")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Histone demethylase LSD1-mediated repression of GATA-2 is critical for erythroid differentiation
Authors Guo Y, Fu X, Jin Y, Sun J, Liu Y, Huo B, Li X, Hu X
Received 30 January 2015
Accepted for publication 26 February 2015
Published 19 June 2015 Volume 2015:9 Pages 3153—3162
DOI https://doi.org/10.2147/DDDT.S81911
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Yidi Guo,1 Xueqi Fu,1,2 Yue Jin,1 Jing Sun,1 Ye Liu,1 Bo Huo,1 Xiang Li,1 Xin Hu1–3
1School of Life Sciences, Jilin University, 2Key Laboratory for Molecular Enzymology and Engineering of Ministry of Education, Jilin University, 3National Engineering Laboratory of AIDS Vaccine, School of Life Sciences, Jilin University, Changchun, People’s Republic of China
Background: The transcription factor GATA-2 is predominantly expressed in hematopoietic stem and progenitor cells and counteracts the erythroid-specific transcription factor GATA-1, to modulate the proliferation and differentiation of hematopoietic cells. During hematopoietic cell differentiation, GATA-2 exhibits dynamic expression patterns, which are regulated by multiple transcription factors.
Methods: Stable LSD1-knockdown cell lines were established by growing murine erythroleukemia (MEL) or mouse embryonic stem cells together with virus particles, in the presence of Polybrene® at 4 µg/mL, for 24–48 hours followed by puromycin selection (1 µg/mL) for 2 weeks. Real-time polymerase chain reaction (PCR)-based quantitative chromatin immunoprecipitation (ChIP) analysis was used to test whether the TAL1 transcription factor is bound to 1S promoter in the GATA-2 locus or whether LSD1 colocalizes with TAL1 at the 1S promoter. The sequential ChIP assay was utilized to confirm the role of LSD1 in the regulation of H3K4me2 at the GATA-2 locus during erythroid differentiation. Western blot analysis was employed to detect the protein expression. The alamarBlue® assay was used to examine the proliferation of the cells, and the absorbance was monitored at optical density (OD) 570 nm and OD 600 nm.
Results: In this study, we showed that LSD1 regulates the expression of GATA-2 during erythroid differentiation. Knockdown of LSD1 results in increased GATA-2 expression and inhibits the differentiation of MEL and embryonic stem cells. Furthermore, we demonstrated that LSD1 binds to the 1S promoter of the GATA-2 locus and suppresses GATA-2 expression, via histone demethylation.
Conclusion: Our data revealed that LSD1 mediates erythroid differentiation, via epigenetic modification of the GATA-2 locus.
Keywords: LSD1, GATA factor switching, histone demethylation
Introduction
GATA-1 and GATA-2 are two important transcription factors that regulate hematopoietic lineage commitment and differentiation. While GATA-1 is mainly expressed in differentiated hematopoietic lineages, such as erythroid,1 megakaryocytic,2 eosinophilic,3 and mast cells,4 GATA-2 is mainly expressed in hematopoietic stem and progenitor cells.5–8 The dynamic expression of GATA-1 and GATA-2 is critical for erythroid differentiation, and this is termed as “GATA factor switching”. Transcription of GATA-2 can be initiated from two different first exons, ie, the proximal first exon (1G promoter) and the distal first exon (1S promoter). While the 1G promoter and 1S promoter are mainly located in hematopoietic and progenitor tissues.9,10 At the early stage of erythroid differentiation, GATA-2 binds to five GATA motifs in the GATA-2 locus (-77 kb, -3.9 kb, -2.8 kb, -1.8 kb, and +9.5 kb from the 1S promoter) and promotes its own expression.11–13 At the late stage of erythroid differentiation, GATA-1 replaces GATA-2 and binds to these GATA motifs in the GATA-2 locus, which inhibits GATA-2 expression.7,8 Thus, GATA-2 expression is regulated by both GATA-2 and GATA-1 during erythroid differentiation. In addition, GATA-2 transcription can be regulated by several other transcription factors, including V-ets ETS1,14 BMP4,15 NOTCH1,16,17 PU.1,18 and EVI1,19,20 and cytokines, such as IL-1 and TNFα.21
Histone modifications, including acetylation, phosphorylation, methylation, and ubiquitination, play important roles in altering chromatin structure and regulating gene expression.22–24 Lysine methylation is an important histone modification that marks either negative or positive expression of genes, depending on the position of the lysine and the number of methyl moieties.25,26 For instance, genes that are repressed are generally associated with methylations at lysine 9 or 27 of histone H3 (H3K9 and H3K27), whereas actively transcribed genes are marked by methylations at the H3K4, H3K36, and H3K79 residues.27 Epigenetic changes are reversible, and chromatin modifiers are often proteins with enzymatic activities. Lysine methylation is regulated by the opposite activities of lysine methyltransferases (KMTs) and lysine demethylases (KDMs).28–33 KDMs consist of the FAD-dependent lysine-specific demethylases, LSD1 and LSD2,34–36 as well as the Jumonji C domain-containing histone demethylases.29 LSD1 is the first identified histone demethylase and forms a core complex with corepressors, including HDAC1/2 and CoREST, and represses the transcription of target genes.37 LSD1 is crucial for a variety of cellular processes, such as embryonic stem (ES) cell pluripotency, cellular proliferation, and differentiation.38–42 In addition, abnormal expression of LSD1 in hematopoietic progenitors perturbs the development of several hematopoietic lineages, suggesting that LSD1 is essential in hematopoietic development.43 We previously demonstrated that LSD1 cooperates with TAL1 to regulate the transcription of the P4.2 gene.44 In this study, we report that LSD1 associates with TAL1 to regulate the expression of the GATA-2 gene during erythroid differentiation.
Materials and methods
Plasmids and antibodies
Plasmids pSUPERretro vector and pSUPERretro-shLSD1 were gifts from Dr Suming Huang at the University of Florida. Anti-LSD1 and anti-TAL1 antibodies (ab17721, ab138276) were purchased from Abcam plc (Cambridge, UK).
Cell culture, transfection, and short hairpin (sh)RNA-mediated knockdown
Murine erythroleukemia (MEL) cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) and induced to differentiation with dimethyl sulfoxide (DMSO) as previously described.45 Mouse ES cells were cultured in DMEM and induced to differentiation with erythropoietin (EPO) as previously described.46 Retroviral packaging and infection were performed as previously described.45 To generate stable LSD1-knockdown cell lines, 5×105 exponentially growing MEL or ES cells were cultured with virus particles in the presence of Polybrene® (4 μg/mL) for 24–48 hours followed by puromycin selection (1 μg/mL) for 2 weeks.
Chromatin immunoprecipitation (ChIP) and Double ChIP
Real-time polymerase chain reaction (RT-PCR)-based quantitative ChIP analysis and Double ChIP were performed as previously described.47 The RT-PCR primer sequences have been listed in Table S1.
RNA isolation and quantitative RT-PCR
Total RNA was isolated using the SV Total RNA Isolation System kit (Promega Corporation, Fitchburg, WI, USA), and reverse transcription was performed according to the manufacturer’s instructions (Invitrogen; Life Technologies Corp, Carlsbad CA, USA). SYBR Green quantitative polymerase chain reaction (PCR) was performed as previously described.48 The RT-PCR primer sequences have been listed in Table S2.
Western blot analyses
For the western blot analysis, whole-cell extracts were prepared by boiling cells for 10 minutes in sodium dodecyl sulfate (SDS) sample buffer (50 mM tris(hydroxymethyl)aminomethane [Tris] [pH 6.8], 100 mM dithiothreitol [DTT], 2% SDS, 0.1% bromophenol blue, 10% glycerol) and were resolved using SDS–polyacrylamide gel electrophoresis (PAGE). Primary antibodies were detected using horseradish peroxidase (HRP)-conjugated anti-rabbit antibodies and visualized using enhanced chemiluminescence detection (ECL) (ECL reagents were from Thermo Fisher Scientific Inc, Waltham, MA, USA). Western blot analysis of β-actin (Thermo Fisher Scientific Inc) was used as a loading control.
Differentiation and proliferation assays
For the differentiation assay, MEL cells were treated with DMSO to induce differentiation and stained with benzidine blue (for hemoglobin), and benzidine blue–positive cells were quantified at day 3 and 5 of DMSO treatment. The proliferation of cells was determined using alamarBlue® assays (Invitrogen; Life Technologies Corp). Briefly, 1×105 cells were seeded in a 96-well plate with 100 μL of DMEM and cultured for 10 days. Next, 10 μL (1/10 volume) of alamarBlue reagent was directly added to the cells in culture medium and incubated for 4 hours at 37°C. The absorbance at optical density (OD) 570 nm and OD 600 nm were monitored, and the relative proliferation was calculated according to the manufacturer’s instructions.
Statistical analysis
The statistical analysis for data was performed employing the SPSS software package 19.0 (IBM Corporation, Armonk, NY, USA). Student t-test and one way analysis of variance (ANOVA) were employed for two groups and multiple group comparison. Data were presented as mean ± standard deviation (SD). P<0.05 represented a statistically significant difference.
Results
Knockdown of LSD1 inhibits the differentiation of MEL cells
To investigate the function of LSD1 in erythroid differentiation, we generated stable LSD1-knockdown clones from MEL and ES cells using shRNA-mediated gene silencing (Figure 1A and B). The LSD1-knockdown cells were induced to differentiate and were stained with benzidine to detect hemoglobin production. In our experiments, we performed at least three technical replicates with the one optimal MEL cell clone and one optimal ES cell clone. Compared with control cells, the LSD1-knockdown cells showed a significantly lower percentage of benzidine-positive cells (P<0.05) (Figure 1C), demonstrating that knockdown of LSD1 inhibited the differentiation of MEL cells into hemoglobin-producing cells. In addition to the inhibition of erythroid differentiation, knockdown of LSD1 also significantly inhibited MEL cell proliferation (P<0.05) (Figure 1D) and resulted in increased cell death (P<0.05) (Figure 1D and E) compared with the control cells.
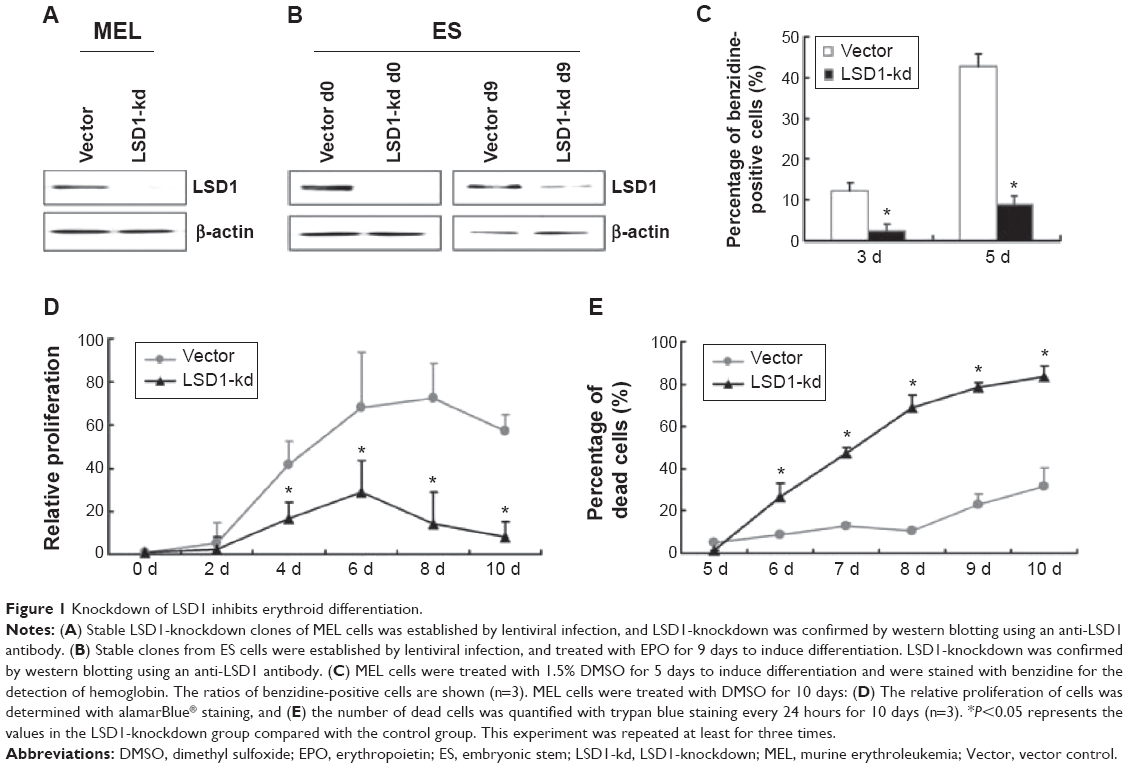 | Figure 1 Knockdown of LSD1 inhibits erythroid differentiation. |
Furthermore, we also detected the effects of LSD1-knockdown in ES cells. Our results also indicated that LSD1-knockdown inhibits EPO-induced ES cell differentiation to erythroid cell lineage (with the same changes in the MEL cells [data not shown]).
Knockdown of LSD1 results in the decreased expression of GATA-1 and increased expression of GATA-2 in differentiated cells
Because GATA-1 and GATA-2 are two master transcription factors in hematopoietic differentiation, we examined the expression of GATA-1 and GATA-2 in MEL and EPO-induced differentiated ES cells. While normal MEL and ES cells showed low levels of GATA-2 and high levels of GATA-1 expression upon induced differentiation, knockdown of LSD1 resulted in an increased expression of GATA-2 and decreased expression of GATA-1 (Figure 2A and B). More importantly, while GATA-1 was the dominant form of the GATA switch in differentiated MEL and ES cells, GATA-2 became the dominant form of the GATA switch in LSD1-knockdown cells, demonstrating an essential role of LSD1 in regulating the expression of GATA switch factors (Figure 2C). Consistent with the decreased expression of the GATA-1 gene in LSD1-knockdown cells, LSD1-knockdown cells showed decreased expression of β-major, a well-established erythroid differentiation marker (P<0.05). Meanwhile, the western blot analysis results indicated that the LSD knockdown (both in MEL and ES cells) also significantly increased the GATA-2 protein levels and decreased the GATA-1 levels compared with the blank vectors (both in MEL and ES cells) (all P<0.05) (Figure S1).
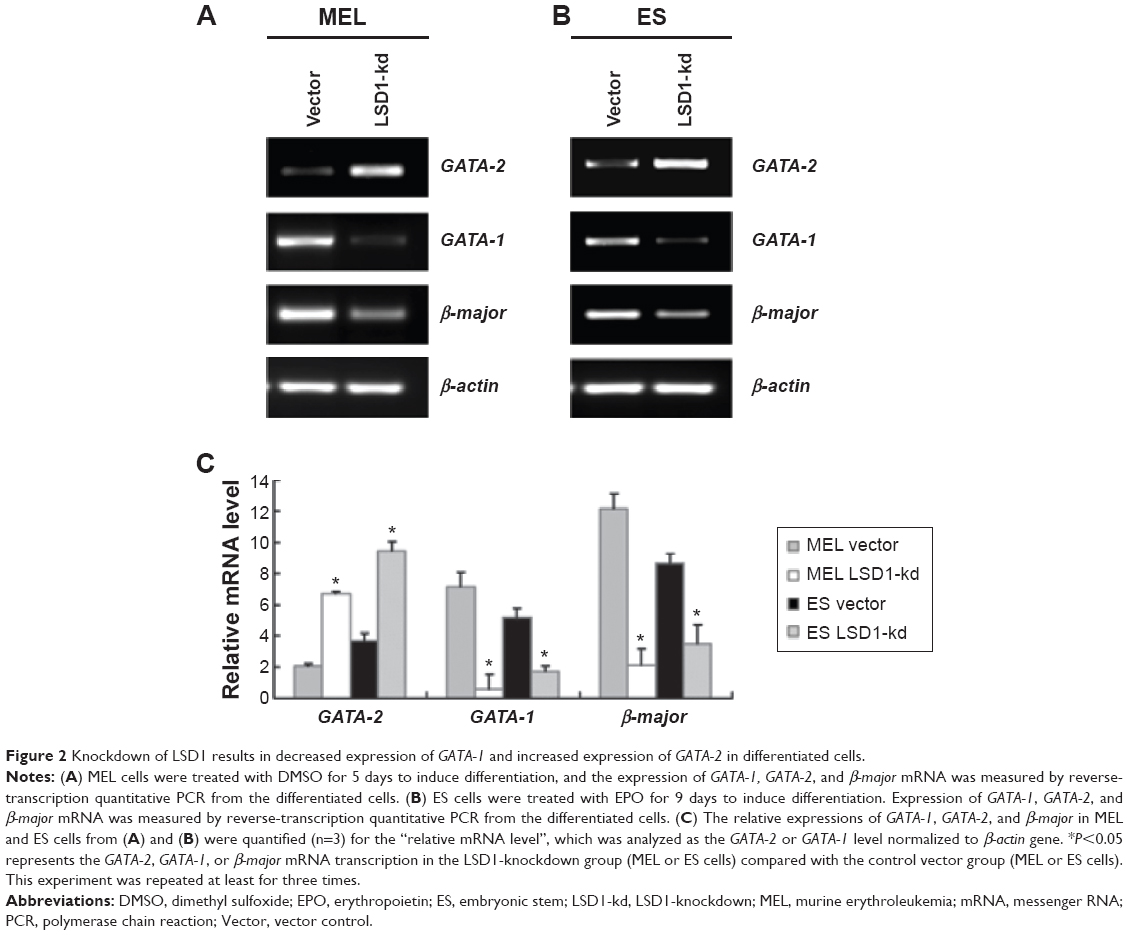 | Figure 2 Knockdown of LSD1 results in decreased expression of GATA-1 and increased expression of GATA-2 in differentiated cells. |
Furthermore, we also detected the GATA-2 or GATA-1 levels in the undifferentiated MEL and ES cells. Our preliminary results indicated that there were no differences in GATA-2 or GATA-1 levels in the MEL and ES cells before induction of differentiation (P>0.05) (Figure S2).
LSD1 colocalizes with TAL1 at the 1S promoter of the GATA-2 locus
Within the GATA-2 locus, multiple sites have been suggested to be involved in the regulation of GATA-2 expression (Figure 3A).9,10 To study the molecular mechanism underlying LSD1 regulation of GATA-2 expression during erythroid differentiation, we examined the enrichment of LSD1 at different regulatory sites in the GATA-2 locus using ChIP assay and found that LSD1 was mainly recruited to the 1S promoter of the GATA-2 locus in MEL cells (Figure 3B). Because LSD1 does not have a DNA-binding domain, we hypothesized that LSD1 is recruited by sequence-specific transcription factors to the GATA-2 locus. It has been reported that TAL1 recruits LSD1 to regulate gene transcription in normal and malignant hematopoiesis.49 Thus, we reasoned that TAL1 might also recruit LSD1 to the 1S promoter of the GATA-2 locus during erythroid differentiation. To test this hypothesis, we performed ChIP assays using the TAL1 antibody and found that TAL1 bound predominantly to the 1S promoter in the GATA-2 locus (Figure 3C). To further test whether LSD1 colocalizes with TAL1 at the 1S promoter, we performed TAL1 and LSD1 sequential ChIP assays. Expectedly, LSD1 and TAL1 co-occupied the 1S promoter, suggesting that TAL1 recruited LSD1 to the GATA-2 1S promoter (Figure 3D).
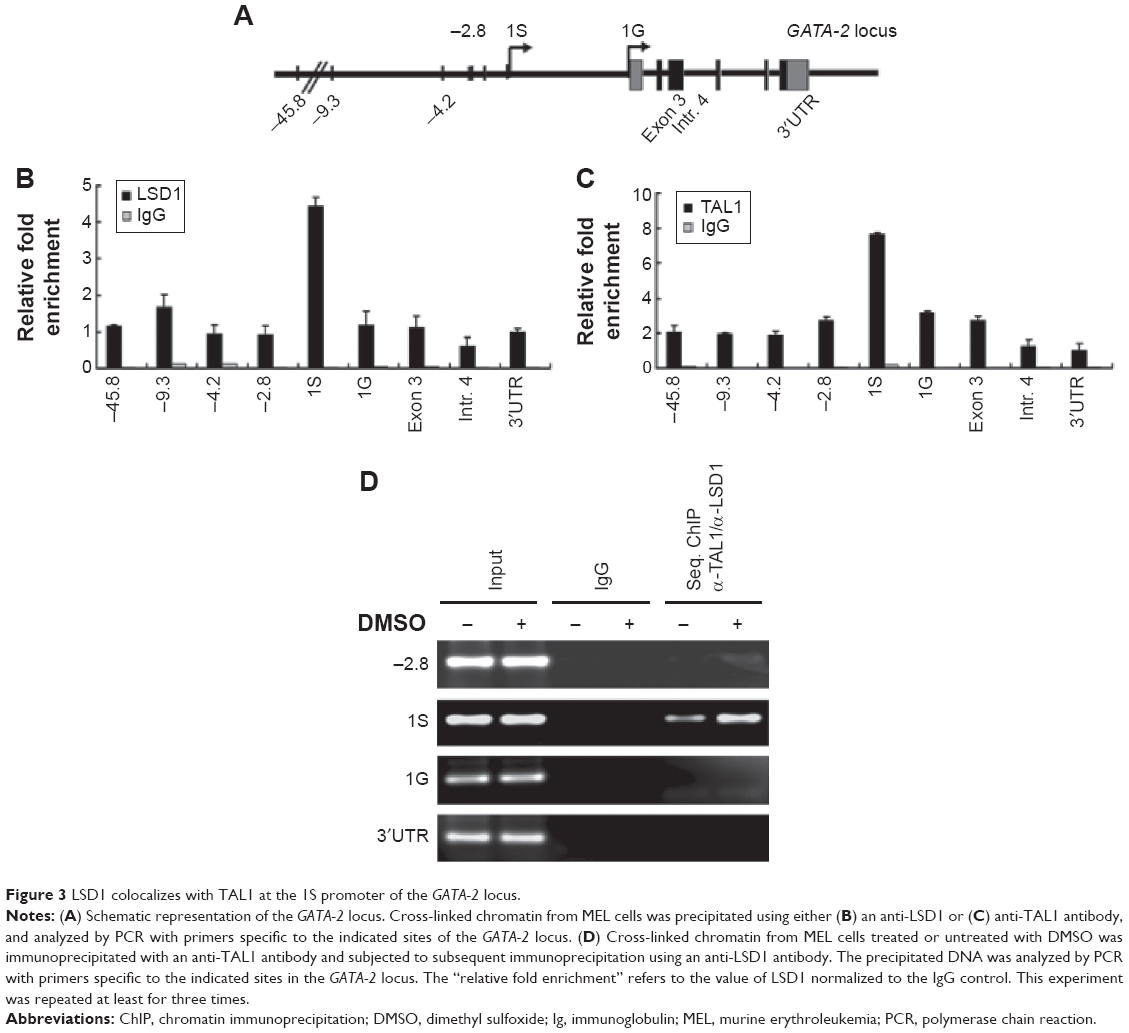 | Figure 3 LSD1 colocalizes with TAL1 at the 1S promoter of the GATA-2 locus. |
LSD1-knockdown increases H3K4me2 at the GATA-2 locus
LSD1 is an important histone demethylase.34 Our results showed that the enrichment of LSD1 was increased at the 1S promoter in the GATA-2 locus (Figure 3D), suggesting that LSD1 might decrease H3K4me2 at the 1S promoter via its lysine demethylases activity. Thus, we investigated the alteration of histone methylation levels at the GATA-2 locus during erythroid differentiation. The result of ChIP assays demonstrated that H3K4me2 was significantly decreased at −2.8 kb, 1S promoter, and 1G promoter in the GATA-2 locus in DMSO-treated MEL cells on day 3 compared with that on day 0 (P<0.05) (Figure 4A). The decreased H3K4me2 at the GATA-2 locus, particularly at the 1S promoter, was correlated with the decreased expression of GATA-2 during erythroid cell differentiation, suggesting that LSD1 might inhibit the expression of GATA-2 via histone modification. To confirm the role of LSD1 in the regulation of H3K4me2 at the GATA-2 locus during erythroid differentiation, we performed ChIP analysis for H3K4me2 enrichment at the GATA-2 locus in LSD1-knockdown MEL cells. H3K4me2 levels were significantly increased in the LSD1-knockdown cells compared with the control vector cells (P<0.05) (Figure 4B), demonstrating that LSD1 is required for the decreased H3K4me2 at the GATA-2 locus during erythroid differentiation.
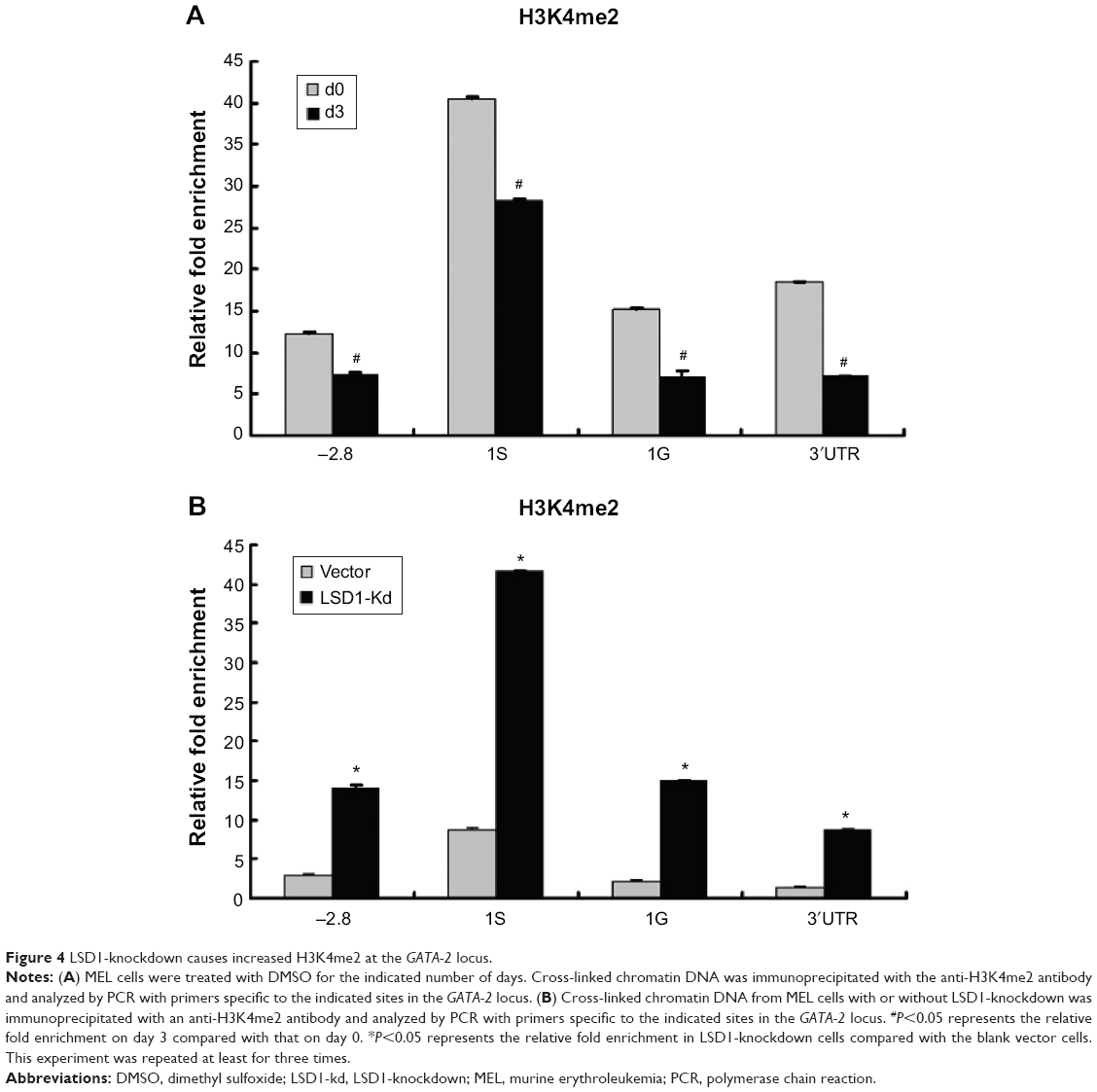 | Figure 4 LSD1-knockdown causes increased H3K4me2 at the GATA-2 locus. |
Discussion
The dynamic expression of GATA-1 and GATA-2 is crucial for erythroid differentiation. Our results, together with previous findings,44,50 led us to propose a novel epigenetic mechanism for the regulation of GATA-2 expression. During erythroid differentiation, LSD1 is recruited to the GATA-2 locus by TAL1. The demethylation activity of LSD1 results in decreased H3K4me2, which subsequently reduces the transcription of GATA-2. Thus, LSD1 is an essential epigenetic regulator of GATA-2 expression.
While this work highlights the significance of LSD1-mediated epigenetic silencing of GATA-2 expression, previous work has demonstrated that GATA-1 also suppresses GATA-2 expression. Genome-wide studies have demonstrated that TAL1 and GATA-1 extensively co-occupy genomic sites in erythroid cells.44,49,50 Thus, it is likely that LSD1 collaborates with TAL1 and GATA-1 to regulate gene expression during erythroid differentiation. Moreover, previous studies have already confirmed the interaction between LSD1 and TAL1.44,49 However, despite repeated efforts, our coimmunoprecipitation results do not show such an interaction between GATA-1 and LSD1 (data not shown), suggesting that LSD1 and GATA-1 regulate GATA-2 expression via different protein complexes.
In addition to increased expression of GATA-2, LSD1-knockdown cells also exhibited decreased expression of GATA-1 during erythroid differentiation. However, the precise mechanisms underlying LSD1-mediated increased expression of GATA-1 is still unknown. It remains to be determined whether and how LSD1 cooperates with TAL1 or GATA-2 to regulate GATA-1 expression.
LSD1 is a component of several histone deacetylase complexes, though it silences genes by functioning as a histone demethylase.34,37,39 Alterative splicing results in multiple transcript variants. The histone modification mediated by LSD1 always plays an important role in some diseases, such as leukocythemia and some cancers. Adamo et al39 reported that LSD1 regulates the balance between self-renewal and differentiation in human ES cells. Therefore, we speculated that the LSD1 may become a clinical therapeutic target for leukocythemia. GATA-2 is expressed in hematopoietic progenitors, including early erythroid cells, mast cells, and megakaryocytes, and also in nonhematopoietic ES cells. In chicken erythroid progenitors, forced expression of GATA-2 promotes proliferation at the expense of differentiation.51 Multiple mutations on the GATA-2 gene have been recently implicated as the cause of primary immunodeficiency in patients with MonoMAC syndrome, and cases of dendritic cell, monocyte, B and natural killer (NK) lymphoid deficiency, and leukocythemia.52 Also, GATA-2 has recently been implicated in non–small cell lung cancer.53 Targeting processes that occur downstream of GATA-2 signaling, with clinically approved drugs, had a significant effect in mouse models of the disease.53 All of these studies showed that the GATA-2 may be a therapeutic target for some kinds of diseases, especially for cancers and leukocythemia. However, no effective and available compounds targeting GATA-2 have been discovered. Our study proved, for the first time, that LSD1 could regulate the GATA-2-mediated erythroid differentiation, which would provide the potential clue for new anticancer drugs.
In conclusion, our data revealed that LSD1 mediates erythroid differentiation via epigenetic modification of the GATA-2 locus.
Acknowledgments
We are grateful to Dr Suming Huang for his suggestions and comments. We also express appreciation to Dr Douglas Linn for his critical reading of this manuscript.
This work was supported by grants obtained from the Natural Science Foundation of China (grant numbers 31100926 and 31471356) and China Postdoctoral Science Foundation (grant numbers 20100481059 and 2012T50290).
Disclosure
The authors report no conflicts of interest in this work.
References
Pevny L, Simon MC, Robertson E, et al. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature. 1991;349(6306):257–260. | ||
Shivdasani RA, Fujiwara Y, McDevitt MA, Orkin SH. A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. EMBO J. 1997;16(13):3965–3973. | ||
Yu C, Cantor AB, Yang H, et al. Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J Exp Med. 2002;195(11):1387–1395. | ||
Tsai FY, Keller G, Kuo FC, et al. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371(6494):221–226. | ||
Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S. GATA switches as developmental drivers. J Biol Chem. 2010;285(41):31087–31093. | ||
Suzuki M, Shimizu R, Yamamoto M. Transcriptional regulation by GATA1 and GATA2 during erythropoiesis. Int J Hematol. 2011;93(2):150–155. | ||
Kaneko H, Shimizu R, Yamamoto M. GATA factor switching during erythroid differentiation. Curr Opin Hematol. 2010;17(3):163–168. | ||
Suzuki M, Kobayashi-Osaki M, Tsutsumi S, et al. GATA factor switching from GATA2 to GATA1 contributes to erythroid differentiation. Genes Cells. 2013;18(11):921–933. | ||
Minegishi N, Ohta J, Suwabe N, et al. Alternative promoters regulate transcription of the mouse GATA-2 gene. J Biol Chem. 1998;273(6):3625–3634. | ||
Pan X, Minegishi N, Harigae H, et al. Identification of human GATA-2 gene distal IS exon and its expression in hematopoietic stem cell fractions. J Biochem. 2000;127(1):105–112. | ||
Grass JA, Boyer ME, Pal S, Wu J, Weiss MJ, Bresnick EH. GATA-1-dependent transcriptional repression of GATA-2 via disruption of positive autoregulation and domain-wide chromatin remodeling. Proc Natl Acad Sci U S A. 2003;100(15):8811–8816. | ||
Martowicz ML, Grass JA, Boyer ME, Guend H, Bresnick EH. Dynamic GATA factor interplay at a multicomponent regulatory region of the GATA-2 locus. J Biol Chem. 2005;280(3):1724–1732. | ||
Grass JA, Jing H, Kim SI, et al. Distinct functions of dispersed GATA factor complexes at an endogenous gene locus. Mol Cell Biol. 2006;26(19):7056–7067. | ||
Lulli V, Romania P, Morsilli O, et al. Overexpression of Ets-1 in human hematopoietic progenitor cells blocks erythroid and promotes megakaryocytic differentiation. Cell Death Differ. 2006;13(7):1064–1074. | ||
Maeno M, Mead PE, Kelley C, et al. The role of BMP-4 and GATA-2 in the induction and differentiation of hematopoietic mesoderm in Xenopus laevis. Blood. 1996;88(6):1965–1972. | ||
Robert-Moreno A, Espinosa L, de la Pompa JL, Bigas A. RBPjkappa-dependent Notch function regulates Gata2 and is essential for the formation of intra-embryonic hematopoietic cells. Development. 2005;132(5):1117–1126. | ||
Kumano K, Chiba S, Shimizu K, et al. Notch1 inhibits differentiation of hematopoietic cells by sustaining GATA-2 expression. Blood. 2001;98(12):3283–3289. | ||
Walsh JC, DeKoter RP, Lee HJ, et al. Cooperative and antagonistic interplay between PU.1 and GATA-2 in the specification of myeloid cell fates. Immunity. 2002;17(5):665–676. | ||
Yatsula B, Lin S, Read AJ, et al. Identification of binding sites of EVI1 in mammalian cells. J Biol Chem. 2005;280(35):30712–30722. | ||
Yuasa H, Oike Y, Iwama A, et al. Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J. 2005;24(11):1976–1987. | ||
La Ferla K, Reimann C, Jelkmann W, Hellwig-Bürgel T. Inhibition of erythropoietin gene expression signaling involves the transcription factors GATA-2 and NF-kappaB. FASEB J. 2002;16(13):1811–1813. | ||
Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. | ||
Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14(11):1008–1016. | ||
Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8(12):983–994. | ||
Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. | ||
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. | ||
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. | ||
Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8(4):307–318. | ||
Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25(1):1–14. | ||
Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22(9):1115–1140. | ||
Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13(5):343–357. | ||
Forneris F, Binda C, Battaglioli E, Mattevi A. LSD1: oxidative chemistry for multifaceted functions in chromatin regulation. Trends Biochem Sci. 2008;33(4):181–189. | ||
Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48(4):491–507. | ||
Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. | ||
Ciccone DN, Su H, Hevi S, et al. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature. 2009;461(7262):415–418. | ||
Fang R, Barbera AJ, Xu Y, et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell. 2010;39(2):222–233. | ||
Lan F, Nottke AC, Shi Y. Mechanisms involved in the regulation of histone lysine demethylases. Curr Opin Cell Biol. 2008;20(3):316–325. | ||
Whyte WA, Bilodeau S, Orlando DA, et al. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482(7384):221–225. | ||
Adamo A, Sesé B, Boue S, et al. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat Cell Biol. 2011;13(6):652–659. | ||
Sprüssel A, Schulte JH, Weber S, et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia. 2012;26(9):2039–2051. | ||
Kerenyi MA, Shao Z, Hsu YJ, et al. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife. 2013;2:e00633. | ||
Amente S, Lania L, Majello B. The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim Biophys Acta. 2013;1829(10):981–986. | ||
Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell. 2007;27(4):562–572. | ||
Hu X, Li X, Valverde K, et al. LSD1-mediated epigenetic modification is required for TAL1 function and hematopoiesis. Proc Natl Acad Sci U S A. 2009;106(25):10141–10146. | ||
Huang S, Brandt SJ. mSin3A regulates murine erythroleukemia cell differentiation through association with the TAL1 (or SCL) transcription factor. Mol Cell Biol. 2000;20(6):2248–2259. | ||
Kitajima K, Tanaka M, Zheng J, Sakai-Ogawa E, Nakano T. In vitro differentiation of mouse embryonic stem cells to hematopoietic cells on an OP9 stromal cell monolayer. Methods Enzymol. 2003;365:72–83. | ||
Crusselle-Davis VJ, Vieira KF, Zhou Z, Anantharaman A, Bungert J. Antagonistic regulation of beta-globin gene expression by helix-loop-helix proteins USF and TFII-I. Mol Cell Biol. 2006;26(18):6832–6843. | ||
Huang S, Li X, Yusufzai TM, Qiu Y, Felsenfeld G. USF1 recruits histone modification complexes and is critical for maintenance of a chromatin barrier. Mol Cell Biol. 2007;27(22):7991–8002. | ||
Li Y, Deng C, Hu X, et al. Dynamic interaction between TAL1 oncoprotein and LSD1 regulates TAL1 function in hematopoiesis and leukemogenesis. Oncogene. 2012;31(48):5007–5018. | ||
Hu X, Ybarra R, Qiu Y, Bungert J, Huang S. Transcriptional regulation by TAL1: a link between epigenetic modifications and erythropoiesis. Epigenetics. 2009;4(6):357–361. | ||
Briegel K, Lim KC, Plank C, Beug H, Engel JD, Zenke M. Ectopic expression of a conditional GATA-2/estrogen receptor chimera arrests erythroid differentiation in a hormone-dependent manner. Genes Dev. 1993;7(6):1097–1109. | ||
Dickinson RE, Griffin H, Bigley V, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118(10):2656–2658. | ||
Kumar MS, Hancock DC, Molina-Arcas M, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149(3):642–655. |
Supplementary materials
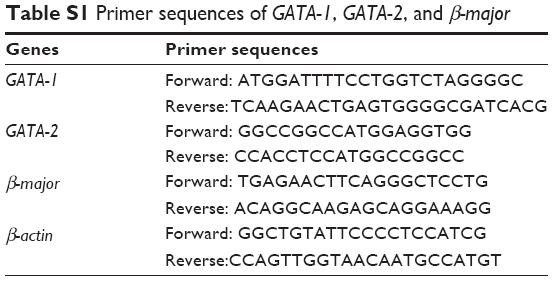 | Table S1 Primer sequences of GATA-1, GATA-2, and β-major |
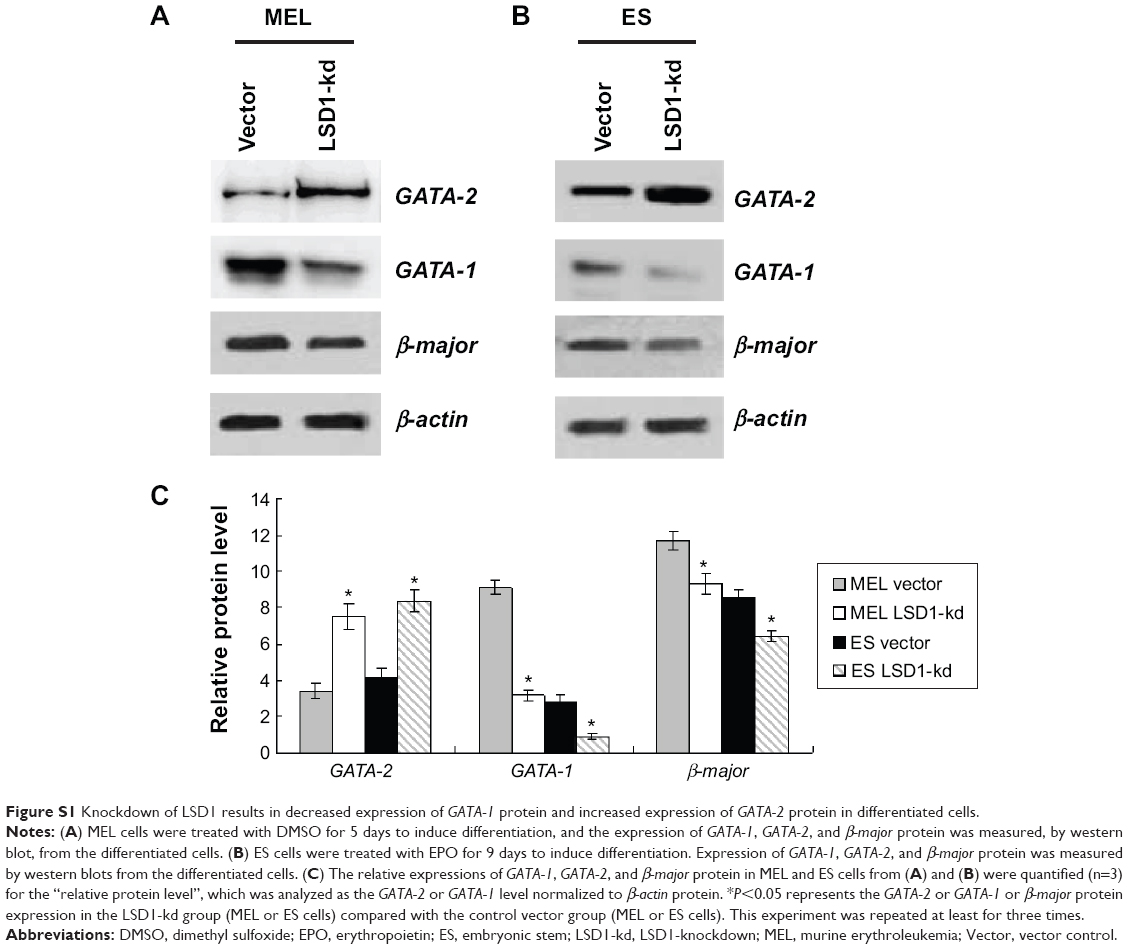 | Figure S1 Knockdown of LSD1 results in decreased expression of GATA-1 protein and increased expression of GATA-2 protein in differentiated cells. |
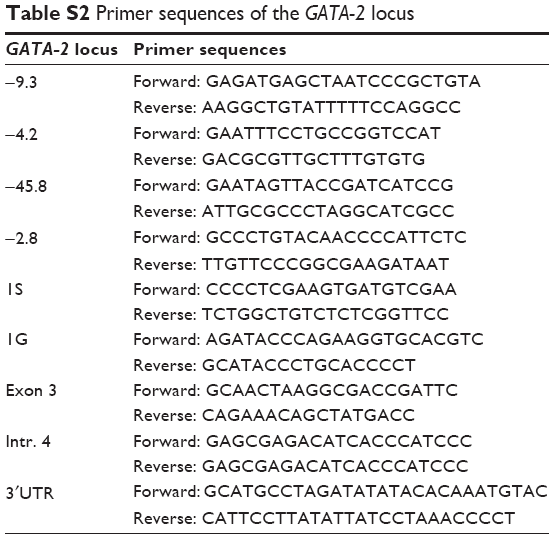 | Table S2 Primer sequences of the GATA-2 locus |
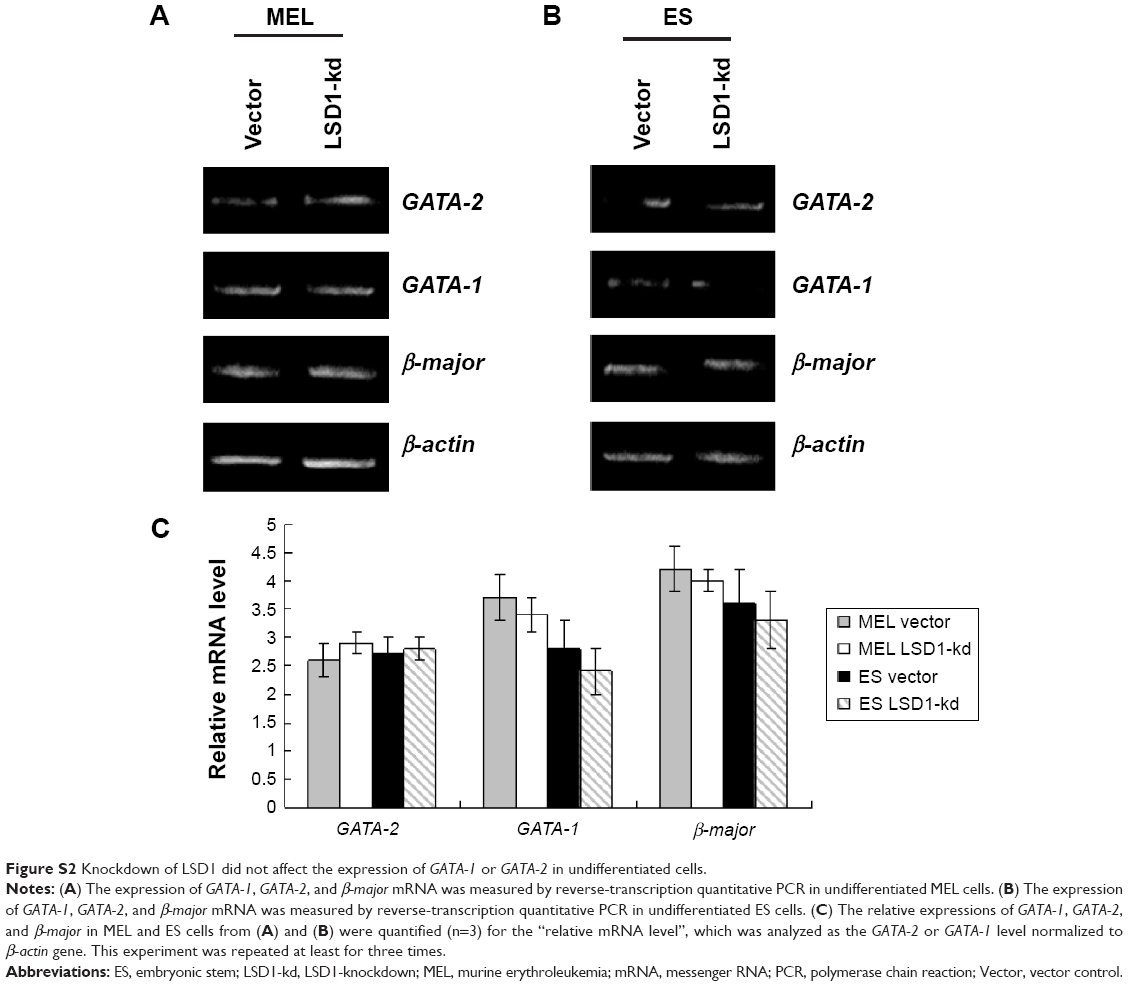 | Figure S2 Knockdown of LSD1 did not affect the expression of GATA-1 or GATA-2 in undifferentiated cells. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.