")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Hydrogen Sulfide Protects Against High Glucose-Induced Human Umbilical Vein Endothelial Cell Injury Through Activating PI3K/Akt/eNOS Pathway
Authors Lin F , Yang Y, Wei S, Huang X, Peng Z, Ke X, Zeng Z, Song Y
Received 16 December 2019
Accepted for publication 5 February 2020
Published 14 February 2020 Volume 2020:14 Pages 621—633
DOI https://doi.org/10.2147/DDDT.S242521
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Anastasios Lymperopoulos
Fengxia Lin,1,* Yiying Yang,2,* Shanyin Wei,1 Xiaojing Huang,1 Zhijian Peng,1 Xiao Ke,3 Zhicong Zeng,1 Yinzhi Song1
1Department of Cardiology, Shenzhen Bao’an Traditional Chinese Medicine Hospital Group, The Affiliated Hospital of Guangzhou University of Chinese Medicine, Shenzhen 518133, People’s Republic of China; 2Department of Cardiology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, People’s Republic of China; 3Department of Cardiology, Fuwai Hospital, Chinese Academy of Medical Sciences, (Shenzhen Sun Yat-sen Cardiovascular Hospital), Shenzhen 518057, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhicong Zeng; Yinzhi Song Email [email protected]; [email protected]
Purpose: Dysfunction of endothelial cells plays a key role in the pathogenesis of diabetic atherosclerosis. High glucose (HG) has been found as a key factor in the progression of diabetic complications, including atherosclerosis. PI3K/Akt/eNOS signaling pathway has been shown to involve in HG-induced vascular injuries. Hydrogen sulfide (H2S) has been found to exhibit protective effects on HG-induced vascular injuries. Moreover, H2S activates PI3K/Akt/eNOS pathway in endothelial cells. Thus, the present study aimed to determine if H2S exerts protective effects against HG-induced injuries of human umbilical vein endothelial cells (HUVECs) via activating PI3K/Akt/eNOS signaling.
Materials and Methods: The endothelial protective effects of H2S were evaluated and compared to the controlled groups. Cell viability, cell migration and tube formation were determined by in vitro functional assays; protein levels were evaluated by Western blot assay and ELISA; cell apoptosis was determined by Hoechst 33258 nuclear staining; Reactive oxygen species (ROS) production was evaluated by the ROS detection kit.
Results: HG treatment significantly inhibited PI3K/Akt/eNOS signaling in HUVECs, which was partially reversed by the H2S treatment. HG treatment inhibited cell viability of HUVECs, which were markedly prevented by H2S or PI3K agonist Y-P 740. HG treatment also induced HUVEC cell apoptosis by increasing the protein levels of cleaved caspase 3, Bax and Bcl-2, which were significantly attenuated by H2S or 740 Y-P. ROS production and gp91phox protein level were increased by HG treatment in HUVECs and this effect can be blocked by the treatment with H2S or Y-P 740. Moreover, HG treatment increased the protein levels of pro-inflammatory cytokines, caspase-1 and phosphorylated JNK, which was significantly attenuated by H2S or Y-P 740. Importantly, the cytoprotective effect of H2S against HG-induced injury was inhibited by LY294002 (an inhibitor of PI3K/Akt/eNOS signaling pathway).
Conclusion: The present study demonstrated that exogenous H2S protects endothelial cells against HG-induced injuries by activating PI3K/Akt/eNOS pathway. Based on the above findings, we proposed that reduced endogenous H2S levels and the subsequent PI3K/Akt/eNOS signaling impairment may be the important pathophysiological mechanism underlying hyperglycemia-induced vascular injuries.
Keywords: endothelial cells, hydrogen sulfide, high glucose, injury, PI3K/Akt/eNOS
Introduction
Diabetes-associated vascular complications are regarded as a big threat to the health of human beings on account of its increased morbidity and mortality1 and are caused by exposure to chronic high glucose (HG).2,3 Increasing evidence has demonstrated that endoplasmic reticulum (ER) stress,4–6 apoptosis,6–8 oxidative stress4,6-8 and inflammation6,9,10 are essential players in controlling the progression of diabetes-associated atherosclerosis. Diabetes-induced atherosclerosis is associated with endothelial dysfunction, referred as reduced endothelium-dependent vascular relaxation.11 Although glucose control represents the basics of diabetes therapy, there is limited improvement in ameliorating cardiovascular complications of diabetic patients.12,13 Therefore, it is urgent to develop effective therapies that control the course of diabetes-induced atherosclerosis. Recently, there is accumulating evidence showing that activation of PI3K/Akt/eNOS signaling orchestrates protective actions against endothelial dysfunction and apoptosis.14,15 As a pro-apoptotic factor, Bax can be passivated by Akt, thus inhibiting cell apoptosis and facilitating cell proliferative ability by mitigating its inhibitory action on Bcl-2.16,17 Hyperglycemia has been well documented to potentiate intracellular reactive oxygen species (ROS) generation, which is considered as another contributor to the dysfunction of endothelial cells.18 ROS accumulation leads to the impairment of antioxidant system and DNA synthesis which results in enhanced inflammatory response.19,20 Evidence from in vitro studies indicated that the PI3K/Akt/eNOS signaling contributes to ROS-associated endothelial injuries, including inflammation.21,22 However, the mechanisms remain unclarified especially concerning the effect of PI3K/Akt/eNOS signaling on endothelial cell injuries under high serum glucose level.
Hydrogen sulfide (H2S) belongs to a type of gas with distinct smell of rotten eggs. However, there is now an abundance of literature indicating that H2S is an endogenous gasotransmitter with multiple functions in the cardiovascular systems.23–26 It has been reported that atherosclerosis acceleration is associated with decreased endogenous production of H2S.27 Inhibition of cystathionine γ-lyase (CSE), a synthase of endogenous H2S, promotes endothelial cell dysfunction induced by hyperglycemia,28 and reduced H2S levels in the streptozotocin-induced diabetic rats may be linked with vascular inflammation.25 In diabetic mice, however, treatment with chronic H2S can restore nitro oxide efficacy and decrease oxidative stress in the mouse aorta, thus improving endothelial function.29 An in vitro study reported that H2S up-regulated the production of NO from eNOS via an Akt-dependent manner in endothelial cells.30 H2S may therefore orchestrate key function in diabetes-associated atherosclerosis and H2S-mediated actions may be via activating PI3K/Akt/eNOS signaling. In the present study, we aimed to determine if H2S can exert protective effects against HG-induced injuries of human umbilical vein endothelial cells (HUVECs) via activating PI3K/Akt/eNOS signaling.
Materials and Methods
Chemicals and Reagents
Sodium hydrosulfide (NaHS; a donor of H2S) was purchased from Gibco-BRL (Grand Island, NY, USA). Fetal bovine serum (FBS), 2′, 7′-dichlorofluorescein diacetate (DCFH-DA), 740 Y-P (a PI3K agonist), LY294002 (a reversible PI3K inhibitor), sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), M200 medium, D-glucose, Hoechst 33258 and mannitol were supplied by Sigma-Aldrich (St Louis, MO, USA). Cell counter kit-8 (CCK-8) was purchased from Dojindo Lab (Kumamoto, Japan). Anti-GAPDH (#8884), anti-ATF6 (#65880), anti-CHOP (#2895), anti-BiP (#3177), anti-phospho (p)-PI3K (#4228), anti-p-Akt (#4060), anti-p-eNOS (#9570), anti-total (t)-PI3K (#4249), anti-t-Akt (#4685), anti-t-eNOS (#9586), anti-Bax (#5023), anti-Bcl2 (#2827), anti-cleaved caspase 3 (#9661), anti-cleaved caspase 1 (#4199), anti-p-JNK(#4668), anti-t-JNK (#9252) and anti-gp91phox (#80897) antibodies were from Cell Signaling Technology (Boston, MA, USA). Enhanced chemiluminescence (ECL) solution was purchased from KeyGen Biotech (Nanjing, China). Interleukin (IL)-1β (#ab46052), IL-6 (#ab46027) and tumor necrosis factor (TNF)-α (#ab10054) ELISA kits were provided by Abcam (Cambridge, UK). Horseradish peroxidase (HRP)-conjugated secondary antibody and BCA protein assay kit were obtained from KangChen Bio-tech, Inc (Shanghai, China).
Cell Culture
HUVECs were purchased from Sigma-Aldrich (#SCCE001; St. Louis, USA) and were routinely cultured in M200 medium containing 2% FBS at 37°C with 5% CO2/95% air. To evaluate the effects of HG on HUVECs, cells were incubated with medium containing either 5 mM glucose (physiological glucose concentration) or 33 mM glucose (HG concentration) for 24 hrs. In some experimental designs, HUVECs in the specified groups were pretreated with 400 μM NaHS for 30 min before HG-treatment. To determine the role of PI3K̸Akt/eNOS signaling pathway in cytoprotection of H2S, HUVECs in the specified groups were either pretreated with 20 μM 740 Y-P or co-treated with 10 μM LY294002 and NaHS for a duration of 30 min prior to treatment with HG. To analyze the osmotic stress effect of HG on cells, mannitol (33 mM) was used as an isotonic group in which some HUVECs were treated for 24 hrs in osmotic pressure similar to those in the HG group.
Western Blot Analysis
After the indicated treatments, HUVECs were harvested for Western blot analysis according to previous studies.31 Briefly, the proteins were extracted using RIPA buffer. Protein concentrations were determined by the BCA protein assay kit. Equal amounts of proteins were resolved on the 10% SDS-PAGE gel followed by transferring to the PVDF membranes. After blocking with 1.5% skimmed milk at room temperature for 1 hr, the membranes were incubated with the primary antibodies specific to anti-GAPDH (1:1000 dilution), anti-ATF6 (1:1000 dilution), anti-CHOP (1:1000 dilution), anti-BiP (1:1000 dilution), anti-p-PI3K (1:1000 dilution), anti-p-Akt (1:1000 dilution), anti-p-eNOS (1:1000 dilution), anti-t-PI3K (1:1000 dilution), anti-t-Akt (1:1000 dilution), anti-t-eNOS (1:1000 dilution), anti-Bax (1:1000 dilution), anti-Bcl2 (1:1000 dilution), anti-cleaved caspase 3 (1:1000 dilution), anti-cleaved caspase 1 (1:1000 dilution), anti-p-JNK (1:1000 dilution), anti-t-JNK (1:1000 dilution) or anti-gp91phox antibody (1:1000 dilution) overnight at 4°C. After that, the membranes were further probed by the HRP-conjugated secondary antibodies. The Western blot bands were visualized by ECL detection kit. Experiments were repeated three times.
Cell Viability Determination
HUVECs viability was determined by the CCK-8 assay kit. After the indicated treatments, the HUVECs were washed with phosphate-buffered saline (PBS), and 10 µL CCK-8 solution was added to each well for 2-hr incubation at room temperature. Optical density (OD) value determination at 450 nm wavelength was performed to measure HUVECs viability. Experiments were repeated three times.
Hoechst 33258 Nuclear Staining for Assessing Apoptosis
Apoptotic cells’ most significant change is chromosome condensation. Hoechst 33258 can bind to the DNA molecule as fluorescent probe. The intake of the Hoechst 33258 is increased in apoptotic cells and the apoptotic cells show strong blue fluorescence. After above-indicated treatments, HUVECs were fixed with 4% paraformaldehyde in 0.1 M PBS (pH 7.4) for 10 min at 4°C. Then, the slides were washed five times with PBS. After staining with 5 mg/mL Hoechst 33258 dye for 10 min, the apoptotic HUVECs were examined using a fluorescent microscope. Experiments were repeated three times.
Determination of Intracellular ROS Generation
ROS generation was confirmed by the oxidative conversion of cell-permeable substrate DCFH-DA, which converts to the detectable fluorescent product 2′,7′-dichlorodihydrofluorescein (DCF) inside cells. After the above treatments, the slides were washed two times with PBS followed by incubating 10 µM DCFH-DA solution at 37°C for 30 min. DCF fluorescence was measured using a fluorescence microscope to determine intracellular ROS production. Experiments were repeated three times.
Pro-Inflammatory Cytokines Levels Evaluated by ELISA
The HUVECs medium was collected after indicated treatments. IL-1β, IL-6 and TNF-α were evaluated by ELISA kits by following the manufacturer’s instructions. Briefly, protein samples were diluted and cytokine standards were added to the ELISA plates. The corresponding antibodies were added to the samples and incubated for 1 hr at room temperature. Followed by incubating with streptavidin-horseradish peroxidase-conjugated secondary antibodies at room temperature for 20 min. The protein levels of the cytokines were determined by measuring optical density values at 450 nm. Experiments were repeated three times.
Tube Formation Assay
In vitro capillary-like structure formation of HUVECs was evaluated using growth factor reduced Matrigel (BD Biosciences). Briefly, Matrigel was added to 96-well plates and incubated at 37°C for 30 min. The cells were seeded on Matrigel and cultured for 8 hrs. Tube formation was quantified using an inverted microscope (Olympus BX51). Experiments were repeated three times.
Transwell Migration Assay
Transwell migration assays were performed using a Transwell system (Corning Costar, Tewksbury, MA, USA) with 8 µM polycarbonate filter inserts in 24-well plates. Briefly, HUVECs were suspended in the upper chamber filled with M200 medium, and the lower chamber was placed in a 24-well culture dish containing full medium. After incubation at 37°C for 24 hrs, cells were fixed with 4% paraformaldehyde and stained by crystal violet. The migrated cells were counted by independent investigators who were blinded to the treatment groups. Experiments were repeated three times.
Statistical Analysis
All data are shown as mean ± standard error of mean. Differences among groups were analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s post-hoc test using SPSS 13.0 (SPSS, Chicago, IL, USA) software. Statistical significance was considered when p < 0.05.
Results
NaHS Alleviates HG-Induced Inactivation of PI3K/AKT/eNOS Pathway in HUVECs
HUVECs were subjected to pretreatment by 400 μM NaHS for 30 min before exposure to HG for 24 hrs. As shown in Figure 1, exposure of cells to HG dramatically reduced p-PI3K, p-Akt and p-eNOS expression levels. On the other hand, the decreased expression of these proteins was enhanced by the pretreatment with NaHS. Neither NaHS nor mannitol alone affected p-PI3K, p-Akt or p-eNOS expression, excluding the influence of the potency of NaHS and osmotic stress on PI3K/AKT/eNOS pathway activity.
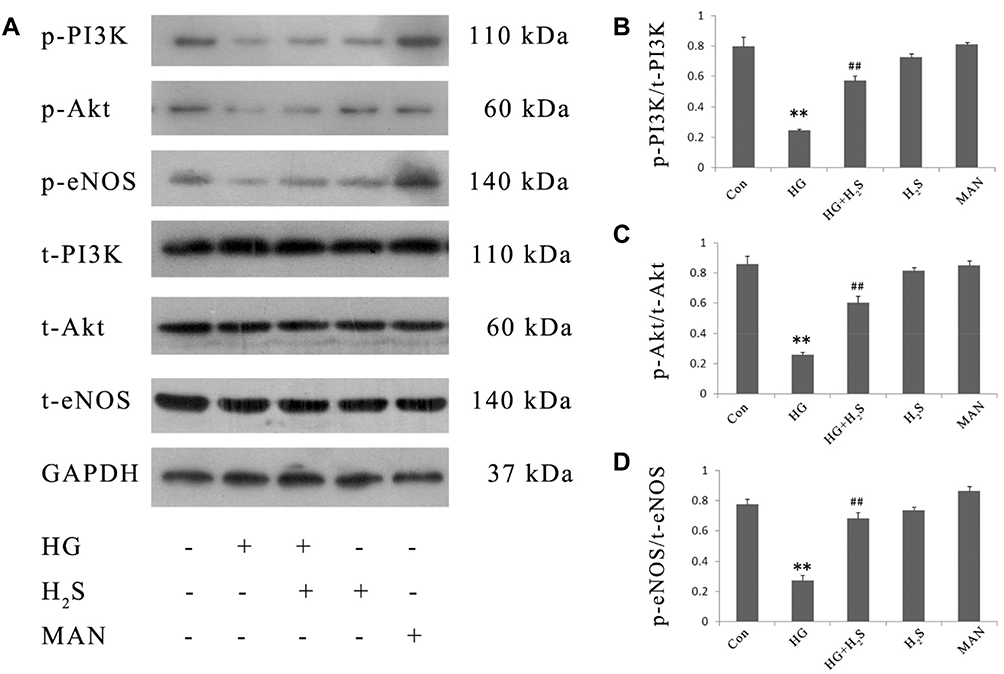 |
Figure 1 H2S mitigates HG-induced inactivation of PI3K/AKT/eNOS pathway in HUVECs. HUVECs were treated with 33 mM glucose (HG concentration), or pretreated with 400 μM NaHS before HG treatment, or treated with 400 μM NaHS alone or treated with 33 mM mannitol alone. The expression levels of p-PI3K, p-Akt and p-eNOS were detected by Western blotting analysis. (A) Variations in the expression levels of p-PI3K, p-Akt and p-eNOS in the indicated groups. (B–D) Densitometry analysis of the results displayed in (A). Experiments were repeated three times. Data are presented as the mean ± standard error of the mean. **p < 0.01 compared with the control group; ##p < 0.01 compared with the HG-treated group. Con, control group; HG, high glucose (33 mM); MAN, mannitol. |
PI3K/AKT/eNOS Signaling Activation Involves the Cytoprotective Effects of H2S Against the HG-Induced HUVEC Injuries
In order to examine whether H2S exerts protective effects on HUVECs against HG-induced cytotoxicity via the activation of PI3K/AKT/eNOS pathway, HUVECs were pre-treated with either NaHS or 740 Y-P prior to HG exposure. Pretreatment with NaHS blunted the HG-induced cytotoxic effect and increased the HUVEC viability, which was similar to pretreatment with 740 Y-P. In addition, HG treatment also suppressed the tube formation and cell migration of HUVECs, which was attenuated by the treatment with NaHS (See Supplemental Figure S1). However, the inhibition of PI3K/AKT/eNOS pathway by LY294002 markedly attenuated the protective effects of NaHS against HG-induced cytotoxicity, resulting in a decrease in HUVEC viability. Respectively, NaHS, 740 Y-P or LY294002 did not significantly alter HUVEC viability (Figure 2). These data suggest that PI3K/AKT/eNOS signaling mediates the protective effects of H2S on HUVECs against cytotoxicity induced by HG. Mannitol had no effect on the HUVEC viability, which excluded the effect of osmotic stress on HUVEC viability.
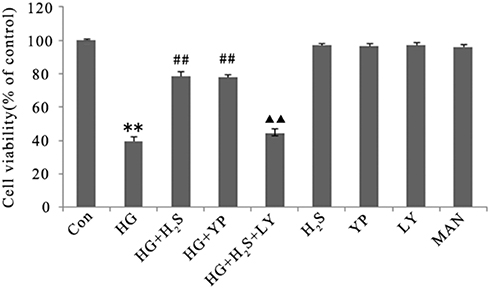 |
Figure 2 H2S protects against HG-induced decrease in viability of HUVECs by activating PI3K/AKT/eNOS pathway. Cell viability was detected using by CCK-8 assay. HUVECs were treated with 33 mM glucose (HG concentration), or pretreated with 400 μM NaHS before HG treatment, or pretreated with 20 μM 740 Y-P before HG treatment, or co-treated with 10 μM LY294002 and 400 μM NaHS prior to HG treatment, or treated with 400 μM NaHS alone, or treated with 20 μM 740 Y-P alone, or treated with 10 μM LY294002 alone or treated with 33 mM mannitol alone. Experiments were repeated three times. Data are presented as the mean ± standard error of the mean. **p < 0.01 compared with the control group; ##p < 0.01 compared with the group treated with HG; ▲▲p < 0.01 compared with the group pretreated with NaHS before HG treatment. Con, control group; HG, high glucose (33 mM); YP, 740 Y-P; LY, LY294002; MAN, mannitol. |
PI3K/AKT/eNOS Signaling Activation Involves the Protective Effects of H2S Against HG-Induced ER Stress in HUVECs
Continuous HG-treatment triggered the ER stress response in HUVECs. As shown in Figure 3, ATF6, CHOP and BiP expression levels were markedly increased in HUVECs with continuous HG-treatment. Importantly, pretreatment of HUVECs with either NaHS or 740 Y-P prior to HG treatment reduced the expression levels of these proteins, indicating a protective role of H2S against HG-induced ER stress in HUVECs. Moreover, LY294002 remarkably reversed the protective effect of H2S against HG-induced ER stress, resulting in the promotion of the aforementioned markers of ER stress response. Respectively, NaHS, 740 Y-P or LY294002 did not apparently alter the expression of ATF6, CHOP or BiP. These findings indicated that PI3K/AKT/eNOS signaling activation was involved in protective effects of H2S on HUVECs against ER stress induced by HG. Mannitol alone did not increase the expression of these markers, which excluded the participation of osmotic stress in the ER stress response.
 |
Figure 3 H2S alleviates HG-induced activation of ER stress response in HUVECs by activating PI3K/AKT/eNOS pathway. HUVECs were treated with 33 mM glucose (HG concentration), or pretreated with 400 μM NaHS before HG treatment, or pretreated with 20 μM 740 Y-P before HG treatment, or co-treated with 10 μM LY294002 and 400 μM NaHS prior to HG treatment, or treated with 400 μM NaHS alone, or treated with 20 μM 740 Y-P alone, or treated with 10 μM LY294002 alone or treated with 33 mM mannitol alone. The expression levels of ER stress markers were detected by Western blotting analysis. (A) Variations in the expression levels of ATF6, CHOP and BiP in the indicated groups. (B–D) Densitometry analysis of the results displayed in (A). Experiments were repeated three times. Data are presented as the mean ± standard error of the mean. **p < 0.01 compared with the control group; ##p < 0.01 compared with the HG-treated group; ▲▲p < 0.01 compared with the group pretreated with NaHS before HG treatment. Con, control group; HG, high glucose (33 mM); YP, 740 Y-P; LY, LY294002; MAN, mannitol. |
PI3K/AKT/eNOS Signaling Activation Involves in the Cytoprotection of H2S Against HG-Induced Apoptosis in HUVECs
In the present study, DNA fragmentation staining with Hoechst 33258 was used to determine HUVEC apoptosis after different treatments. Apoptosis was calculated in conformity to the method used in a previous study.32 Figure 4 showed that treatment of HUVECs with HG for 24 hrs significantly increased apoptosis. However, the increase can be clearly reversed by pretreatment with either NaHS or 740 Y-P. Interestingly, LY294002 remarkably weakened the protective effects of H2S against HG-induced HUVEC apoptosis.
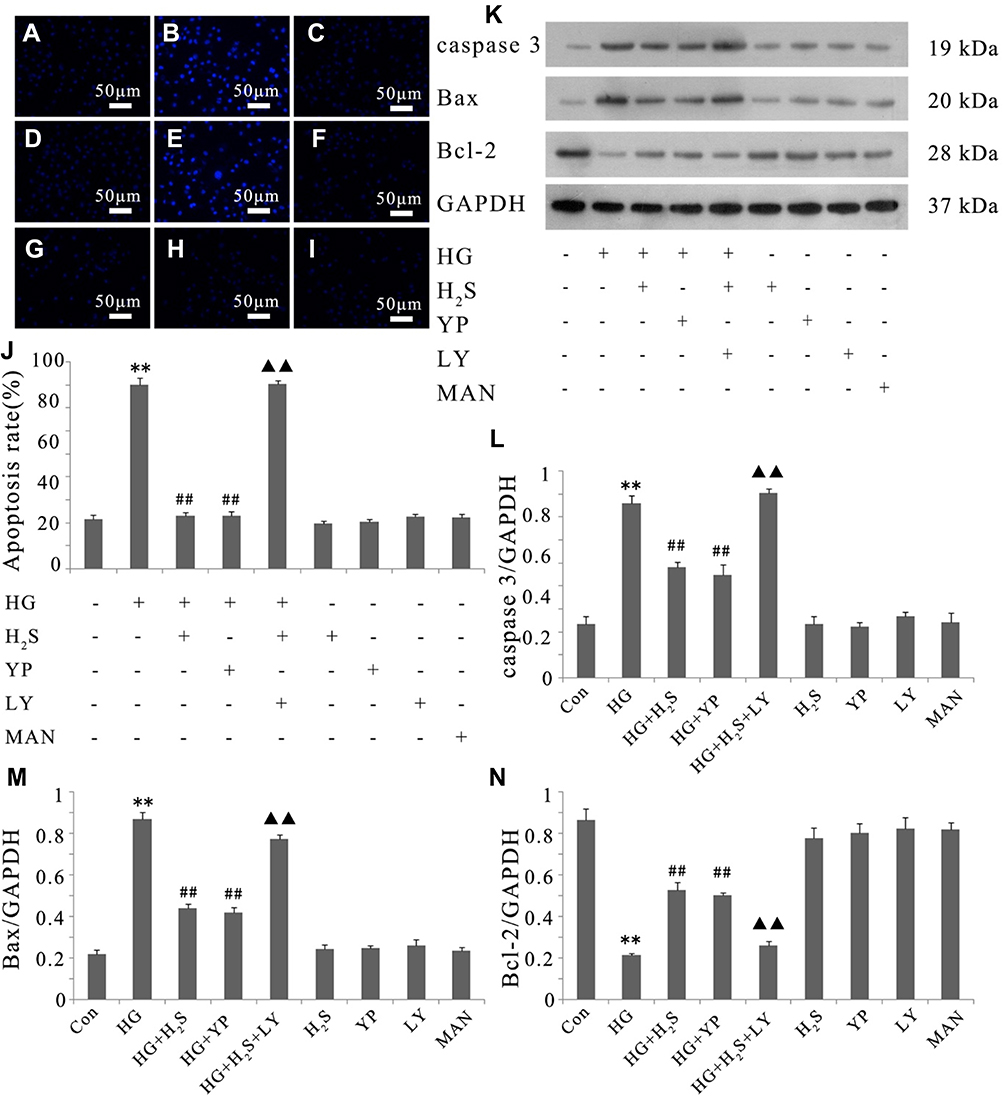 |
Figure 4 Activation of PI3K/AKT/eNOS pathway is related to the protective effect of H2S against HG-induced apoptosis in HUVECs. (A–I) After the indicated treatments, HUVEC apoptosis was assessed by Hoechst 33258 staining followed by photofluorography. (A) Control group. (B) HUVECs were exposed to 33 mM glucose (HG concentration) for 24 hrs. (C) HUVECs were pretreated with 400 μM NaHS before HG treatment. (D) HUVECs were pretreated with 20 μM 740 Y-P before HG treatment. (E) HUVECs were co-treated with 10 μM LY294002 and 400 μM NaHS prior to HG treatment. (F) HUVECs were treated with 400 μM NaHS alone. (G) HUVECs were treated with 20 μM 740 Y-P alone. (H) HUVECs were treated with 10 μM LY294002 alone. (I) HUVECs were treated with 33 mM mannitol alone. (J) The apoptosis rate was analyzed by cell counting kit 8 assay and ImageJ 1.41o software. (K) Variations in the expression levels of active (cleaved) caspase 3, Bax and Bcl-2 in the indicated groups. (L–N) Densitometric analysis of the results in (K). Experiments were repeated three times. Data are presented as the mean ± standard error of the mean. **p < 0.01 compared with the control group; ##p < 0.01 compared with the HG-treated group; ▲▲p < 0.01 compared with the group pretreated with NaHS before HG treatment. Con, control group; HG, high glucose (33 mM); YP, 740 Y-P; LY, LY294002; MAN, mannitol. |
We further evaluated the expression of active (cleaved) caspase 3, Bax and Bcl-2 in HUVECs by Western blotting. As shown in Figure 4, the exposure of HUVECs to HG for 24 hrs induced a noteworthy decline in the expression of Bcl-2 but promoted the expression of Bax and cleaved caspase 3. Whereas pretreatment with either NaHS or 740 Y-P can reverse the variation in the expression of these apoptotic regulatory protein after HG-stimulation. In addition, LY294002 notably attenuated the protective effects of H2S against HG-induced apoptosis, which was demonstrated by increase in the expression of Bax and cleaved caspase 3 along with the decrease in Bcl-2 expression. These results showed that the protective effect of H2S against HG-mediated apoptosis might be related to the activation of PI3K/AKT/eNOS pathway.
Either NaHS, 740 Y-P or LY294002 did not exert any effect on endothelial apoptosis. Additionally, mannitol alone did not affect endothelial apoptosis, which excluded the participation of osmotic stress in the HUVEC apoptosis.
PI3K/AKT/eNOS Signaling Activation Involves the Cytoprotection of H2S Against HG-Induced ROS Production in HUVECs
As shown in Figure 5, continuous exposure to HG-induced noticeable ROS generation in HUVECs compared to the negative control, and the increased production of ROS was reduced by pretreatment with either NaHS or 740 Y-P. Moreover, LY294002 significantly attenuated the protective effects of H2S against HG-induced ROS generation in HUVECs.
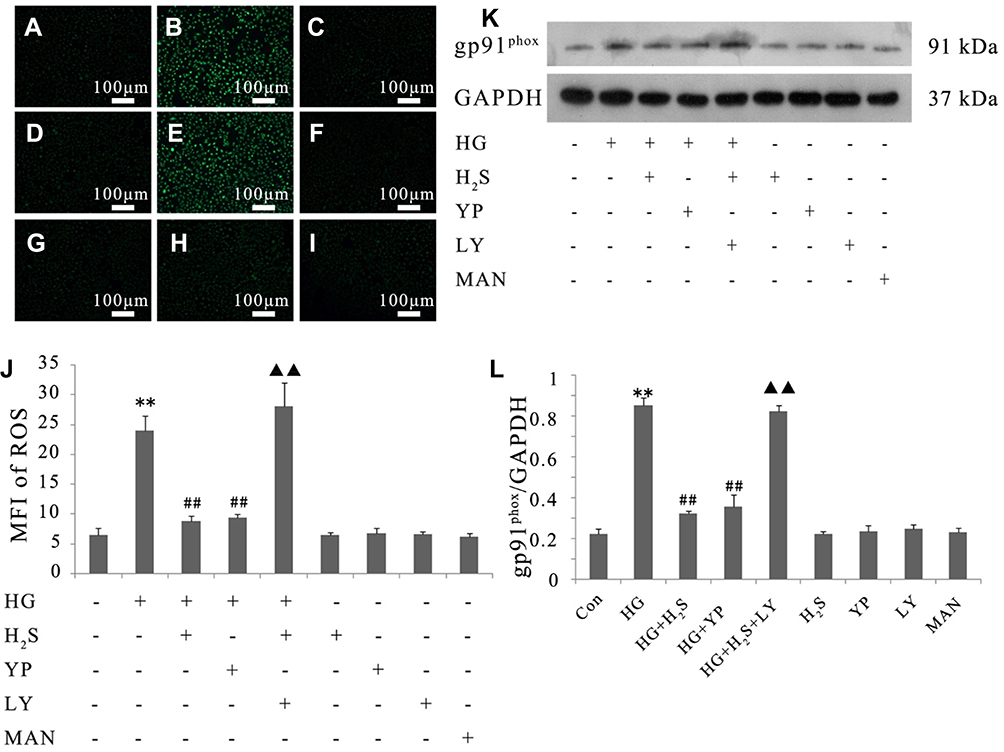 |
Figure 5 Activation of PI3K/AKT/eNOS pathway is implicated in the protective effect of H2S against HG-induced ROS generation in HUVECs. (A–I) Intracellular ROS levels were observed with DCFH-DA staining followed by photofluorography. (A) Control group. (B) HUVECs were exposed to 33 mM glucose (HG concentration) for 24 hrs. (C) HUVECs were pretreated with 400 μM NaHS before HG treatment. (D) HUVECs were pretreated with 20 μM 740 Y-P before HG treatment. (E) HUVECs were co-treated with 10 μM LY294002 and 400 μM NaHS prior to HG treatment. (F) HUVECs were treated with 400 μM NaHS alone. (G) HUVECs were treated with 20 μM 740 Y-P alone. (H) HUVECs were treated with 10 μM LY294002 alone. (I) HUVECs were treated with 33 mM mannitol alone. (J) Quantitative analysis of the MFI of DCFH-DA in (A–I) with ImageJ 1.41o software. (K) Variations in the expression level of gp91phox in the indicated groups. (L) Densitometric analysis of the results in (K). Experiments were repeated three times. Data are presented as the mean ± standard error of the mean. **p < 0.01 compared with the control group; ##p < 0.01 compared with the HG-treated group; ▲▲p < 0.01 compared with the group pretreated with NaHS before HG treatment. Con, control group; HG, high glucose (33 mM); YP, 740 Y-P; LY, LY294002; MAN, mannitol. |
In order to confirm the protective influence of H2S against HG-induced ROS production in HUVECs, we further tested the activity of gp91phox, the main component of the NADPH oxidase system as well as an important source of ROS production.33 As demonstrated in Figure 5, continuous treatment of HUVECs with HG significantly increased gp91phox expression compared to the negative control. Pretreatment with either NaHS or 740 Y-P, however, observably reversed this adverse effect of HG. Additionally, LY294002 alleviated NaHS-induced inactivation of gp91phox in HUVECs, underlying that PI3K/AKT/eNOS signaling activation might involve in the protective effect of H2S against HG-mediated oxidative stress.
Either NaHS, 740 Y-P, LY294002 or mannitol did not exert any influence on oxidative stress in HUVECs. In addition, mannitol alone did not affect the production of ROS and the expression of gp91phox in HUVECs excluding the participation of osmotic stress.
Activation of PI3K/AKT/eNOS Pathway Is Implicated in the Protective Effect Exerted by H2S Against HG-Induced Inflammation in HUVECs
The activation of caspase 1 and JNK, the sensitive markers of the early inflammatory response, is associated with the generation of pro-inflammatory cytokines. As demonstrated in Figure 6, cleaved caspase 1 and JNK phosphorylation were remarkably increased compared to the negative control after treating HUVECs with HG for 24 hrs, and the increase could be restrained by pretreatment with either NaHS or 740 Y-P. Besides, LY294002 significantly attenuated the NaHS-induced inactivation of both caspase 1 and JNK in HUVECs.
 |
Figure 6 Activation of PI3K/AKT/eNOS pathway is involved in the protective effect of H2S against HG-induced inflammatory responses in HUVECs. HUVECs were treated with 33 mM glucose (HG concentration), or pretreated with 400 μM NaHS before HG treatment, or pretreated with 20 μM 740 Y-P before HG treatment, or co-treated with 10 μM LY294002 and 400 μM NaHS prior to HG treatment, or treated with 400 μM NaHS alone, or treated with 20 μM 740 Y-P alone, or treated with 10 μM LY294002 alone or treated with 33 mM mannitol alone. After the indicated treatments, the release of (A) IL-1β, (B) IL-6 and (C) TNF-α was assessed by ELISA. (D) Variations in the expression levels of p-JNK cleaved caspase 1 in the indicated groups. (E, F) Densitometry analysis of the results shown in (D). Experiments were repeated three times. Data are presented as the mean ± standard error of the mean. **p < 0.01 compared with the control group; ##p < 0.01 compared with the HG-treated group; ▲▲p < 0.01 compared with the group pretreated with NaHS before HG treatment. Con, control group; HG, high glucose (33 mM); YP, 740 Y-P; LY, LY294002; MAN, mannitol. |
We further assessed the expression of several key pro-inflammatory cytokines, including IL-1β, IL-6 and TNF-α. As shown in Figure 6, the secretion of these pro-inflammatory cytokines was markedly increased after treatment of HUVECs with HG for 24 hrs compared to the negative control group. The increase was dramatically reversed by pretreatment with either NaHS or 740 Y-P. Moreover, LY294002 alleviated NaHS-induced decrease in IL-1β, IL-6 and TNF-α levels, indicating that H2S plays a protective role in the HG-triggered inflammatory response via the activation of PI3K/AKT/eNOS pathway in HUVECs.
Either NaHS, 740 Y-P or LY294002 did not exert any influence on inflammation in HUVECs. Additionally, mannitol alone did not affect inflammatory response in HUVECs excluding the participation of osmotic stress.
Discussion
Although it is well known that HG plays a key role in diabetic endothelial dysfunction and atherosclerosis, the mechanisms responsible for HG-induced endothelial damage remain to be elucidated. The HG (33 mM)-induced HUVECs injury model was employed to investigate the underlying mechanisms of endothelial injury, and further investigated cytoprotection of H2S. In the present study, HG treatment for 24 hrs caused a significant reduction in the cell viability of HUVECs, which was consistent with the results from Han et al,34 However, several studies demonstrated that the inhibitory effects of HG on HUVEC viability were observed after 48-hr35 and 72-hr treatment.36–39 The inconsistency regarding the HG treatment duration across different studies may be due to the different culture conditions, different sources for obtaining HUVECs, or different experimental protocols, which may require further examination.
Endothelial cell apoptosis can be triggered by multiple factors for atherosclerosis.40 One classical type of apoptosis is mediated by mitochondria. Cytochrome c is released from mitochondria after the irritation of a series of pro-apoptotic factors, including HG, ROS and ox‐LDL. Subsequently, Cytoplasmic c activates apoptosis initiator caspase 9, which can hydrolyze and activate downstream apoptosis executioner caspases to initiate apoptosis.41 Experiments in vivo and in vitro confirmed that HG-treatment induces cell apoptosis and cleaved caspase 3 level in HUVECs,42 which is consistent with our findings.
ER stress regulated by blood glucose level may play another important role in endothelial dysfunction and ultimately contribute to the originating of vascular complications in diabetes mellitus.43 Once ER is subjected to external stimulus, PERK/eIF2α pathway will be activated and then mediate the attenuation of protein translation, which is involved in the regulation of the pro-apoptotic factor, CHOP. In addition, PERK/eIF2α pathway activation can prompt ATF6 to be active, which is the primary ER membrane–associated protein and engages complex downstream signaling pathways. As ER stress arises, BiP, the ER chaperone, dissociates from unfolded proteins within the ER and becomes active.44,45 It is demonstrated that in type 2 diabetic mice, ER stress was associated tightly with vascular dysfunction.5 Consistently, we observed in our study that the increase of ER stress markers in HUVECs was induced by HG-stimulation
The protein-folding load increases during the ER stress response and activates ROS generation, which initiates oxidative stress.46 It may be the possible mechanism that misfolded proteins bind protein chaperones which expend ATP and trigger mitochondrial oxidative phosphorylation, then producing ROS as a byproduct.47 ROS accumulation may result in the damages to proteins and cytomembrane, subsequently activating inflammatory signaling.22 Recent studies have shown that ROS production in cardiac microvascular endothelial cells was markedly increased after HG incubation,48 and in streptozotocin-induced diabetic mice, vascular inflammation occurs in association with exposure to HG.49 Consistent with previous reports, the increases of ROS level and inflammation factors in HUVECs were observed after HG-incubation in the present study.
It is worth noting that PI3K/Akt/eNOS signaling activation in HUVECs was suppressed by HG-treatment. PI3K/Akt pathway may act as a positive regulator of endothelial NO synthase in endothelial cells and play a key role in the process of endothelial cell survival, mobilization, migration and homing.8 Increasing researches indicated that PI3K/Akt/eNOS signaling pathway possesses a crucial role in the repair of endothelial damage under HG condition.50,51 Once activated, phosphorylated PI3K/Akt can directly phosphorylate eNOS followed by the generation of NO, which acts as an endothelial cell survival factor.52 Yan et al found that the impaired vascular regeneration was partially ascribed to the deficiency of NO accounts in diabetic ischemic injury animal model.53 In agreement with previous reports, the agonist of PI3K/Akt/eNOS, 740 Y-P, obviously alleviated the HG-induced damages in HUVECs including cytotoxicity, apoptosis, ER stress, oxidative stress and inflammation, which was observed in our study.
Another novel finding is connected with PI3K/Akt/eNOS signaling activation on the protective effects of H2S against HG-induced HUVECs injuries. H2S is a newly found gas transmitter, which is key in mediating vascular inflammation, proliferation and cell death as well as endothelial protection.54 It was reported that in diabetic patients and streptozotocin-induced diabetic rats, vascular inflammation may be partly associated with low levels of H2S in the blood.25 Recently, Liu et al55 have demonstrated that exogenous H2S significantly prevented cell death, decreased the generation of apoptotic markers and suppressed mitochondrial ROS production in rat aortic endothelial cells under HG situation. Additionally, H2S has been proved to be beneficial to accelerate the wound healing in rats with diabetes by promoting angiogenesis, which may be associated with the effect of anti-inflammation.56 In a recent research, it was demonstrated that exogenous H2S can effectively attenuate HG-induced multiple injuries in endothelial cells.57 Consistent with previous studies, we observed in the present research that NaHS-treatment can distinctly reduce HG-induce cytotoxicity, apoptosis, oxidative stress and inflammation in HUVECs. The present study is the first to demonstrate that HG-triggered ER stress in HUVECs can be suppressed by treatment with exogenous H2S. There is a recent report showing that exogenous H2S can alleviate cardiovascular injury by inhibiting ER stress in diabetic rats, which is consistent with our observation.58 Moreover, it was demonstrated in our study that exogenous H2S can clearly activate the PI3K/Akt/eNOS signaling in HUVECs which was weakened by HG-treatment. In a recent research, Predmore et al revealed that endogenous H2S can up-regulate NO generation via Akt-dependent phosphorylation of eNOS in endothelial cells.30 In CSE-KO mice, it was observed that acute H2S therapy can restore eNOS function and NO bioavailability, and then attenuate myocardial ischemia/reperfusion injury. In eNOS phospho-dead mutant mice, however, ischemia/reperfusion injury cannot be alleviated by exogenous H2S.59 Similarly, Cai et al found that NaHS-treatment increased Akt phosphorylation, while the beneficial effects of NaHS on endothelial wound healing and tube-like structure formation were prevented by LY294002, the inhibitor of PI3K̸Akt/eNOS signaling pathway.60 In agreement with these studies, we noticed in the present research that the protective effects of endogenous H2S against HG-induced injuries on the endothelial cells were immensely blocked by LY294002. Taking into consideration with our finding that PI3K/Akt/eNOS pathway mediated HG-induced HUVEC damages, the present study supports a novel hypothesis that activation of PI3K/Akt/eNOS pathway may be one of the key mechanisms underlying the protective effect of H2S against the HG-induced endothelial injuries.
There are several concerns that should be addressed in this study. The cell apoptosis of HUVECs was evaluated by the Hoechst 33258 staining, and the results showed the relatively high proportion of apoptotic cells, which is due to the fact that the fluorescent signaling represents the estimated apoptotic rates. Future studies may employ the flow cytometry analysis to measure the cell apoptosis in a more accurate manner. The PI3K/AKT/eNOS pathway may be not the only pathway responsible for the protective role of NaHS on glucotoxicity, and necroptosis signaling has been shown to involve in the protective role of NaHS.57 On the other hand, PI3K/AKT/eNOS pathway is an important signaling pathway for ER stress, ROS production, inflammation and apoptosis in endothelial cells.61–64 LY294002 had no effects on the functions of the HUVECs, which is consistent with previous findings.65 We are speculating that the PI3K/AKT/eNOS signaling is activated by detrimental/protective stimulus, which can be effectively inhibited by LY294002; while at normal conditions, the PI3K/ATK/eNOS signaling may not be activated. The underlying mechanisms may require further investigations in future studies.
To summarize, this study provides the novel evidence that the PI3K/Akt/eNOS signaling impairment is correlated with HG-induced multiple endothelial injuries. Thus, inhibition of PI3K/Akt/eNOS signaling should be considered as a key risk factor for atherosclerosis in diabetes. Importantly, the present study demonstrated that exogenous H2S protects endothelial cells against HG-induced injuries by activating PI3K/Akt/eNOS pathway. Based on the above findings, we proposed that reduced endogenous H2S levels and the subsequent PI3K/Akt/eNOS signaling impairment may be the important pathophysiological mechanism underlying hyperglycemia-induced vascular injuries.
Data Sharing Statement
All the data are available upon reasonable request.
Acknowledgments
The study was supported by the Science and Technology Project of Shenzhen City of China (JCYJ20160426100250466, JCYJ20170306152620264 and JCYJ20170307161535847) and the Bao’an Science and Technology Innovation Department of Shenzhen City of China (2016CX024).
Disclosure
Dr. Yiying Yang, Zhicong Zeng, Yinzhi Song, Fengxia Lin and Xiao Ke report grants from Science and Technology Project of Shenzhen City of China, grants from Bao’an Science and Technology Innovation Department of Shenzhen City of China, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Dokken B. Mechanisms of cardiovascular injury in type 2 diabetes and potential effects of dipeptidyl peptidase-4 inhibition. J Cardiovasc Nurs. 2016;31(3):274–283. doi:10.1097/JCN.0000000000000245
2. Lee WS, Kim J. Diabetic cardiomyopathy: where we are and where we are going. Korean J Intern Med. 2017;32(3):404–421. doi:10.3904/kjim.2016.208
3. Ren J, Davidoff AJ. Diabetes rapidly induces contractile dysfunctions in isolated ventricular myocytes. Am J Physiol. 1997;272(1 Pt 2):H148–H158. doi:10.1152/ajpheart.1997.272.1.H148
4. Cheang WS, Tian XY, Wong WT, et al. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5ʹ adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor delta pathway. Arterioscler Thromb Vasc Biol. 2014;34(4):830–836. doi:10.1161/ATVBAHA.113.301938
5. Choi SK, Lim M, Yeon SI, Lee YH. Inhibition of endoplasmic reticulum stress improves coronary artery function in type 2 diabetic mice. Exp Physiol. 2016;101(6):768–777. doi:10.1113/EP085508
6. Fiorentino TV, Prioletta A, Zuo P, Folli F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr Pharm Des. 2013;19(32):5695–5703. doi:10.2174/1381612811319320005
7. Bitar MS, Ayed AK, Abdel-Halim SM, Isenovic ER, Al-Mulla F. Inflammation and apoptosis in aortic tissues of aged type II diabetes: amelioration with alpha-lipoic acid through phosphatidylinositol 3-kinase/Akt- dependent mechanism. Life Sci. 2010;86(23–24):844–853. doi:10.1016/j.lfs.2010.03.019
8. Chu P, Han G, Ahsan A, et al. Phosphocreatine protects endothelial cells from methylglyoxal induced oxidative stress and apoptosis via the regulation of PI3K/Akt/eNOS and NF-kappaB pathway. Vascul Pharmacol. 2017;91:26–35. doi:10.1016/j.vph.2016.08.012
9. Jung CH, Lee WJ, Hwang JY, et al. Vaspin protects vascular endothelial cells against free fatty acid-induced apoptosis through a phosphatidylinositol 3-kinase/Akt pathway. Biochem Biophys Res Commun. 2011;413(2):264–269. doi:10.1016/j.bbrc.2011.08.083
10. Nakamura N, Naruse K, Kobayashi Y, et al. High glucose impairs the proliferation and increases the apoptosis of endothelial progenitor cells by suppression of Akt. J Diabetes Investig. 2011;2(4):262–270. doi:10.1111/j.2040-1124.2010.00093.x
11. Yang Z, Ming XF. Recent advances in understanding endothelial dysfunction in atherosclerosis. Clin Med Res. 2006;4(1):53–65. doi:10.3121/cmr.4.1.53
12. Mannucci E, Dicembrini I, Lauria A, Pozzilli P. Is glucose control important for prevention of cardiovascular disease in diabetes? Diabetes Care. 2013;36(Suppl 2):S259–S263. doi:10.2337/dcS13-2018
13. Giorgino F, Leonardini A, Laviola L. Cardiovascular disease and glycemic control in type 2 diabetes: now that the dust is settling from large clinical trials. Ann N Y Acad Sci. 2013;1281:36–50. doi:10.1111/nyas.12044
14. Hu L, Sun Y, Hu J. Catalpol inhibits apoptosis in hydrogen peroxide-induced endothelium by activating the PI3K/Akt signaling pathway and modulating expression of Bcl-2 and Bax. Eur J Pharmacol. 2010;628(1–3):155–163. doi:10.1016/j.ejphar.2009.11.046
15. Kim F, Tysseling KA, Rice J, et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler Thromb Vasc Biol. 2005;25(5):989–994. doi:10.1161/01.ATV.0000160549.60980.a8
16. Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231–241. doi:10.1016/S0092-8674(00)80405-5
17. Romorini L, Coso OA, Pecci A. Bcl-XL mediates epidermal growth factor dependent cell survival in HC11 mammary epithelial cells. Biochim Biophys Acta. 2009;1793(3):496–505. doi:10.1016/j.bbamcr.2008.12.002
18. Wei W, Liu Q, Tan Y, Liu L, Li X, Cai L. Oxidative stress, diabetes, and diabetic complications. Hemoglobin. 2009;33(5):370–377. doi:10.3109/03630260903212175
19. Naudi A, Jove M, Ayala V, et al. Cellular dysfunction in diabetes as maladaptive response to mitochondrial oxidative stress. Exp Diabetes Res. 2012;2012:696215. doi:10.1155/2012/696215
20. Huang X, Zhang J, Liu J, et al. C-reactive protein promotes adhesion of monocytes to endothelial cells via NADPH oxidase-mediated oxidative stress. J Cell Biochem. 2012;113(3):857–867. doi:10.1002/jcb.v113.3
21. Liu D, Wu M, Lu Y, et al. Protective effects of 6-Gingerol on vascular endothelial cell injury induced by high glucose via activation of PI3K-AKT-eNOS pathway in human umbilical vein endothelial cells. Biomed Pharmacother. 2017;93:788–795. doi:10.1016/j.biopha.2017.07.037
22. Zhang D, Gao X, Wang Q, et al. Kakkalide ameliorates endothelial insulin resistance by suppressing reactive oxygen species-associated inflammation. J Diabetes. 2013;5(1):13–24. doi:10.1111/1753-0407.12017
23. Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6(11):917–935. doi:10.1038/nrd2425
24. Li L, Moore PK. Putative biological roles of hydrogen sulfide in health and disease: a breath of not so fresh air? Trends Pharmacol Sci. 2008;29(2):84–90. doi:10.1016/j.tips.2007.11.003
25. Jain SK, Bull R, Rains JL, et al. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid Redox Signal. 2010;12(11):1333–1337. doi:10.1089/ars.2009.2956
26. Whiteman M, Gooding KM, Whatmore JL, et al. Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia. 2010;53(8):1722–1726. doi:10.1007/s00125-010-1761-5
27. Mani S, Li H, Untereiner A, et al. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation. 2013;127(25):2523–2534. doi:10.1161/CIRCULATIONAHA.113.002208
28. Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. doi:10.1038/nchembio711
29. Ng HH, Yildiz GS, Ku JM, Miller AA, Woodman OL, Hart JL. Chronic NaHS treatment decreases oxidative stress and improves endothelial function in diabetic mice. Diabetes Vasc Dis Res. 2017;14(3):246–253. doi:10.1177/1479164117692766
30. Predmore BL, Julian D, Cardounel AJ. Hydrogen sulfide increases nitric oxide production from endothelial cells by an akt-dependent mechanism. Front Physiol. 2011;2:104. doi:10.3389/fphys.2011.00104
31. Ke X, Chen J, Peng L, et al. Heat shock protein 90/Akt pathway participates in the cardioprotective effect of exogenous hydrogen sulfide against high glucose-induced injury to H9c2 cells. Int J Mol Med. 2017;39(4):1001–1010. doi:10.3892/ijmm.2017.2891
32. Chen J, Guo R, Yan H, et al. Naringin inhibits ROS-activated MAPK pathway in high glucose-induced injuries in H9c2 cardiac cells. Basic Clin Pharmacol Toxicol. 2014;114(4):293–304. doi:10.1111/bcpt.2014.114.issue-4
33. Yu L, Quinn MT, Cross AR, Dinauer MC. Gp91(phox) is the heme binding subunit of the superoxide-generating NADPH oxidase. Proc Natl Acad Sci U S A. 1998;95(14):7993–7998. doi:10.1073/pnas.95.14.7993
34. Han X, Wang B, Sun Y, et al. Metformin modulates high glucose-incubated human umbilical vein endothelial cells proliferation and apoptosis through AMPK/CREB/BDNF pathway. Front Pharmacol. 2018;9:1266. doi:10.3389/fphar.2018.01266
35. Li Q, Lin Y, Wang S, Zhang L, Guo L. GLP-1 inhibits high-glucose-induced oxidative injury of vascular endothelial cells. Sci Rep. 2017;7(1):8008. doi:10.1038/s41598-017-06712-z
36. Rezabakhsh A, Ahmadi M, Khaksar M, et al. Rapamycin inhibits oxidative/nitrosative stress and enhances angiogenesis in high glucose-treated human umbilical vein endothelial cells: role of autophagy. Biomed Pharmacother. 2017;93:885–894. doi:10.1016/j.biopha.2017.07.044
37. Rezabakhsh A, Rahbarghazi R, Malekinejad H, Fathi F, Montaseri A, Garjani A. Quercetin alleviates high glucose-induced damage on human umbilical vein endothelial cells by promoting autophagy. Phytomedicine. 2019;56:183–193. doi:10.1016/j.phymed.2018.11.008
38. An X, Li L, Chen Y, et al. Mesenchymal stem cells ameliorated glucolipotoxicity in HUVECs through TSG-6. Int J Mol Sci. 2016;17(4):483. doi:10.3390/ijms17040483
39. Yuan Y, Shi M, Li L, et al. Mesenchymal stem cell-conditioned media ameliorate diabetic endothelial dysfunction by improving mitochondrial bioenergetics via the Sirt1/AMPK/PGC-1alpha pathway. Clin Sci (Lond). 2016;130(23):2181–2198. doi:10.1042/CS20160235
40. Stoneman VE, Bennett MR. Role of apoptosis in atherosclerosis and its therapeutic implications. Clin Sci (Lond). 2004;107(4):343–354. doi:10.1042/CS20040086
41. Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science (New York, NY). 1997;275(5303):1132–1136. doi:10.1126/science.275.5303.1132
42. Yu S, Liu X, Men L, Yao J, Xing Q, Du J. Selenoprotein S protects against high glucose-induced vascular endothelial apoptosis through the PKCbetaII/JNK/Bcl-2 pathway. J Cell Biochem. 2018;120(5):8661–8675.
43. Basha B, Samuel SM, Triggle CR, Ding H. Endothelial dysfunction in diabetes mellitus: possible involvement of endoplasmic reticulum stress? Exp Diabetes Res. 2012;2012:481840. doi:10.1155/2012/481840
44. Maamoun H, Zachariah M, McVey JH, Green FR, Agouni A. Heme oxygenase (HO)-1 induction prevents endoplasmic reticulum stress-mediated endothelial cell death and impaired angiogenic capacity. Biochem Pharmacol. 2017;127:46–59. doi:10.1016/j.bcp.2016.12.009
45. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467–478. doi:10.1016/j.cmet.2010.04.005
46. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454(7203):455–462. doi:10.1038/nature07203
47. Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9(12):2277–2293. doi:10.1089/ars.2007.1782
48. Peng C, Ma J, Gao X, Tian P, Li W, Zhang L. High glucose induced oxidative stress and apoptosis in cardiac microvascular endothelial cells are regulated by FoxO3a. PLoS One. 2013;8(11):e79739. doi:10.1371/journal.pone.0079739
49. Wang Q, Zhang M, Torres G, et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. 2017;66(1):193–205. doi:10.2337/db16-0915
50. Zhu KX, Nie SP, Li C, Gong D, Xie MY. Ganoderma atrum polysaccharide improves aortic relaxation in diabetic rats via PI3K/Akt pathway. Carbohydr Polym. 2014;103:520–527. doi:10.1016/j.carbpol.2013.12.080
51. Liu M, Xiang G, Lu J, Xiang L, Dong J, Mei W. TRAIL protects against endothelium injury in diabetes via Akt-eNOS signaling. Atherosclerosis. 2014;237(2):718–724. doi:10.1016/j.atherosclerosis.2014.10.013
52. Hoffmann J, Haendeler J, Aicher A, et al. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ Res. 2001;89(8):709–715. doi:10.1161/hh2001.097796
53. Yan J, Tie G, Park B, Yan Y, Nowicki PT, Messina LM. Recovery from hind limb ischemia is less effective in type 2 than in type 1 diabetic mice: roles of endothelial nitric oxide synthase and endothelial progenitor cells. J Vasc Surg. 2009;50(6):1412–1422. doi:10.1016/j.jvs.2009.08.007
54. Whiteman M, Le Trionnaire S, Chopra M, Fox B, Whatmore J. Emerging role of hydrogen sulfide in health and disease: critical appraisal of biomarkers and pharmacological tools. Clin Sci (Lond). 2011;121(11):459–488. doi:10.1042/CS20110267
55. Liu J, Wu J, Sun A, et al. Hydrogen sulfide decreases high glucose/palmitate-induced autophagy in endothelial cells by the Nrf2-ROS-AMPK signaling pathway. Cell Biosci. 2016;6:33. doi:10.1186/s13578-016-0099-1
56. Wang G, Li W, Chen Q, Jiang Y, Lu X, Zhao X. Hydrogen sulfide accelerates wound healing in diabetic rats. Int J Clin Exp Pathol. 2015;8(5):5097–5104.
57. Lin J, Chen M, Liu D, et al. Exogenous hydrogen sulfide protects human umbilical vein endothelial cells against high glucose-induced injury by inhibiting the necroptosis pathway. Int J Mol Med. 2018;41(3):1477–1486. doi:10.3892/ijmm.2017.3330
58. Guo R, Wu Z, Jiang J, et al. New mechanism of lipotoxicity in diabetic cardiomyopathy: deficiency of endogenous H2S production and ER stress. Mech Ageing Dev. 2017;162:46–52. doi:10.1016/j.mad.2016.11.005
59. King AL, Polhemus DJ, Bhushan S, et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc Natl Acad Sci U S A. 2014;111(8):3182–3187. doi:10.1073/pnas.1321871111
60. Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76(1):29–40. doi:10.1016/j.cardiores.2007.05.026
61. Zhou J, Abid MD, Xiong Y, Chen Q, Chen J. ox-LDL downregulates eNOS activity via LOX-1-mediated endoplasmic reticulum stress. Int J Mol Med. 2013;32(6):1442–1450. doi:10.3892/ijmm.2013.1513
62. Chen L, Qin L, Liu X, Meng X. CTRP3 alleviates Ox-LDL-induced inflammatory response and endothelial dysfunction in mouse aortic endothelial cells by activating the PI3K/Akt/eNOS pathway. Inflammation. 2019;42(4):1350–1359. doi:10.1007/s10753-019-00996-1
63. Cheng Q, Zhao Y, Li J. Sodium tanshinone IIA sulfonate suppresses heat stress-induced endothelial cell apoptosis by promoting NO production through upregulating the PI3K/AKT/eNOS pathway. Mol Med Rep. 2017;16(2):1612–1618. doi:10.3892/mmr.2017.6760
64. Wang Y, Cui L, Xu H, et al. TRPV1 agonism inhibits endothelial cell inflammation via activation of eNOS/NO pathway. Atherosclerosis. 2017;260:13–19. doi:10.1016/j.atherosclerosis.2017.03.016
65. Sheu ML, Ho FM, Yang RS, et al. High glucose induces human endothelial cell apoptosis through a phosphoinositide 3-kinase-regulated cyclooxygenase-2 pathway. Arterioscler Thromb Vasc Biol. 2005;25(3):539–545. doi:10.1161/01.ATV.0000155462.24263.e4
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.