered valence of center metal ions to release ligands. In addition, various metal complex prodrug nanocarriers have also been designed.67
Redox-responsive drug delivery nanocarriers
Nitroimidazole derivatives
The first strategy to design a redox-responsive system was to use photo-induced electron transfer (PET) between fluorescent molecules and nitroimidazole (Figure 5A). 2-Nitroimidazole was commonly conjugated with (7-mithoxycoumarin-4-yl) methyl carboxylates for assessing the photo-SN1-dependent cleavage process. In the presence of 2-nitroimidazole, there was no photo cleavage and consequently no fluorescence emission, due to the PET effects. Therefore, release of the antitumor drug etoposide (a topoisomerase inhibitor) could not be achieved in normal cells. However, in cancer cells, the hypoxia specificity of nitro to amino reduction attenuated PET and hence activated drug release, which was accompanied by blue fluorescence emission.74
 | Figure 5 Targeting hypoxia drug delivery and release strategies. |
Comparable strategies have been applied for the development of self-assembled nanocarriers to release hydrophobic drugs under hypoxic conditions. In these nanocarriers, the carboxymethyl dextran was treated as a backbone and conjugated to 2-nitroimidazole. In effect, the nanocarriers could release DOX more rapidly under a low oxygen environment rather than in normoxic conditions, due to nitro- to amino-group reduction. This induced phase changes occurred in nanocarriers and resulted in higher accumulation of DOX in hypoxic cells (Figure 5B). In addition, nanocarriers have been reported to selectively accumulate in hypoxic tumor regions as a result of the enhanced permeability and retention (EPR) effects. Therefore, these nanocarriers provide a potent drug delivery method for redox-responsive release of active drugs.75
Azo-group-based chemicals
Azo-groups are commonly introduced in the process of hypoxic prodrugs fabrication. Nanocarriers have been specifically designed for hypoxia-induced siRNA uptake and silencing, and these include PEG2000, azobenzene, PEI (Mw: ~1.8 kDa), and 1,2-dioleyl-sn-glycero-3-phosphoethanolamine (DOPE) (Figure 5C). PEG2000 has been shown to extend the blood circulation time of NPs, which promotes their accumulation in tumors via EPR effects, but restricts their therapeutic effects. Therefore, to overcome this drawback effect, azo-benzenes were introduced to cleave PEG2000 from the PEI-DOPE NPs. The release of conjugated GFP was used as an indicator of siRNA targeting tumor hypoxia.76
Redox-responsive ferrocene
Bioresponsive and self-healing supramolecular hydrogels have been synthesized based on host–guest inclusion for loading β-cyclodextrin as a host and propenoic acid conjugated to ferrocene as a guest (Figure 5D).77 The phase transformation of supramolecular hydrogels could be changed by oxidation-reduction due to interactions with ferrocene-β-cyclodextrin, which could promote the release of active drugs. The morphology of nanocarriers, which were composed of a poly(vinylferrocene)-bl-poly(methacrylate) shell and a hydrophobic core, could be altered by selective oxidation and further conversion of the polarity of their shells. Hence, the drug-controlled release can be triggered by the transition of hydrophilic to hydrophobic phase.78
Metal oxide NPs
Metal oxide NPs were a series of stimuli-responsive-based delivery systems in biomedical applications,79 such as magnetic NPs based on ferric oxide, cobalt, or nickel, which were usually used in magnetic resonance imaging but also played significant roles in redox-sensitive drug delivery vehicles.80 For instance, superparamagnetic iron oxide NPs were introduced to deliver O6-BG to brain tumors with redox-responsive coating layer through the peptide chlorotoxin receptor-mediated transcytosis.81 Folate-(PLGA-PEG-PLGA-urethane-ss) encapsulating DOX and γFe2O3 showed synergistic cytotoxic effects in tumor cells that were overexpressing folate receptors. These multiblock polymeric NPs could be triggered under redox environment and achieve excellent chemotherapeutic effects rather than serious side effects.82 Manganese dioxide could be used as the core coated with nanoscale coordination polymers that were constructed based on HF ions and a cisplatin prodrug, which could release cisplatin in reduction/acidic pH/H2O2-responsive multi-triggered conditions.83
Selenium nanocarriers
Mesoporous selenium NPs have been synthesized to load DOX and to enhance their targeting effects by introducing redox-responsive properties for GSH-activated drug release. The synergistic effects induced by MSe and DOX enhanced the antitumor efficiency and decreased the drug side effects.84 Cross-linking of three diselenide-rich polymers (PEG-b-PESe, PEG-b-PUSe, and PEG-b-PBSe) to nanomicelles containing camptothecin (CPT) and DOX resulted in synergistic inhibition of tumor growth (Figure 5E). Visible light could induce diselenide metathesis, regeneration, and lipid bilayer formation, with increased sensitivity to GSH and ROS. In vitro and in vivo studies showed that tumor growth suppression could be achieved at low drug dosages due to their synergistic effects.85 A nanomicelle system delivering PTX was composed with diselenide-containing triblock copolymer, poly(ε-caprolactone)-bis(diselenide-methoxy poly(ethylene glycol)/poly(ethylene glycol)-folate) [PCL-(SeSe-mPEG/PEG-FA)2]. Their cell internalization indicated that they showed an enhanced cell uptake through folate receptor-mediated endocytosis pathway in folate overexpressing 4T1 breast cancer cells rather than other cell lines that lacked FA.86
Nanocarriers based on disulfide bonds
Reductive chemicals such as GSH have a significant role under redox conditions during tumor therapy, and dithiothreitol (DTT) is frequently used in vitro to replicate the redox environment.87 Disulfide bonds are often introduced to redox-responsive prodrugs and nanocarriers because they can be cleaved in a redox environment (Table 1).88,89 For instance, PEG, PEO, PCL, and PEI/HA are commonly used as nanocarrier fabrication materials (Figure 6).
 | Table 1 The properties of redox-responsive nanomicelles |
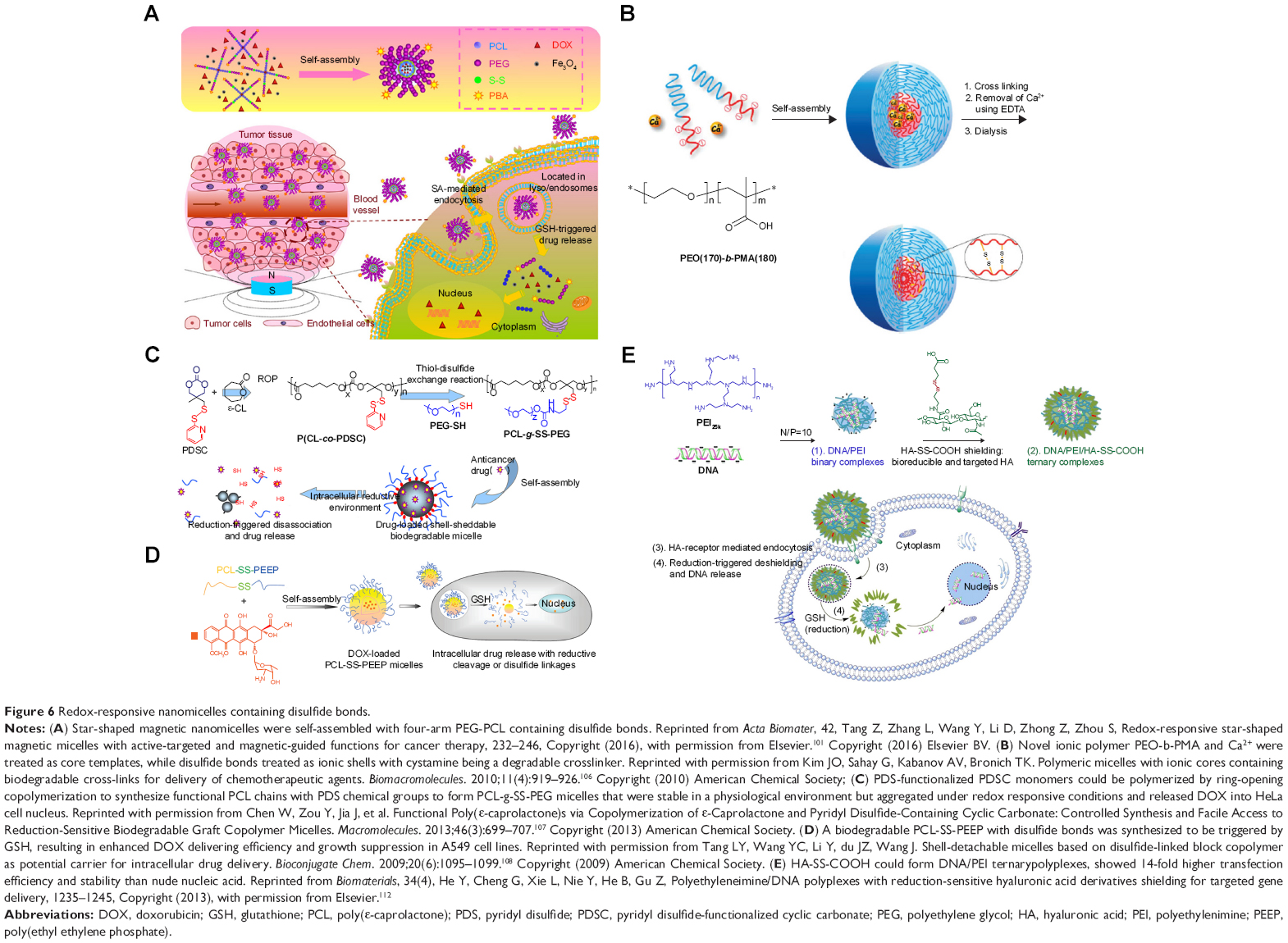 | Figure 6 Redox-responsive nanomicelles containing disulfide bonds. |
PEG
Disulfide bonds are frequently used in redox-responsive drug delivery micelles made from various PEG derivatives. Low-molecular-weight PEG, with amide group and disulfide bonds, was dissociated to free PEG gels under bioreductive status.90 PEG has been introduced to increase nanographene oxide (NGO) surface biocompatibility and biostability in PEGylated nanographene oxides (NGO-SS-mPEG), in which DOX release could be triggered by GSH.91,92
Disulfide bond cleavage could cause nanomicelle phase changes. Hence, cleavage of the new near-infrared (NIR) prodrug, DCM-S-CPT, can release fluorescence, which can be used in NIR optical imaging, and CPT which can be applied in chemotherapy. Adicyanomethylene-4H-pyran derivative has been used as an NIR fluorophore and conjugated by disulfide bonds to CPT as an antitumor drug. Hybrids of these molecules with PEG-PLA micelles have demonstrated good performance in tumor diagnosis and chemotherapy.93
Redox-sensitive self-assembly of the disulfide bond containing micelles mPEG-S-S-C16 could effectively deliver DOX, and the release was faster than that of the mPEG-C-C-C16 control, which did not include any redox-sensitive disulfide bonds.94 DOX release mainly occurred in the cytoplasm and then entered the cell nucleus, as observed by fluorescence confocal microscopy (FCFM). In contrast, DOX seldom entered the nucleus in the control. Tioguanine and tioguanine-generating prodrugs could be treated as antitumor and immunosuppressive agents. PEG-bl-poly (propylene sulfide) copolymers could form micelles by self-assembling with a free terminal thiol, causing propylene sulfide monomer ring-opening polymerization, and prodrug copolymers could be conjugated to tioguanine with disulfide bonds. Tioguanine prodrug micelles with the size of 18–40 nm were fabricated by self-assembly and responded to cysteine and serum simultaneously.95 Additionally, a series of shell-core structure disulfide-containing PEG micelles have been fabricated. For instance, micelles were synthesized with disulfides cross-linked P-(VCL-s-s-MAA)-PEG shells and carboxylic modified mesoporous silica (MSN) cores.96
Circular redox-responsive micelles that incorporated PEG-A and PLA-B formed spherical micelles with a size ranging from 34 to 107 nm. These micelles could be triggered by DTT, and in 14 hours almost 60% of DOX was released, as compared to the control which released <30% of DOX. They were also triggered to release DOX in the HeLa cell line in the presence of GSH.97 Hypoxia-responsive 100 nm nanomicelles were fabricated by mPEG-PBLG-SS-DTX, which could be triggered to release 40% of docetaxel (DTX) when treated with 50 mM of DTT within 120 hours, while the control groups released only 10% of DTX. Cytotoxicity was observed in MCF-7/ADR and A549 cells with IC50 values 15-fold lower than the free DTX.98 Redox-responsive nanomicelles-loading DOX were constructed with PEGylated hyperbranched polyphosphoester (hbPPE), in which PEGylation could promote stability of nanomicelles. More than 50% of DOX was released from the DOX@ss-hbPPE within 72 hours when treated with 10 mM of DTT in vitro, in contrast to 23% release in the control group.99
A redox-responsive stomatocyte has been used for drug release triggered under reducing conditions. The disulfide bonds linked the hydrophilic PEG with the hydrophobic PS chains, which could be cleaved by GSH in order to release DOX.100 Star-shaped magnetic nanomicelles were self-assembled with four-arm PEG-PCL containing disulfide bonds. Both DOX and Fe3O4 were encapsulated, and PBA was treated as the active ligand to target salic acid (SA)-mediated endocytosis. Both PBA and external magnetic field could enhance cell uptake of star-shaped magnetic nanomicelles in vitro. These dual-targeting micelles could accumulate in tumor tissues by both magnetic guidance and SA-mediated endocytosis, and DOX release could be triggered by the redox environment to inhibit tumor growth.101
Internalization of redox-responsive PLGA-PEG NPs was studied based on FRET fluorophore pairs (DiO/DiL). The cell uptake efficiency of redox-responsive NPs on 3D cell culture models of lung cancer was higher than that of non-redox-responsive NPs, and the extracellular release of PEG fringe could promote the internalization of NPs.102 Redox-responsive PTX-loaded PEG micelles were modified with ABD035, a peptide with albumin-binding domain, targeting overexpressing albumin-binding proteins such as secreted protein acidic and rich in cysteine (SPARC) and gp60. The cellular internalization results indicated that the accelerated transport rate was due to SPARC- and caveolin-1-mediated pathways and therefore improved therapeutic effects on representative drug resistance model: triple-negative breast cancer by increasing cellular internalization as well as drug distribution.103 Polycarbonate-based NPs composed of PEG-b-poly(MPC)n, azide-terminated α-lipoic acid, and 6-bromohexanoic acid were used in the redox-responsive drug delivery. The cellular internalization was investigated by CLSM and flow cytometry, indicating a high internalization eficiency of DOX-encapsulated NP systems.104
PEO
PEO is a similar polymer to PEG. Novel PEO-b-PMABC diblock copolymer has been synthesized by using dithioester-capped PEO in a reversible addition-fragmentation chain transfer polymerization process. The results demonstrated that PEO-b-PMABC micelles were stable under physiological conditions but were cleaved into unbound copolymers with rapid DOX release under bioreductive conditions.105 The novel ionic polymer PEO-b-PMA and Ca2+ were treated as the core templates, while disulfide bonds were treated as ionic shells with cystamine being a degradable cross-linker. These gelatinous micelles with 50% of DOX encapsulation rate have nanocarrier surface- and time-dependent degradation effects in cells, demonstrating increased release of DOX in human A2780 ovarian cells along with increased cytotoxicity.106
PCL
Various redox-responsive PCL micelles with disulfide bonds have been investigated because of their favorable biocompatibility and biodegradability. Pyridyl disulfide-functionalized cyclic carbonate monomers could be polymerized by ring-opening copolymerization within four steps, during which the synthesized functional PCL chains with pendant pyridyl disulfide (PDS) chemical groups derive from 3-methyl-3-oxetanemethanol. PCL-g-SS-PEG micelles were stable in a physiological environment but aggregated under redox-responsive conditions and released 82.1% of DOX within 12 hours, while only 17.5% of DOX was released without stimulation. FCFM showed that the DOX loaded in PCL-g-SS-PEG micelles could be delivered into the HeLa cell nucleus.107 A biodegradable ε-PCL-co-poly(ethyl ethylene phosphate) with disulfide bonds was synthesized to be triggered by GSH, resulting in enhanced DOX delivering efficiency and growth suppression in A549 cell lines.108
PEI/HA
Both PEI and HA are suitable for nucleic acid delivery and gene therapy. Therefore, HA/PEI nanocarriers have been vastly used for delivering nucleic acids and antitumor agents. The intercellular PTX drug-controlled release from HA-ss-DOCA nanomicelles depended on HA-deoxycholic acid, whose drug encapsulation rate and drug loading rate were as high as 34.1% and 93.2%, respectively. HA-ss-DOCA micelles were stable under physiological conditions but dissociated in 20 mM of reducing agents. Furthermore, a conspicuous faster release rate of PTX was observed, resulting in increased in vitro cytotoxicity, while Nile Red release provided a specialized drug delivery monitoring system. HA-ss-DOCA micelles possessed higher uptake rates in vivo, especially in MDA-MB-231 cells overexpressing the HA surface receptor.109
Bioresponsive NIR fluorescence and PDT agents were synthesized by conjugation of a photosensitizer and HA by disulfide bonds, which upon GSH triggering resulted in NIR fluorescence emission and cytotoxic compound release.110 Shell-core CC-HAM drug delivery micelles could also be triggered by GSH, and in vivo distribution analysis revealed tumor-targeting characteristic, improved DOX pharmacokinetics, and increased tumor accumulation.111 HA-SS-COOH could form DNA/PEI ternary polyplexes, and the fabricated procedure was accompanied by increases in size and zeta-potential. These HA-SS-COOH-coated ternary complexes showed 14-fold higher transfection efficiency and stability than nude nucleic acids.112 In addition, they showed higher accumulation in tumors of C57BL/6 tumor-bearing mice due to HA-targeting ligands, which also supported that HA derivatives can be applied in targeting gene therapy, as indicated by strong fluorescence intensity assessments. HA-PEI copolymers with disulfide bonds have been synthesized as gene nanocarriers, which were sensitive to acidic pH values and redox responsive conditions. These HA-ss-PEI/pEndo nanocarriers could transport nucleic acids to human mesenchymal stem cells and generate endostatin protein.113 Redox/pH-responsive MnO2 nanosheets have also been fabricated and synergistically functioned with HA to enhance biocompatibility. Cisplatin was absorbed by the nanosheets and delivered into tumor cells. The complexes were triggered in a low pH environment, and GSH could reduce MnO2 to Mn2+, whose biodistribution could be monitored by T1-MRI.114
It is well known that HA can bind specifically to CD44. Responsive glycosaminoglycans (GAG) with alkane thiol were introduced to GAG:HA amphiphiles to form self-assembly micellar NPs, and CD44-mediated internalization was proven by loaded Nile Red among the three groups such as without blocking, specific CD44 blocking, and unspecific IgG1 blocking.115
Other materials
Besides macromolecules such as PEG, PEO, PCL, and HA/PEI, comparable materials have also been investigated for drug loading in redox-responsive micelles. DOX-SS-CSO-SA nanomicelles with size of 62.8 nm could release DOX through disulfide bond cleavage and exhibited 34.8-fold higher cytotoxicity than the free DOX, in which stearic acid was grafted on the low-molecular-weight chitosan.116 Moreover, novel redox-responsive hyperbranched polymers have been prepared by reversible addition-fragmentation chain transfer and self-condensing vinyl polymerization, with the incorporation of DNA aptamer for targeting tumors. 10 mM of GSH could trigger almost 90% of DOX release, unlike the control which only released 5% of DOX within 60 hours. In HeLa cells, these polymer NPs were degraded in the cytoplasm and rapidly released DOX.117 Nanomicelles loading dual antitumor drugs were constructed by co-loading of DOX and POEG-b-PSSDas on prodrug polymers attached to the oncogenic tyrosine kinase inhibitor dasatinib, which could be redox-responsive and showed enhanced antitumor effects both in vitro and in vivo.118
Monodispersed mesoporous organosilica NPs cooperating with thioether bonds exhibited redox-responsive biodegradation phenomenon and improved the drug delivery efficiency as well as tumor suppressing ability.119 Mesoporous organosilica redox-responsive HMONs-ss-PAE has been synthesized to deliver P-gp modulator siRNA and chemotherapeutic DOX releasing drugs to overcome tumor drug resistance.120 Degradable silica NPs loading DOX (DS-DOX) were synthesized to co-deliver plasmid DNA (pDNA) and DOX. The surface of DS-DOX was modified with cyclodextrin-poly(glycidyl methacrylate) (CD-PGEA), including one β-CD core and two ethanolamine-functionalized PGEA arms in order to absorb pDNA. After degradation of surface CD-PGEA, disulfide bonds in the center of DS-DOX were exposed and cleaved under hypoxia to release the pDNA and the active drug, which synergistically acted against tumor cells.121 Figure 7 illustrates the construction of hypoxia-responsive drug release of NPs by the introduction of the alkyl chain of stearic acid (C18) with thiol group in mesoporous silica NPs, in which DOX release could be prevented by the formed disulfide bonds under normoxia but could be triggered under hypoxia. In addition, the targeting RGD motifs have been modified to enhance tumor accumulation.122 Redox-responsive MSN capping with therapeutic and targeting peptides has been developed to enhance the RGD-induced cell endocytosis and release the loaded DOX by GSH activation, in order to induce tumor cells apoptosis.123 Transferrin can function as the gatekeeper of MSN for both active-targeting and controlled drug release. GSH was the key enzyme to trigger DOX release, and these nanocarriers demonstrated enhanced inhibition of tumor growth.124 In addition, MSNs were used to deliver siRNA and DOX. The surface of MSNs was modified by MPTMS/Py-SS-Py, and then DOX was loaded into the channels of MSNs. HS-siRNA was used to replace Py-SH in order to form a new disulfide linkage as a gatekeeper. In vitro, GSH could trigger siRNA and DOX release, and 49.4% and 78.8% of DOX was released when treated with 2 and 5 mM of GSH for 24 hours, respectively, while in the control group, only 14.8% of DOX was released. The siRNA also knocked down the expression of the target protein and demonstrated a synergistic effect with the chemotherapeutic agent.125
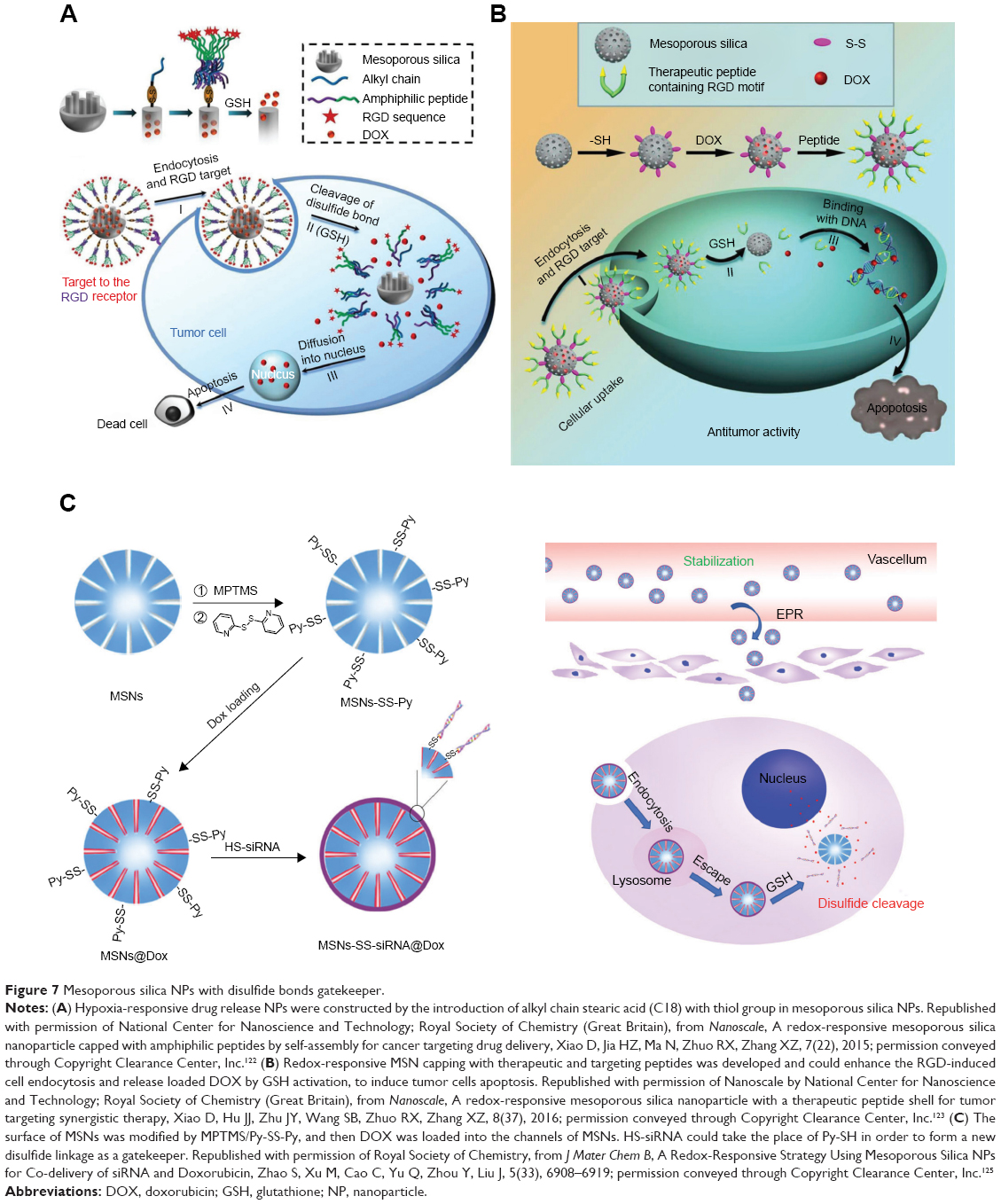 | Figure 7 Mesoporous silica NPs with disulfide bonds gatekeeper. |
Multiple functional sensitive nanomicelles
Various dual- and even tri-functional reductively selective nanomicelles have been obtained by different methods (Figure 8). Using the layer-by-layer method, self-fluorescence microcapsules were fabricated from the polysaccharide alginic acid and cystamine dihydrochloride, demonstrating pH- and redox-sensitivity.126 Experiments on both human and animal tumor cells illustrated that the dual-sensitive HP-ICM could transport drugs in tumors. Comparable pH- and redox-responsive micelles were able to transport and release DOX under low pH and highly reductive conditions. Novel poly[2-(pyridine-2-yldisulfanly)ethyl acrylate] nanocarriers possessed a free-radical polymerization side chains, which were coupled with PEG and cyclic RGD (cRGD) polypeptides via disulfide bonds cross-linking to form RPDSG polymers. These 100 nm submicelles were stable in a physiological environment but released active drugs under reductive conditions.127 Reduction and pH dual-sensitive polypeptide micelles modified with copolymer PEG-b-poly(L-lysine) [PEG-P(LL-CCA/LA)] could be synthesized with lipoic acid (LA) and cis-1,2-cyclohexanedicarboxylic acid (CCA). Both PEG-P(LL18-CCA4/LA14) and PEG-P(LL18-CCA8/LA10) nanomicelles loaded with DOX were triggered by 10 mM of DTT at pH 7.4. In vitro experiments demonstrated that DOX could be released in the endosome at pH 5.0 due to CCA cleavage, or released in the presence of 10 mM of GSH. PEG-P(LL18-CCA4/LA14) was able to release more DOX into the cell nucleus than PEG-P(LL14-/LA14), as demonstrated by FCFM assays.128 PAA-SS-AD, PEG-AD, and PAA-CD, three constituents of self-assembled NPs loading DOX, containing disulfide bonds, released DOX through triggering by the reducing agent and the acidic medium. These DOX-loaded NPs were used to treat the animal model zebrafish and illustrated improved viability.129 A type of pH/redox dual responsive p(L-histidine)n-SS-PU-SS-p(His)n polymer was synthesized with pH-sensitive tiles p(His)n and redox-responsive disulfide bonds. The polymer chains could be self-assembled to form nanomicelles for loading DOX with 19% loading efficiency. Further DOX release could be triggered by low pH or by treatment with 10 mM of GSH, and fluorescence indicated that the nanomicelles could be biodistributed in the animal model within 72 hours and inhibit CT26 tumors.130 Reduction- and pH-responsive nanomicelles, composed of poly(styrene-alt-maleic anhydride) and short PEG chains, were used for delivering drugs and PLK-1 siRNA. These micelles were coated with BSA to enhance their stability. Micelle dissociation could be triggered by low pH and by 10 mM of GSH, resulting in tumor regression both in vitro and in vivo.131 DOX was grafted onto HA by using cis-aconityl and disulfide bond linkage and demonstrated both pH- and redox-responsiveness. These 229 nm micelles released 95% of the active drugs in 150 hours, in the presence of low pH and reductive agents, while the control release was just below 20%. Certain peptide conjugation enhanced the micelle uptake by OVCAR-3 tumor cells, resulting in the suppression of cell growth both in vitro and in vivo.132 A dual-responsive supramolecular PEGylated dendrimer has been investigated for delivering platinum-based drug and NIR indictors. Low pH values and reducing agents could trigger the drug release, and these nanoplatforms showed enhanced antitumor activity, and at the same time reduced the renal toxicity of free drugs.133 pH/redox dual-responsive 100 nm nanomicelles fabricated from diblock copolymers PEG-(MTC-BnCl) loading DOX showed optimized antitumor effects. DOX release could be enhanced by both pH of 5.0 and endo-lysosomal GSH.134
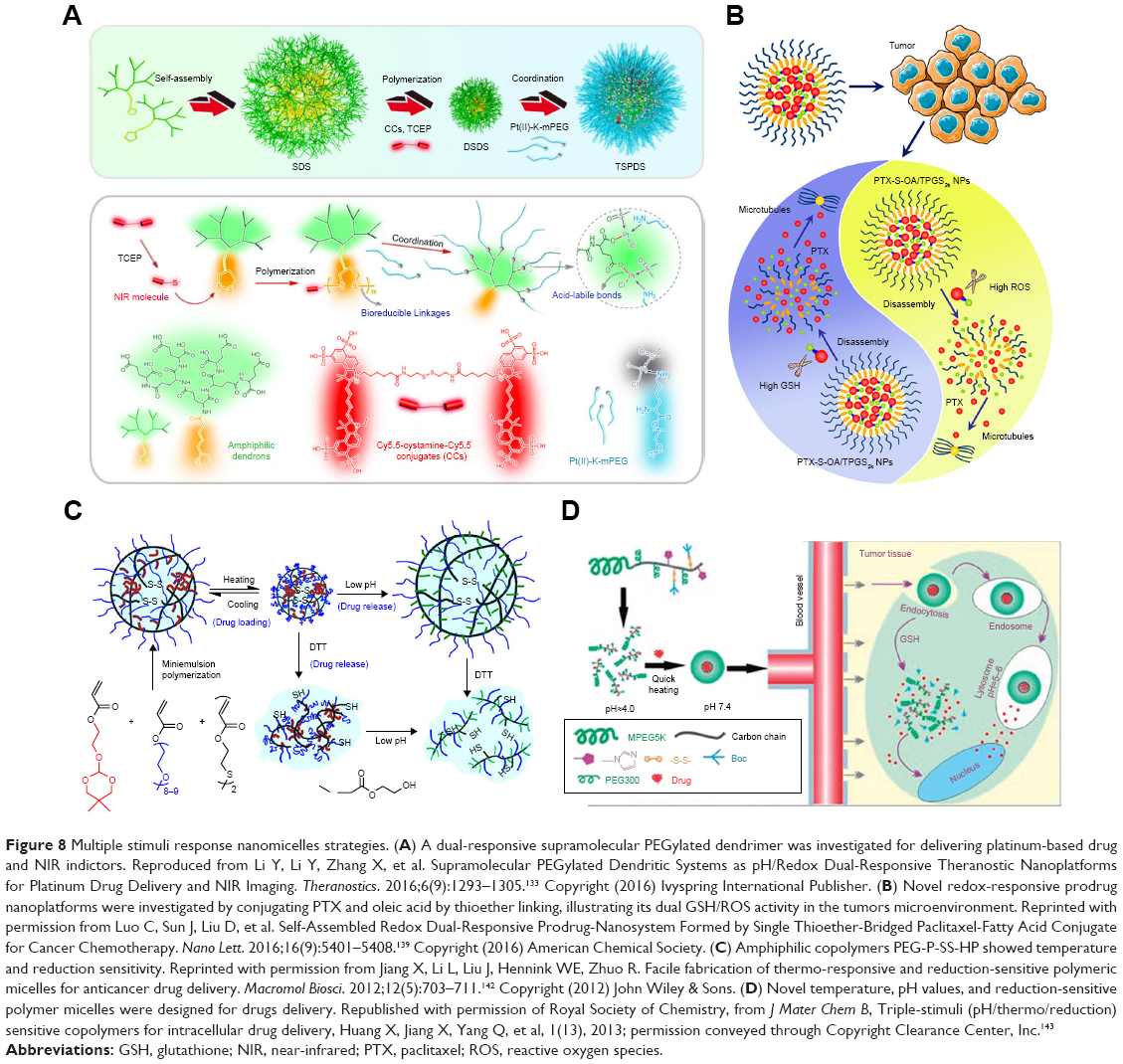 | Figure 8 Multiple stimuli response nanomicelles strategies. (A) A dual-responsive supramolecular PEGylated dendrimer was investigated for delivering platinum-based drug and NIR indictors. Reproduced from Li Y, Li Y, Zhang X, et al. Supramolecular PEGylated Dendritic Systems as pH/Redox Dual-Responsive Theranostic Nanoplatforms for Platinum Drug Delivery and NIR Imaging. Theranostics. 2016;6(9):1293–1305.133 Copyright (2016) Ivyspring International Publisher. (B) Novel redox-responsive prodrug nanoplatforms were investigated by conjugating PTX and oleic acid by thioether linking, illustrating its dual GSH/ROS activity in the tumors microenvironment. Reprinted with permission from Luo C, Sun J, Liu D, et al. Self-Assembled Redox Dual-Responsive Prodrug-Nanosystem Formed by Single Thioether-Bridged Paclitaxel-Fatty Acid Conjugate for Cancer Chemotherapy. Nano Lett. 2016;16(9):5401–5408.139 Copyright (2016) American Chemical Society. (C) Amphiphilic copolymers PEG-P-SS-HP showed temperature and reduction sensitivity. Reprinted with permission from Jiang X, Li L, Liu J, Hennink WE, Zhuo R. Facile fabrication of thermo-responsive and reduction-sensitive polymeric micelles for anticancer drug delivery. Macromol Biosci. 2012;12(5):703–711.142 Copyright (2012) John Wiley & Sons. (D) Novel temperature, pH values, and reduction-sensitive polymer micelles were designed for drugs delivery. Republished with permission of Royal Society of Chemistry, from J Mater Chem B, Triple-stimuli (pH/thermo/reduction) sensitive copolymers for intracellular drug delivery, Huang X, Jiang X, Yang Q, et al, 1(13), 2013; permission conveyed through Copyright Clearance Center, Inc.143 |
Core-shell NPs loading DOX are composed of a hydrophobic core containing DOX and dendrimer PAMAM modified with SPDP, and a conjugated PEG-SH hydrophilic shell. These NPs have been modified with cRGD to enhance their targeting activity and could be triggered by redox and low pH values to enhance antitumor efficiency both in vivo and in vitro.135 A novel copolymer named TPGS-SS-PLA was developed to form 130 nm micelles to encapsulate PTX. cRGD was used to target tumor and enhance cell uptake. The PTX-loaded NPs showed dual pH-sensitivity and redox-responsiveness and could be triggered to release >80% of PTX in 7 days, in the presence of DTT, at pH 5.5. These NPs inhibited tumor growth and reduced cytotoxicity side effects in vivo.136 Dual pH- and hypoxia-responsive liposomes have been constructed by Chol-SS-COOH with modification of the CD-44 targeting HA. The liposome degradation and DOX release were triggered by both mild acidic conditions and reductive agents. Liposome administration with HA modification showed more intensive in vitro cytotoxicity than liposomes, and longer in vivo half-life time release of DOX than free DOX. Finally, in vivo tests revealed CD-44-mediated biodistribution, GSH triggered controlled release and more effective antitumor activity.137 A dual pH- and redox-responsive NP system was fabricated with charge-reversible pullulan derivative and poly(β-amino ester) for both gene and methotrexate delivery. Human hepatoma cells were treated with these NPs that entered cells through endocytosis but finally escaped from the endo/lysosomes and located the cytoplasm, and tetramethylrhodamine (TAMRA)-DNA was proven significantly internalized compared with naked DNA control, because the surface coating of pullulan showed specific affinity-binding ability to asialoglycoprotein receptors.138
Novel redox-responsive prodrug nanoplatforms have been synthesized by conjugating PTX and oleic acid by thioether linking. PEGylated PTX-S-OA micelles showed 57.4% drug-loading efficiency and inhibited tumor growth in vivo. In contrast to the disulfide bond cleavage, PTX could be cleaved by both intracellular GSH and ROS, illustrating its dual GSH/ROS activity in the tumor microenvironment.139 α-Amylase and hydroxyethyl starch have been incorporated into redox-responsive NPs to deliver PTX via disulfide bond conjugation. HES-SS-PTX NPs could be degraded by α-amylase. Pharmacokinetic analysis demonstrated that these NPs had a longer half-life-enhanced accumulation in tumor regions and were triggered by reducing agents, thus illustrating higher antitumor efficiency than free PTX.140 A redox/enzyme dual-responsive nanomedicine system was triggered to release NO to kill specifically tumor cells, whose procedure was monitored by flow cytometry, cooperating with a GSH S-transferases π responsive drug as NO donor, and its cellular internalization was confirmed by a time-dependent red fluorescent dye QM-2.141
Amphiphilic PEG-P-SS-HP copolymers showed temperature and reduction sensitivity with 90% of PTX encapsulation rate and drug content >35%.142 Furthermore, DTT was required for rapid drug release, and the vehicles could decrease the PTX cytotoxicity side effects. Based on p(PEG-MEMA-co-Boc-Cyst-MMAm-co-VI)-b-PEG (PPBV), novel temperature, pH values, and reduction responsive polymer micelles were designed for drug delivery. These PTX-loaded nanomicelles demonstrated stability in PBS but the release of active drugs could only be triggered at certain temperatures, under sub-acidic or reductive conditions.143 Amphiphilic PEG-PCL chains with three disulfide bonds were incorporated in the triple-sensitive cleavable polymeric nanocarriers. Triple disulfide bonds improved the redox responsive sensitivity, and GSH could trigger the DOX release to inhibit tumor cell proliferation.144 Triple-responsive (protease/redox/pH) porous silica NPs have been developed as drug delivery vehicles. Oxidized GSH GSSG was conjugated on the surface of silica NPs to promote redox and protease sensitivity. Acid-triggered fluorescent molecules, such as ZnO quantum dots were attached to the channels of nanopores of porous silica NPs as imaging probes and drug release gatekeepers.145
Summary and perspective
Since hypoxia is one of the marked features of malignant tumors, which is associated with several adaptation changes in the microenvironment of tumor cells, the exploitation of targeting tumor hypoxia is a research hotspot for cancer therapy. In this review, we described current hypoxia-activated therapeutic systems in two aspects: several hypoxia-activated chemotherapeutic prodrugs and redox-responsive nanocarriers. Hypoxia-activated prodrugs can be activated in tumor hypoxic tissue, which can avoid nonspecificity of current antineoplastic agents to minimize side effects and benefit to clinical propulsion. Currently available biomaterials or drugs have a significant role in targeting hypoxia tissue and curing human malignant tumors and other types of cancer. Because of the highly complicated microenvironment of tumors and drug cytotoxicity side effects, few drugs could easily pass the tests required for drug discovery and applied in clinical chemotherapy. In this stage, the hypoxia selectivity of many candidate drugs is not high enough to pass clinical trials. Therefore, few hypoxia-sensitive prodrugs have been circulated in the markets. It needs high throughput methodology to screen ideal candidates for clinical phases, and a deeper understanding of interactions between small molecules and targeted biomacromolecules is required, which can indicate the suitable conditions of the hypoxia-activated molecular structure transformations. But until now, limitations of medicinal development are the disadvantages of hypoxia-activated prodrugs. Hypoxia-active prodrugs with more sensitivity to tumor microenvironment should be designed to increase their specificity to tumor tissue. Hydrophilic hypoxia-activated prodrugs are more significant in clinical trails.
However, at this time, nanotechnology has provided a useful approach for delivering drugs that had been previously eliminated, by increasing their tumor-specific cytotoxicity, hypoxia sensitivity, and preventing drug side effects. Accompanied with in-depth studies on the molecular regulation of the HIF-1 cell signaling pathway, hypoxia tumor regions can be targeted more accurately.146
It was widely known that the drug delivery systems could only deliver <5% of injected dose to tumor region.147 The redox-responsive drug delivery nanocarriers mainly use nanomicelles containing disulfide bonds to target hypoxia, in which the hypoxic cells are identified by various redox molecules such as GSH, cysteine, and homocysteine. Biocompatible macromolecules such as PEG, PEO, PCL, and HA/PEI are frequently used to fabricate these redox-responsive nanomicelles. Comparable active groups, such as nitroimidazole or azo-groups, can also be reduced by specific reducible enzymes in hypoxic cells. Besides, because all nanomicelles have a loose structure and show low encapsulation rates, research studies on redox-responsive nanomicelles are still at the experimental stage, and more clinical trials are required to assess their applicability in clinical practice. It is challenging to apply micro/nano system on patients rather than animal models. In addition, the release mechanism of nanomicelles was still not very clear and should be explored in vivo, in order to reveal the molecular process of inter- and/or intracellular responses to prodrugs. Nanoplatforms for multi-responsibility and theragnostic integration should be developed to provide tumor therapy with more specificity and precision. The reductive microenvironment of tumor cells can also be targeted by developing novel redox-responsive drug delivery nanocarriers. In conclusion, these hypoxia- targeting strategies have potential therapeutic applications and should be further investigated in experimental and clinical research studies.
Acknowledgments
This work was supported in part by the National Key R&D Program of China under Grant No. 2016YFC1300300, the National Natural Science Foundation of China under Grant No. 81627807, 11727813, 81571725, 81701852, 81530058, the Fok Ying-Tong Education Foundation of China under Grant 161104, the Natural Science Basic Research Plan in Shaanxi Province of China under Grant No. 2017JM8057, 2018JM7072, the Natural Science Basic Research Plan in Ningxia Province of China under Grant No. NZ17156; the Natural Science Basic Research Plan in Ningbo of China under Grant No 2017A610144; and the Fundamental Research Funds for the Central Universities (JB171202, JB171203, JB171204, JB171206, JB171207, JB181203, JB181204).
Disclosure
The authors report no conflicts of interest in this work.
References
Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11(6):393–410. | ||
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. | ||
Pentea M, Butu M, Samfira I, Cristina RT, Butnariu M. Extraction and analytical study of salvinorin from leaves of Salvia divinorum. Dig J Nanomater Bios. 2015;10(1):291–297. | ||
Putnoky S, Caunii A, Butnariu M. Study on the stability and antioxidant effect of the Allium ursinum watery extract. Chem Cent J. 2013;7(1):21. | ||
Chen Y, Hu L. Design of anticancer prodrugs for reductive activation. Med Res Rev. 2009;29(1):29–64. | ||
Butnariu M. Detection of the polyphenolic components in Ribes nigrum L. Ann Agric Environ Med. 2014;21(1):11–14. | ||
Butnariu M, Caunii A, Putnoky S. Reverse phase chromatographic behaviour of major components in Capsicum Annuum extract. Chem Cent J. 2012;6(1):146. | ||
Rodino S, Butu M, Negoescu C, Caunii A, Cristina RT, Butnariu M. Spectrophotometric method for quantitative determination of nystatin antifungal agent in pharmaceutical formulations. Dig J Nanomaterials Bios. 2014;9(3):1215–1222. | ||
Barbat C, Rodino S, Petrache P, Butu M, Butnariu M. Microencapsulation of the allelochemical compounds and study of their release from different products. Dig J Nanomater Bios. 2013;8(3):945–953. | ||
Butu AL, Rodino S, Golea D, et al. Liposomal nanodelivery system for proteasome inhibitor anticancer drug bortezomib. Farmacia. 2015;63(2):224–229. | ||
Meng F, Hennink WE, Zhong Z. Reduction-sensitive polymers and bioconjugates for biomedical applications. Biomaterials. 2009;30(12):2180–2198. | ||
Butnariu M, Grozea I. Antioxidant (Antiradical) Compounds. J Bioequiv Availab. 2012;4(6):17–19. | ||
Negrea P, Caunii A, Sarac I, Butnariu M. The study of infrared spectrum of chitin and chitosan extract as potential sources of biomass. Dig J Nanomater Bios. 2015;10(4):1129–1138. | ||
Denny WA. Hypoxia-activated prodrugs in cancer therapy: progress to the clinic. Future Oncol. 2010;6(3):419–428. | ||
Siegel D, Yan C, Ross D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem Pharmacol. 2012;83(8):1033–1040. | ||
Xu S, Yao H, Pei L, et al. Design, synthesis, and biological evaluation of NAD(P)H: Quinone oxidoreductase (NQO1)-targeted oridonin prodrugs possessing indolequinone moiety for hypoxia-selective activation. Eur J Med Chem. 2017;132:310–321. | ||
Inman M, Moody CJ. Synthesis of Indolequinones from Bromoquinones and Enamines Mediated by Cu(OAc)2·H2O. J Org Chem. 2010;75(17):6023–6026. | ||
Hall BS, Meredith EL, Wilkinson SR. Targeting the substrate preference of a type I nitroreductase to develop antitrypanosomal quinone-based prodrugs. Antimicrob Agents Chemother. 2012;56(11):5821–5830. | ||
Lajiness JP, Robertson WM, Dunwiddie I, et al. Design, synthesis, and evaluation of duocarmycin O-amino phenol prodrugs subject to tunable reductive activation. J Med Chem. 2010;53(21):7731–7738. | ||
Sharma K, Iyer A, Sengupta K, Chakrapani H. INDQ/NO, a bioreductively activated nitric oxide prodrug. Org Lett. 2013;15(11):2636–2639. | ||
Huang B, Desai A, Tang S, Thomas TP, Baker JR. The synthesis of a c(RGDyK) targeted SN38 prodrug with an indolequinone structure for bioreductive drug release. Org Lett. 2010;12(7):1384–1387. | ||
Kumar R, Kim E-J, Han J, et al. Hypoxia-directed and activated theranostic agent: Imaging and treatment of solid tumor. Biomaterials. 2016;104:119–128. | ||
Zhu R, Baumann RP, Penketh PG, Shyam K, Sartorelli AC. Hypoxia-Selective O6-Alkylguanine-DNA Alkyltransferase Inhibitors: Design, Synthesis, and Evaluation of 6-(Benzyloxy)-2-(aryldiazenyl)-9 H-purines as Prodrugs of O6-Benzylguanine. J Med Chem. 2013;56(3):1355–1359. | ||
Zhu R, Liu M-C, Luo M-Z, et al. 4-Nitrobenzyloxycarbonyl Derivatives of O6-Benzylguanine as Hypoxia-Activated Prodrug Inhibitors of O6-Alkylguanine-DNA Alkyltransferase (AGT), Which Produces Resistance to Agents Targeting the O-6 Position of DNA Guanine. J Med Chem. 2011;54(21):7720–7728. | ||
Zhu R, Seow HA, Baumann RP, et al. Design of a hypoxia-activated prodrug inhibitor of O6-alkylguanine-DNA alkyltransferase. Bioorg Med Chem Lett. 2012;22(19):6242–6247. | ||
Cazares-Körner C, Pires IM, Swallow ID, et al. CH-01 is a hypoxia-activated prodrug that sensitizes cells to hypoxia/reoxygenation through inhibition of Chk1 and Aurora A. ACS Chem Biol. 2013;8(7):1451–1459. | ||
Wolfe AL, Duncan KK, Parelkar NK, Brown D, Vielhauer GA, Boger DL. Efficacious cyclic N-acyl O-amino phenol duocarmycin prodrugs. J Med Chem. 2013;56(10):4104–4115. | ||
Heapy AM, Patterson AV, Smaill JB, et al. Synthesis and cytotoxicity of pyranonaphthoquinone natural product analogues under bioreductive conditions. Bioorg Med Chem. 2013;21(24):7971–7980. | ||
Mendoza MF, Hollabaugh NM, Hettiarachchi SU, Mccarley RL. Human NAD(P)H:quinone oxidoreductase type I (hNQO1) activation of quinone propionic acid trigger groups. Biochemistry. 2012;51(40):8014–8026. | ||
Bae J, Nael MA, Jiang L, et al. Quinone propionic acid-based redox-triggered polymer nanoparticles for drug delivery: Computational analysis and in vitro evaluation. J Appl Polym Sci. 2014;131(13):40461. | ||
Cho H, Bae J, Garripelli VK, Anderson JM, Jun HW, Jo S. Redox-sensitive polymeric nanoparticles for drug delivery. Chem Commun. 2012;48(48):6043–6045. | ||
Çelik A, Yetiş G. An unusually cold active nitroreductase for prodrug activations. Bioorg Med Chem. 2012;20(11):3540–3550. | ||
Benito J, Shi Y, Szymanska B, et al. Pronounced hypoxia in models of murine and human leukemia: high efficacy of hypoxia-activated prodrug PR-104. PLoS One. 2011;6(8):e23108. | ||
Karnthaler-Benbakka C, Groza D, Koblmüller B, et al. Targeting a targeted drug: an approach toward hypoxia-activatable tyrosine kinase inhibitor prodrugs. ChemMedChem. 2016;11(21):2410–2421. | ||
Meng F, Evans JW, Bhupathi D, et al. Molecular and cellular pharmacology of the hypoxia-activated prodrug TH-302. Mol Cancer Ther. 2012;11(3):740–751. | ||
Liu Q, Sun JD, Wang J, et al. TH-302, a hypoxia-activated prodrug with broad in vivo preclinical combination therapy efficacy: optimization of dosing regimens and schedules. Cancer Chemother Pharmacol. 2012;69(6):1487–1498. | ||
Hu J, van Valckenborgh E, Xu D, et al. Synergistic induction of apoptosis in multiple myeloma cells by bortezomib and hypoxia-activated prodrug TH-302, in vivo and in vitro. Mol Cancer Ther. 2013;12(9):1763–1773. | ||
Wang J, Foehrenbacher A, Su J, et al. The 2-nitroimidazole EF5 is a biomarker for oxidoreductases that activate the bioreductive prodrug CEN-209 under hypoxia. Clin Cancer Res. 2012;18(6):1684–1695. | ||
Hunter FW, Wang J, Patel R, et al. Homologous recombination repair-dependent cytotoxicity of the benzotriazine di-N-oxide CEN-209: comparison with other hypoxia-activated prodrugs. Biochem Pharmacol. 2012;83(5):574–585. | ||
Dragovich PS, Broccatelli F, Chen J, et al. Design, synthesis, and biological evaluation of pyrrolobenzodiazepine-containing hypoxia-activated prodrugs. Bioorg Med Chem Lett. 2017;27(23):5300–5304. | ||
Yamazaki H, Lai YC, Tateno M, et al. Hypoxia-activated prodrug TH-302 decreased survival rate of canine lymphoma cells under hypoxic condition. PLoS One. 2017;12(5):e0177305. | ||
Takakusagi Y, Kishimoto S, Naz S, et al. Radiotherapy Synergizes with the Hypoxia-Activated Prodrug Evofosfamide: In Vitro and In Vivo Studies. Antioxid Redox Signal. 2018;28(2):131–140. | ||
Wojtkowiak JW, Cornnell HC, Matsumoto S, et al. Pyruvate sensitizes pancreatic tumors to hypoxia-activated prodrug TH-302. Cancer Metab. 2015;3(1):2. | ||
Lohse I, Rasowski J, Cao P, et al. Targeting hypoxic microenvironment of pancreatic xenografts with the hypoxia-activated prodrug TH-302. Oncotarget. 2016;7(23):33571–33580. | ||
Hajj C, Russell J, Hart CP, et al. A Combination of Radiation and the Hypoxia-Activated Prodrug Evofosfamide (TH-302) is Efficacious against a Human Orthotopic Pancreatic Tumor Model. Transl Oncol. 2017;10(5):760–765. | ||
Liapis V, Labrinidis A, Zinonos I, et al. Hypoxia-activated pro-drug TH-302 exhibits potent tumor suppressive activity and cooperates with chemotherapy against osteosarcoma. Cancer Lett. 2015;357(1):160–169. | ||
Baumann RP, Penketh PG, Ishiguro K, Shyam K, Zhu YL, Sartorelli AC. Reductive activation of the prodrug 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)-2-[[1-(4-nitrophenyl)ethoxy]carbonyl]hydrazine (KS119) selectively occurs in oxygen-deficient cells and overcomes O6-alkylguanine-DNA alkyltransferase mediated KS119 tumor cell resistance. Biochem Pharmacol. 2010;79(11):1553–1561. | ||
Lindquist KE, Cran JD, Kordic K, et al. Selective radiosensitization of hypoxic cells using BCCA621C: a novel hypoxia activated prodrug targeting DNA-dependent protein kinase. Tumor Microenvironment and Therapy. 2013;1:46–55. | ||
Fu J, Liu L, Huang Z, et al. Hybrid Molecule from O2-(2,4-Dinitrophenyl)diazeniumdiolate and Oleanolic Acid: A Glutathione S-Transferase π-Activated Nitric Oxide Prodrug with Selective Anti-Human Hepatocellular Carcinoma Activity and Improved Stability. J Med Chem. 2013;56(11):4641–4655. | ||
Cheng W, Yuan Y, Qiu N, Peng P, Sheng R, Hu Y. Identification of novel 4-anilinoquinazoline derivatives as potent EGFR inhibitors both under normoxia and hypoxia. Bioorg Med Chem. 2014;22(24):6796–6805. | ||
Stevenson RJ, Denny WA, Tercel M, Pruijn FB, Ashoorzadeh A. Nitro seco analogues of the duocarmycins containing sulfonate leaving groups as hypoxia-activated prodrugs for cancer therapy. J Med Chem. 2012;55(6):2780–2802. | ||
Johnson KM, Parsons ZD, Barnes CL, Gates KS. Toward hypoxia-selective DNA-alkylating agents built by grafting nitrogen mustards onto the bioreductively activated, hypoxia-selective DNA-oxidizing agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine). J Org Chem. 2014;79(16):7520–7531. | ||
Hicks KO, Siim BG, Jaiswal JK, et al. Pharmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogues with improved tissue penetration and hypoxic cell killing in tumors. Clin Cancer Res. 2010;16(20):4946–4957. | ||
Shen X, Rajapakse A, Gallazzi F, et al. Isotopic labeling experiments that elucidate the mechanism of DNA strand cleavage by the hypoxia-selective antitumor agent 1,2,4-benzotriazine 1,4-di-N-oxide. Chem Res Toxicol. 2014;27(1):111–118. | ||
Williams KJ, Albertella MR, Fitzpatrick B, et al. In vivo activation of the hypoxia-targeted cytotoxin AQ4N in human tumor xenografts. Mol Cancer Ther. 2009;8(12):3266–3275. | ||
Nesbitt H, Worthington J, Errington RJ, et al. The unidirectional hypoxia-activated prodrug OCT1002 inhibits growth and vascular development in castrate-resistant prostate tumors. Prostate. 2017;77(15):1539–1547. | ||
Nesbitt H, Byrne NM, Williams SN, et al. Targeting Hypoxic Prostate Tumors Using the Novel Hypoxia-Activated Prodrug OCT1002 Inhibits Expression of Genes Associated with Malignant Progression. Clin Cancer Res. 2017;23(7):1797–1808. | ||
Feng L, Cheng L, Dong Z, et al. Theranostic Liposomes with Hypoxia-Activated Prodrug to Effectively Destruct Hypoxic Tumors Post-Photodynamic Therapy. ACS Nano. 2017;11(1):927–937. | ||
Shen S, Wu Y, Li K, et al. Versatile hyaluronic acid modified AQ4N-Cu(II)-gossypol infinite coordination polymer nanoparticles: Multiple tumor targeting, highly efficient synergistic chemotherapy, and real-time self-monitoring. Biomaterials. 2018;154:197–212. | ||
Lavaggi ML, Cabrera M, Celano L, Thomson L, Cerecetto H, González M. Biotransformation of Phenazine 5,10-Dioxides under Hypoxic Conditions as an Example of Activation of Anticancer Prodrug: An Interdisciplinary Experiment for Biochemistry or Organic Chemistry. J Chem Educ. 2013;90(10):1388–1391. | ||
Chowdhury G, Sarkar U, Pullen S, et al. DNA strand cleavage by the phenazine di-N-oxide natural product myxin under both aerobic and anaerobic conditions. Chem Res Toxicol. 2012;25(1):197–206. | ||
Lavaggi ML, Cabrera M, Pintos C, et al. Novel Phenazine 5,10-Dioxides Release ·OH in Simulated Hypoxia and Induce Reduction of Tumour Volume In Vivo. ISRN Pharmacol. 2011;2011(6):1–11. | ||
Gonda M, Nieves M, Nunes E, et al. Phenazine N,N′-dioxide scaffold as selective hypoxic cytotoxin pharmacophore. Structural modifications looking for further DNA topoisomerase II-inhibition activity. MedChemComm. 2013;4(3):595–607. | ||
Lavaggi ML, Nieves M, Cabrera M, et al. Structural modifications on the phenazine N,N′-dioxide-scaffold looking for new selective hypoxic cytotoxins. Eur J Med Chem. 2010;45(11):5362–5369. | ||
Yadav P, Marshall AJ, Reynisson J, Denny WA, Hay MP, Anderson RF. Fragmentation of the quinoxaline N-oxide bond to the ˙OH radical upon one-electron bioreduction. Chem Commun. 2014;50(89):13729–13731. | ||
Heffern MC, Yamamoto N, Holbrook RJ, Eckermann AL, Meade TJ. Cobalt derivatives as promising therapeutic agents. Curr Opin Chem Biol. 2013;17(2):189–196. | ||
Graf N, Lippard SJ. Redox activation of metal-based prodrugs as a strategy for drug delivery. Adv Drug Deliv Rev. 2012;64(11):993–1004. | ||
Yamamoto N, Renfrew AK, Kim BJ, Bryce NS, Hambley TW. Dual targeting of hypoxic and acidic tumor environments with a cobalt(III) chaperone complex. J Med Chem. 2012;55(24):11013–11021. | ||
Kim BJ, Hambley TW, Bryce NS. Visualising the hypoxia selectivity of cobalt(III) prodrugs. Chem Sci. 2011;2(11):2135–2142. | ||
Karnthaler-Benbakka C, Groza D, Kryeziu K, et al. Tumor-targeting of EGFR inhibitors by hypoxia-mediated activation. Angew Chem Int Edit. 2014;53(47):12930–12935. | ||
Chang JY-C, Lu G-L, Stevenson RJ, et al. Cross-Bridged Cyclen or Cyclam Co(III) Complexes Containing Cytotoxic Ligands as Hypoxia-Activated Prodrugs. Inorg Chem. 2013;52(13):7688–7698. | ||
Bustamante FLS, Miranda FS, Castro FAV, Resende JALC, Pereira MD, Lanznaster M. A study on the properties and reactivity of naphthoquinone–cobalt(III) prototypes for bioreductive prodrugs. J Inorg Biochem. 2014;132:37–44. | ||
G-L L, Stevenson RJ, Chang JY-C, et al. Tercel M. N-alkylated cyclen cobalt (III) complexes of 1-(chloromethyl)-3-(5,6,7-trimethoxyindol-2-ylcarbonyl)-2,3-dihydro-1H-pyrrolo [3,2-f] quinolin-5-ol DNA alkylating agent as hypoxia-activated prodrugs. Bioorg Med Chem. 2011;19(16):4861–4867. | ||
Lin Q, Bao C, Yang Y, et al. Highly Discriminating Photorelease of Anticancer Drugs Based on Hypoxia Activatable Phototrigger Conjugated Chitosan Nanoparticles. Adv Mater. 2013;25(14):1981–1986. | ||
Thambi T, Deepagan VG, Yoon HY, et al. Hypoxia-responsive polymeric nanoparticles for tumor-targeted drug delivery. Biomaterials. 2014;35(5):1735–1743. | ||
Perche F, Biswas S, Wang T, Zhu L, Torchilin V. Hypoxia-Targeted siRNA Delivery. Angew Chem. 2014;126(13):3430–3434. | ||
Nakahata M, Takashima Y, Yamaguchi H, Harada A. Redox-responsive self-healing materials formed from host–guest polymers. Nat Commun. 2011;2(1):511. | ||
Staff RH, Gallei M, Mazurowski M, et al. Patchy Nanocapsules of Poly(vinylferrocene)-Based Block Copolymers for Redox-Responsive Release. ACS Nano. 2012;6(10):9042–9049. | ||
Han L, Zhang X-Y, Wang Y-L, et al. Redox-responsive theranostic nanoplatforms based on inorganic nanomaterials. J Control Release. 2017;259:40–52. | ||
Yang HY, Li Y, Lee DS. Multifunctional and Stimuli-Responsive Magnetic Nanoparticle-Based Delivery Systems for Biomedical Applications. Advanced Therapeutics. 2018;1(2):1800011. | ||
Stephen ZR, Kievit FM, Veiseh O, et al. Redox-Responsive Magnetic Nanoparticle for Targeted Convection-Enhanced Delivery of O6-Benzylguanine to Brain Tumors. ACS Nano. 2014;8(10):10383–10395. | ||
Nehate C, Aji Alex MR, Kumar A, Koul V. Combinatorial delivery of superparamagnetic iron oxide nanoparticles (γFe2 O3 ) and doxorubicin using folate conjugated redox sensitive multiblock polymeric nanocarriers for enhancing the chemotherapeutic efficacy in cancer cells. Mater Sci Eng C Mater Biol Appl. 2017;75:1128–1143. | ||
Liu J, Chen Q, Zhu W, et al. Nanoscale-Coordination-Polymer-Shelled Manganese Dioxide Composite Nanoparticles. A Multistage Redox/pH/H2O2-Responsive Cancer Theranostic Nanoplatform. Adv Funct Mater. 2017;27(10):1605926. | ||
Zhao S, Yu Q, Pan J, et al. Redox-responsive mesoporous selenium delivery of doxorubicin targets MCF-7 cells and synergistically enhances its anti-tumor activity. Acta Biomater. 2017;54:294–306. | ||
Zhai S, Hu X, Hu Y, Wu B, Xing D. Visible light-induced crosslinking and physiological stabilization of diselenide-rich nanoparticles for redox-responsive drug release and combination chemotherapy. Biomaterials. 2017;121:41–54. | ||
Behroozi F, Abdkhodaie MJ, Abandansari HS, et al. Engineering Folate-Targeting Diselenide-containing Triblock Copolymer as a Redox-Responsive Shell-sheddable Micelle for Antitumor Therapy In Vivo. Acta Biomater. 2018. | ||
Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011;152(1):2–12. | ||
Lee MH, Yang Z, Lim CW, et al. Disulfide-cleavage-triggered chemosensors and their biological applications. Chem Rev. 2013;113(7):5071–5109. | ||
Wei H, Zhuo R-X, Zhang X-Z. Design and development of polymeric micelles with cleavable links for intracellular drug delivery. Prog Polym Sci. 2013;38(3–4):503–535. | ||
Dispinar T, van Camp W, de Cock LJ, de Geest BG, du Prez FE. Redox-Responsive Degradable PEG Cryogels as Potential Cell Scaffolds in Tissue Engineering. Macromol Biosci. 2012;12(3):383–394. | ||
Wen H, Dong C, Dong H, et al. Engineered Redox-Responsive PEG Detachment Mechanism in PEGylated Nano-Graphene Oxide for Intracellular Drug Delivery. Small. 2012;8(5):760–769. | ||
Xiong H, Guo Z, Zhang W, Zhong H, Liu S, Ji Y. Redox-responsive biodegradable PEGylated nanographene oxide for efficiently chemo-photothermal therapy: a comparative study with non-biodegradable PEGylated nanographene oxide. J Photochem Photobiol B. 2014;138:191–201. | ||
Wu X, Sun X, Guo Z, et al. In vivo and in situ tracking cancer chemotherapy by highly photostable NIR fluorescent theranostic prodrug. J Am Chem Soc. 2014;136(9):3579–3588. | ||
Cui C, Xue Y-N, Wu M, et al. Cellular uptake, intracellular trafficking, and antitumor efficacy of doxorubicin-loaded reduction-sensitive micelles. Biomaterials. 2013;34(15):3858–3869. | ||
van der Vlies AJ, Hasegawa U, Hubbell JA. Reduction-sensitive tioguanine prodrug micelles. Mol Pharm. 2012;9(10):2812–2818. | ||
Chang B, Chen D, Wang Y, et al. Bioresponsive Controlled Drug Release Based on Mesoporous Silica Nanoparticles Coated with Reductively Sheddable Polymer Shell. Chem Mater. 2013;25(4):574–585. | ||
Yang Q, Tan L, He C, et al. Redox-responsive micelles self-assembled from dynamic covalent block copolymers for intracellular drug delivery. Acta Biomater. 2015;17:193–200. | ||
Zhang P, Zhang H, He W, Zhao D, Song A, Luan Y. Disulfide-Linked Amphiphilic Polymer-Docetaxel Conjugates Assembled Redox-Sensitive Micelles for Efficient Antitumor Drug Delivery. Biomacromolecules. 2016;17(5):1621–1632. | ||
Chen C, Zheng P, Cao Z, et al. PEGylated hyperbranched polyphosphoester based nanocarriers for redox-responsive delivery of doxorubicin. Biomater Sci. 2016;4(3):412–417. | ||
Tu Y, Peng F, White PB, Wilson DA. Redox-Sensitive Stomatocyte Nanomotors: Destruction and Drug Release in the Presence of Glutathione. Angew Chem Int Edit. 2017;56(26):7620–7624. | ||
Tang Z, Zhang L, Wang Y, Li D, Zhong Z, Zhou S. Redox-responsive star-shaped magnetic micelles with active-targeted and magnetic-guided functions for cancer therapy. Acta Biomater. 2016;42:232–246. | ||
Conte C, Mastrotto F, Taresco V, et al. Enhanced uptake in 2D- and 3D-lung cancer cell models of redox responsive PEGylated nanoparticles with sensitivity to reducing extra- and intracellular environments. J Control Release. 2018;277:126–141. | ||
Zhang Y, Guo Z, Cao Z, et al. Endogenous albumin-mediated delivery of redox-responsive paclitaxel-loaded micelles for targeted cancer therapy. Biomaterials. 2018;183:243–257. | ||
Xia Y, Wang N, Qin Z, et al. Polycarbonate-based core-crosslinked redox-responsive nanoparticles for targeted delivery of anticancer drug. J Mater Chem B. 2018;6(20):3348–3357. | ||
Sun P, Zhou D, Gan Z. Novel reduction-sensitive micelles for triggered intracellular drug release. J Control Release. 2011;155(1):96–103. | ||
Kim JO, Sahay G, Kabanov AV, Bronich TK. Polymeric micelles with ionic cores containing biodegradable cross-links for delivery of chemotherapeutic agents. Biomacromolecules. 2010;11(4):919–926. | ||
Chen W, Zou Y, Jia J, et al. Functional Poly(ε-caprolactone)s via Copolymerization of ε-Caprolactone and Pyridyl Disulfide-Containing Cyclic Carbonate: Controlled Synthesis and Facile Access to Reduction-Sensitive Biodegradable Graft Copolymer Micelles. Macromolecules. 2013;46(3):699–707. | ||
Tang LY, Wang YC, Li Y, Du JZ, Wang J. Shell-detachable micelles based on disulfide-linked block copolymer as potential carrier for intracellular drug delivery. Bioconjugate Chem. 2009;20(6):1095–1099. | ||
Li J, Huo M, Wang J, et al. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33(7):2310–2320. | ||
Kim H, Mun S, Choi Y. Photosensitizer-conjugated polymeric nanoparticles for redox-responsive fluorescence imaging and photodynamic therapy. J Mater Chem B. 2013;1(4):429–431. | ||
Han HS, Choi KY, Ko H, et al. Bioreducible core-crosslinked hyaluronic acid micelle for targeted cancer therapy. J Control Release. 2015;200:158–166. | ||
He Y, Cheng G, Xie L, Nie Y, He B, Gu Z. Polyethyleneimine/DNA polyplexes with reduction-sensitive hyaluronic acid derivatives shielding for targeted gene delivery. Biomaterials. 2013;34(4):1235–1245. | ||
Yeh P-H, Sun J-S, Wu H-C, Hwang L-H, Wang T-W. Stimuli-responsive HA-PEI nanoparticles encapsulating endostatin plasmid for stem cell gene therapy. RSC Adv. 2013;3(31):12922–12932. | ||
Hao Y, Wang L, Zhang B, et al. Manganese dioxide nanosheets-based redox/pH-responsive drug delivery system for cancer theranostic application. Int J Nanomedicine. 2016;11:1759–1778. | ||
Carvalho AM, Teixeira R, Novoa-Carballal R, Pires RA, Reis RL, Pashkuleva I. Redox-responsive micellar nanoparticles from glycosaminoglycans for CD44 targeted drug delivery. Biomacromolecules. 2018;19(7):2991–2999. | ||
Su Y, Hu Y, du Y, et al. Redox-responsive polymer-drug conjugates based on doxorubicin and chitosan oligosaccharide-g-stearic acid for cancer therapy. Mol Pharm. 2015;12(4):1193–1202. | ||
Zhuang Y, Deng H, Su Y, et al. Aptamer-Functionalized and Backbone Redox-Responsive Hyperbranched Polymer for Targeted Drug Delivery in Cancer Therapy. Biomacromolecules. 2016;17(6):2050–2062. | ||
Sun J, Liu Y, Chen Y, et al. Doxorubicin delivered by a redox-responsive dasatinib-containing polymeric prodrug carrier for combination therapy. J Control Release. 2017;258:43–55. | ||
Yu L, Chen Y, Lin H, du W, Chen H, Shi J. Ultrasmall mesoporous organosilica nanoparticles: Morphology modulations and redox-responsive biodegradability for tumor-specific drug delivery. Biomaterials. 2018;161:292–305. | ||
Wu M, Meng Q, Chen Y, et al. Large pore-sized hollow mesoporous organosilica for redox-responsive gene delivery and synergistic cancer chemotherapy. Adv Mater. 2016;28(10):1963–1969. | ||
Zhang Q, Shen C, Zhao N, Xu F-J. Redox-Responsive and Drug-Embedded Silica Nanoparticles with Unique Self-Destruction Features for Efficient Gene/Drug Codelivery. Adv Funct Mater. 2017;27(10):1606229. | ||
Xiao D, Jia HZ, Ma N, Zhuo RX, Zhang XZ. A redox-responsive mesoporous silica nanoparticle capped with amphiphilic peptides by self-assembly for cancer targeting drug delivery. Nanoscale. 2015;7(22):10071–10077. | ||
Xiao D, Hu JJ, Zhu JY, Wang SB, Zhuo RX, Zhang XZ. A redox-responsive mesoporous silica nanoparticle with a therapeutic peptide shell for tumor targeting synergistic therapy. Nanoscale. 2016;8(37):16702–16709. | ||
Tian Y, Guo R, Jiao Y, et al. Redox stimuli-responsive hollow mesoporous silica nanocarriers for targeted drug delivery in cancer therapy. Nanoscale Horiz. 2016;1(6):480–487. | ||
Zhao S, Xu M, Cao C, Yu Q, Zhou Y, Liu J. A Redox-Responsive Strategy Using Mesoporous Silica NPs for Co-delivery of siRNA and Doxorubicin. J Mater Chem B. 2017;5(33):6908–6919. | ||
Gao L, Fei J, Zhao J, Cui W, Cui Y, Li J. pH- and redox-responsive polysaccharide-based microcapsules with autofluorescence for biomedical applications. Chem Eur J. 2012;18(11):3185–3192. | ||
Remant Bahadur KC, Thapa B, Xu P. pH and redox dual responsive nanoparticle for nuclear targeted drug delivery. Mol Pharm. 2012;9(9):2719–2729. | ||
Wu L, Zou Y, Deng C, Cheng R, Meng F, Zhong Z. Intracellular release of doxorubicin from core-crosslinked polypeptide micelles triggered by both pH and reduction conditions. Biomaterials. 2013;34(21):5262–5272. | ||
Ang CY, Tan SY, Teh C, et al. Redox and pH Dual Responsive Polymer Based Nanoparticles for In Vivo Drug Delivery. Small. 2017;13(7):1602379. | ||
John JV, Uthaman S, Augustine R, Chen H, Park I-K, Kim I. pH/redox dual stimuli-responsive sheddable nanodaisies for efficient intracellular tumour-triggered drug delivery. J Mater Chem B. 2017;5(25):5027–5036. | ||
Aji Alex MR, Nehate C, Veeranarayanan S, Kumar DS, Kulshreshtha R, Koul V. Self assembled dual responsive micelles stabilized with protein for co-delivery of drug and siRNA in cancer therapy. Biomaterials. 2017;133:94–106. | ||
Lin CJ, Kuan CH, Wang LW, et al. Integrated self-assembling drug delivery system possessing dual responsive and active targeting for orthotopic ovarian cancer theranostics. Biomaterials. 2016;90:12–26. | ||
Li Y, Li Y, Zhang X, et al. Supramolecular PEGylated Dendritic Systems as pH/Redox Dual-Responsive Theranostic Nanoplatforms for Platinum Drug Delivery and NIR Imaging. Theranostics. 2016;6(9):1293–1305. | ||
Teo JY, Chin W, Ke X, et al. pH and redox dual-responsive biodegradable polymeric micelles with high drug loading for effective anticancer drug delivery. Nanomed Nanotechnol Biol Med. 2017;13(2):431–442. | ||
Hu W, Qiu L, Cheng L, et al. Redox and pH dual responsive poly(amidoamine) dendrimer-poly(ethylene glycol) conjugates for intracellular delivery of doxorubicin. Acta Biomater. 2016;36:241–253. | ||
Guo Y, Niu B, Song Q, et al. RGD-decorated redox-responsive d-α-tocopherol polyethylene glycol succinate–poly(lactide) nanoparticles for targeted drug delivery. J Mater Chem B. 2016;4(13):2338–2350. | ||
Chi Y, Yin X, Sun K, et al. Redox-sensitive and hyaluronic acid functionalized liposomes for cytoplasmic drug delivery to osteosarcoma in animal models. J Control Release. 2017;261:113–125. | ||
Zhang S, Wang D, Li Y, et al. pH- and redox-responsive nanoparticles composed of charge-reversible pullulan-based shells and disulfide-containing poly(β-amino ester) cores for co-delivery of a gene and chemotherapeutic agent. Nanotechnology. 2018;29(32):325101. | ||
Luo C, Sun J, Liu D, et al. Self-Assembled Redox Dual-Responsive Prodrug-Nanosystem Formed by Single Thioether-Bridged Paclitaxel-Fatty Acid Conjugate for Cancer Chemotherapy. Nano Lett. 2016;16(9):5401–5408. | ||
Li Y, Hu H, Zhou Q, et al. α-Amylase- and Redox-Responsive Nanoparticles for Tumor-Targeted Drug Delivery. ACS Appl Mater Interfaces. 2017;9(22):19215–19230. | ||
Jia X, Zhang Y, Zou Y, et al. Dual Intratumoral Redox/Enzyme-Responsive NO-Releasing Nanomedicine for the Specific, High-Efficacy, and Low-Toxic Cancer Therapy. Adv Mater. 2018;30(30):e1704490. | ||
Jiang X, Li L, Liu J, Hennink WE, Zhuo R. Facile fabrication of thermo-responsive and reduction-sensitive polymeric micelles for anticancer drug delivery. Macromol Biosci. 2012;12(5):703–711. | ||
Huang X, Jiang X, Yang Q, et al. Triple-stimuli (pH/thermo/reduction) sensitive copolymers for intracellular drug delivery. J Mater Chem B. 2013;1(13):1860–1868. | ||
Zhao C, Shao L, Lu J, et al. Triple Redox Responsive Poly(Ethylene Glycol)-Polycaprolactone Polymeric Nanocarriers for Fine-Controlled Drug Release. Macromol Biosci. 2017;17(4):1600295. | ||
Qiu L, Zhao Y, Li B, Wang Z, Cao L, Sun L. Triple-stimuli (protease/redox/pH) sensitive porous silica nanocarriers for drug delivery. Sens Actuators B Chem. 2017;240:1066–1074. | ||
Alimoradi H, Matikonda SS, Gamble AB, Giles GI, Greish K. Hypoxia responsive drug delivery systems in tumor therapy. Curr Pharm Des. 2016;22(19):2808–2820. | ||
Thambi T, Park JH, Lee DS. Hypoxia-responsive nanocarriers for cancer imaging and therapy: recent approaches and future perspectives. Chem Commun. 2016;52(55):8492–8500. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.