")
Back to Journals » Drug Design, Development and Therapy » Volume 11
High-dose atorvastatin versus moderate dose on early vascular protection after ST-elevation myocardial infarction
Authors Gavazzoni M, Gorga E, Derosa G , Maffioli P, Metra M , Raddino R
Received 20 February 2017
Accepted for publication 16 August 2017
Published 4 December 2017 Volume 2017:11 Pages 3425—3434
DOI https://doi.org/10.2147/DDDT.S135173
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr James Janetka
Mara Gavazzoni,1 Elio Gorga,1 Giuseppe Derosa,2–5 Pamela Maffioli,2,4 Marco Metra,1 Riccardo Raddino1
1Cardiology Department, University of Brescia, Spedali Civili di Brescia, Brescia, 2Centre of Diabetes and Metabolic Diseases, Department of Internal Medicine and Therapeutics, University of Pavia, Fondazione IRCCS Policlinico San Matteo, 3Centre for the Study of Endocrine-Metabolic Pathophysiology and Clinical Research, University of Pavia, 4Centre for Prevention, Surveillance, Diagnosis, and Treatment of Rare Diseases, Fondazione IRCCS Policlinico San Matteo, 5Laboratory of Molecular Medicine, University of Pavia, Pavia, Italy
Background and aim: Clinical benefits of early high-dose statin therapy after acute coronary syndromes are widely known; however, there is poor evidence on the specific setting of ST-elevation myocardial infarction (STEMI) and dose-dependent effects of this therapy on endothelial function and inflammatory biomarkers in the most vulnerable phase after acute coronary syndromes: the postdischarge period. In our study, we compared the short-term effects of high (80 mg) vs moderate doses of atorvastatin (20 mg) in patients with STEMI undergoing primary percutaneous coronary intervention on endothelial function and vascular inflammation. The aim of our study was the evaluation of dose-dependent short-term effects.
Subjects and methods: We enrolled 52 patients within 48 hours of a STEMI to atorvastatin 80 mg (n=26) or 20 mg (n=26). Every patient underwent endothelial function evaluation by the reactive hyperemia–peripheral arterial tonometry (RH-PAT) index on the first day and 1 month after the STEMI. At the same time, we measured lipid profile and serum levels of high-sensitivity CRP, IL6, TNFα, and oxidized LDL.
Results: After 1 month of therapy, we observed differences in high-sensitivity CRP levels (0.04±0.02 mg/dL vs 0.36±0.3 mg/dL, P=0.001), IL6 (1.12±0.93 pg/mL vs 3.13±2.84 pg/mL, P=0.03), and improvement in RH-PAT index (1.96±0.16 vs 1.72±0.19, P=0.002) in the group treated with high-dose vs moderate-dose atorvastatin. There was no significant difference in levels of TNFα or oxidized LDL with atorvastatin 20 mg, while there was a reduction in these variables in the group treated with atorvastatin 80 mg. We observed a correlation between high-sensitivity polymerase chain reaction and RH-PAT index on the 30th day after STEMI (r=0.5, P=0.001).
Conclusion: Higher dose statin therapy in patients with STEMI undergoing primary percutaneous coronary intervention showed early greater vascular protective effects that moderate dose.
Keywords: endothelial dysfunction, endo-PAT, vascular inflammation, acute coronary syndrome
Introduction
Recently published trials, such as IMPROVE-IT,1 have confirmed that LDL-cholesterol (LDL-C) reduction lower the currently recommended target has a protective role on cardiovascular events, but improved end-point mortality has not been achieved. Given the results on cardiovascular and total mortality in large clinical trials in the setting of primary and secondary prevention, statin therapy remains the cornerstone of lipid-lowering therapy.2
Since they were used in the setting of acute coronary syndrome (ACS), statins have shown clear efficacy in reducing the risk of cardiovascular events in both chronic and acute phases,3–5 and it is known that their effects are related not only to the precocity of treatment but also to its intensity. The ARMYDA-ACS6 trial and others7,8 suggested that statins could reduce arterial wall mechanical injury induced by percutaneous coronary intervention (PCI); moreover, the precocity of observed clinical advantages on statin therapy led to the hypothesis that they could have a role in limiting inflammation,9–12 oxidative stress,13 and endothelial dysfunction,14 thus contributing to plaque stabilization earlier than the mere reduction in LDL-C.
Among patients with ACS, patients presenting with ST-elevation myocardial infarction (STEMI) have higher levels of inflammation.15 Moreover, primary PCI and stenting throughout mechanical injury contribute to plaque instability in the entire coronary tree,16 and outcomes of revascularization could be affected by adverse events, such as the “no-reflow phenomenon”, in which endothelial dysfunction seems to have a central role.17 Despite these important issues, little evidence has been published at this point to evaluate the role of statin therapy in this specific subgroup of patients with ACS and persistent STEMI. In the last few years, one trial revealed that high-dose atorvastatin before primary PCI in patients presenting with STEMI was able to improve coronary flow downstream of the treated culprit vessel.18 Other small pilot studies have demonstrated intensive statin therapy in acute phase after STEMI reduce oxidized LDL (ox-LDL), inflammation markers, and inflammatory cytokines,19 reduce ex vivo platelet activation20 in a few hours, and improve myocardial function recovery 6 months after an index event.21
In our study, we compared the short-term effects (after 1 month) of early administration of high-dose atorvastatin (80 mg) and a moderate dose of the same molecule (atorvastatin 20 mg) in patients with STEMI undergoing primary PCI on endothelial function, markers of vascular inflammation, and plaque instability. The aim of our study was evaluation of dose-dependent short-term effects.
Subjects and methods
Study design
The present study was a single-center, prospective, randomized, open-label trial with blinded evaluation of end points. Patients were randomized (1:1) to receive 80 mg of atorvastatin or 20 mg of atorvastatin once a day. This protocol was designed and approved in 2010 by the provincial ethics committee of our hospital (Spedali Civili of Brescia).
Population
We randomized patients admitted to the Intensive Coronary Care Unit of the Spedali Civili with a diagnosis of STEMI. Patients included should be 18–80 years old, admitted to the Intensive Care Unit of our Cardiology Department with a diagnosis of STEMI and should be able to get written consent before starting any investigational activity. We excluded patients with STEMI not undergoing primary PCI, those with statin intolerance or contraindications to statin therapy, such as severe liver disease or muscle diseases (muscular dystrophy, previous rhabdomyolysis), patients yet to receive statin therapy, those with cardiogenic shock, chronic severe renal failure (glomerular filtration rate <30 mL/min), documented current infections (community or in-hospital infections), chronic inflammatory disease (ie, systemic lupus erythematosus, rheumatoid arthritis, chronic inflammatory gastrointestinal disorders), coagulation abnormalities, abusing alcohol or drugs, participating simultaneously in another clinical trial, and those with concomitant disease that could interfere with therapy evaluation or reduce life expectancy and alterations in cognitive function.
Protocol treatment
Once eligible, patients were randomly assigned to receive either a moderate 20 mg daily dose of atorvastatin or an intensive 80 mg daily dose for the first month. At the end of this period, patients were revaluated for dose adjustment according to the target (in order to reach LDL-C <70 mg/dL) (Figure 1).
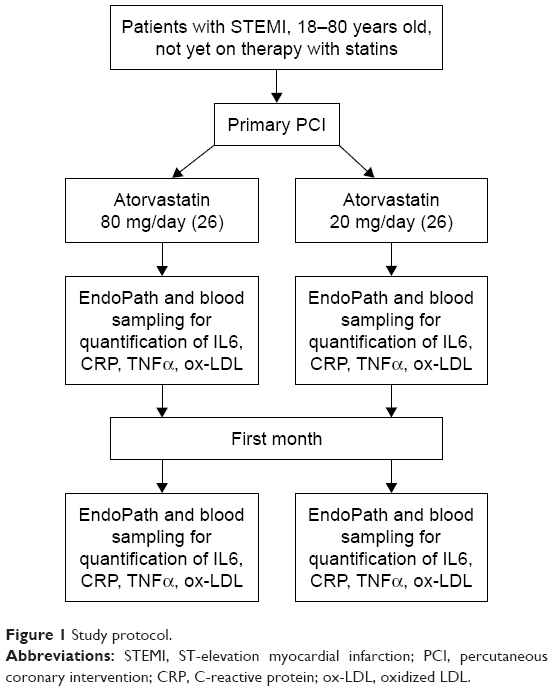 | Figure 1 Study protocol. |
The first dose of atorvastatin was administered within 24 hours of first medical contact. Blood sampling and endothelial function (with EndoPat device) were performed at the randomization and after 1 month of treatment. Both groups were treated with pharmacological optimal therapy according to European Society of Cardiology (ESC) guidelines for the diagnosis and management of STEMI.22 After discharge, patients were reevaluated at the 30th day with physical examination, electrocardiography, echocardiography, blood samples, assessment of adverse events, targeted therapies, and evaluation of endothelial function with the EndoPAT system.
Effectiveness evaluation
Primary end points were variation after 30 days of treatment of endothelium-dependent vasodilation during hyperemia (measured with the reactive hyperemia index [RHI]) and changes in plasma levels of high-sensitivity CRP (HS-CRP), IL6, TNFα, and ox-LDL.
Primary PCI procedure and angiographic analysis
For all patients, diagnosis and treatment of STEMI were performed according to ESC guidelines.22 Before angioplasty, they were treated with intravenous unfractionated heparin (adjusted for corporeal weight bolus), in order to obtain an activated clotting time >300 seconds (or between 200 and 300 seconds when glycoprotein IIB/IIIA inhibitors were used). Glycoprotein IIB/IIIA inhibitors were used at the discretion of operators. Procedural success was measured as TIMI (thrombolysis in MI) 3 flow recovery. Morphology of culprit lesions and their complexity were analyzed using two X-ray orthogonal projections according to American College of Cardiology/American Heart Association guidelines.23
Blood sampling and laboratory analysis
We obtained blood samples at randomization (time 0) and at 30 days (time 1). These blood samples were divided into two test tubes containing citrate 3.8% (green Vacutainer, 5 mL), two test tubes for complete blood count (red Vacutainer, 5 mL), and one test tube for serology (white Vacutainer, 10 mL, without anticoagulant). All test tubes were conserved in crushed ice for 3 hours and then subjected to centrifugation at 3,500 rpm for 20 minutes at 4°C, thereby obtaining plasma separation from cell pellets. Plasma was divided into 0.5 mL parts and collected in test tubes (Eppendorf). Then, samples were stored at −80°C. We measured total cholesterol, LDL-C, HDL-C, TNFα, triglycerides, CK-MM (muscle isoenzyme), AST, and ALT. CRP was measured with a nephelometric assay (Dade Behring Diagnostics), and the inferior limit of measurements was 0.15 mg/L ox-LDL. TNFα and IL6 levels were measured with an enzyme-linked immunosorbent-assay kit (R&D Systems, Minneapolis, MN, USA).
Evaluation of endothelial function
All patients were subjected to evaluation testing of endothelial function within 48 hours of onset of ACS and after 1 month. Endothelial function was expressed using the RHI and assessed using the EndoPAT 2000 device (Itamar Medical, Caesarea, Israel). Measurements were performed according to the manufacturer’s instructions and calculated using an automated algorithm provided with the device. Patients were supine for a minimum of 20 minutes before measurements in a quiet, temperature-controlled (21°C–24°C) room with dimmed lights.
The EndoPAT quantifies endothelium-mediated changes in vascular tone, elicited by a 5-minute occlusion of the brachial artery (using a standard blood-pressure cuff). When the cuff is released, the surge in blood flow causes endothelium-dependent flow-mediated dilatation. The dilatation, manifested as RH, is captured by the EndoPAT as an increase in peripheral arterial tone (PAT)-signal amplitude. A postocclusion:preocclusion ratio is calculated by the EndoPAT software, providing the EndoPAT index. The threshold for a good EndoPAT result is an RHI value of 1.67 and above. This was determined following the study of Bonetti et al,24 which was performed at the Mayo Clinic. In this study, EndoPAT readings were found to be correlated with coronary endothelial function using the gold-standard method of assessment, which is injection of acetylcholine during coronary catheterization.
Statistical analysis
Results are expressed as mean ± SD. Baseline characteristics and posttreatment changes were compared between groups and within groups. Nonparametric variables were compared using χ2 tests, while parametric variables were compared using Student’s t-tests within groups. Absolute changes were calculated by subtracting baseline values from posttreatment values, while percentage changes were calculated by dividing the difference calculated by baseline value. Absolute and percentage changes were compared between groups using Student’s t-test. For all tests, P<0.05 was considered statistically significant. Correlations among continuous variables were analyzed with Pearson’s r-coefficient.
Results
Baseline characteristics of study population
From February 2011 to February 2012, 52 patients with STEMI were enrolled: 26 were randomized to atorvastatin 80 mg and 26 to atorvastatin 20 mg. The average age was 58.4±11 years, and the majority of patients were male (88.4%). Baseline characteristics did not differ significantly between the two groups, and are shown in Table 1.
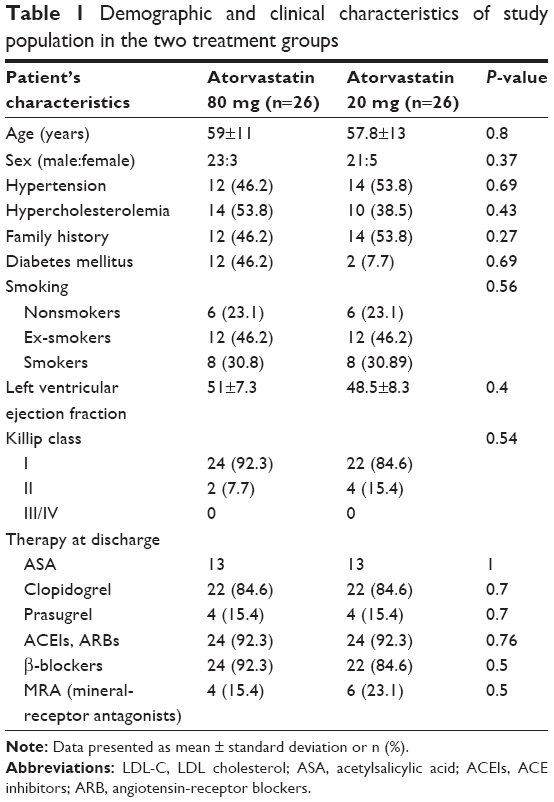 | Table 1 Demographic and clinical characteristics of study population in the two treatment groups |
Procedural features
There were no significant differences related to the site of the culprit lesion: the left anterior descendant was involved in most cases (46.2% in atorvastatin 80 mg vs 45.7% in the 20 mg group, P=0.69) (Table 2).
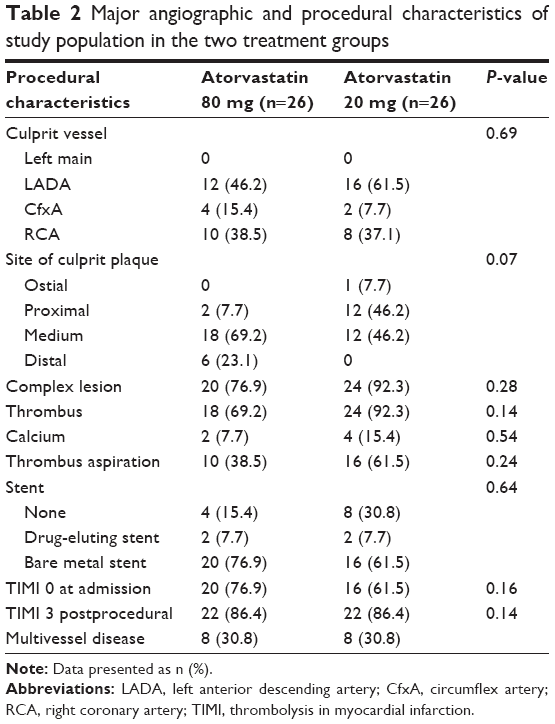 | Table 2 Major angiographic and procedural characteristics of study population in the two treatment groups |
Effects on lipid profiles
Absolute and percentage changes in lipid parameters after 4-week treatment in both groups are listed in Figure 2. Total cholesterol levels after 30 days of therapy were more reduced in the group treated with an intensive daily dose of atorvastatin (reduction of 76±19 mg/dL, 38% in group treated with atorvastatin 80 mg vs 61±26 mg/dL, 27% in the group receiving atorvastatin 20 mg; P=0.001). Levels of LDL-C were more reduced in the first group than in the second: −72±14 mg/dL (55%) vs −55±28 mg/dL 37% (P=0.001). HDL-C increased slightly throughout the population without showing significant differences between the two treatment groups. Triglycerides decreased more in the group treated with atorvastatin 80 mg, though there was no statistically significant difference with the group on low dosage. Figure 2 shows the changes in lipid profiles between the two groups, expressed as a percentage of basal levels.
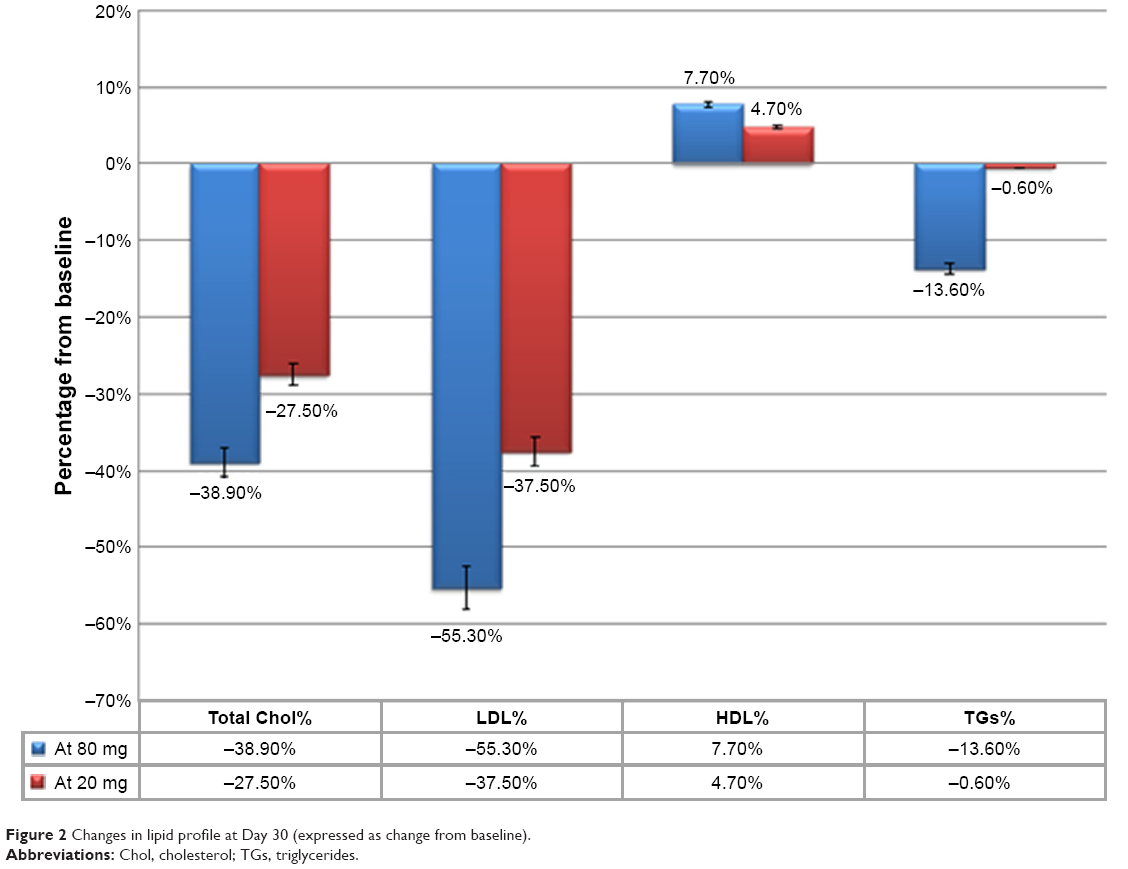 | Figure 2 Changes in lipid profile at Day 30 (expressed as change from baseline). |
Cytokines, inflammatory markers, and oxidized LDL
Plasma levels of HS-CRP, IL6, and TNFα were not significantly different at baseline between the two groups (Tables 3 and 4, Figure 3). HS-CRP levels were reduced in the whole population after 30 days of treatment: 1.88±1.51–0.22±0.31 mg/dL (P=0.0001), but more markedly reduced in the high-dose group than in the moderate-dose group (0.04±0.02 mg/dL vs 0.36±0.3 mg/dL, respectively; P=0.001). IL6 decreased from 7.71±7.44 pg/mL to 2.16±1.2 pg/mL (P=0.0001) in the whole population, and patients treated with atorvastatin 80 mg daily achieved a greater decrease in IL6 (1.12±0.93 pg/mL vs 3.13±2.84 pg/mL, P=0.03). There was a reduction in TNFα in the entire study population (10.36±7.97–8.23±4.26 pg/mL, P=0.07), driven by reduction in the group treated with the higher dose (10.9±6.01–7.54±3.56 pg/mL in atorvastatin 80 mg daily patients [P=0.03] and 9.86±8.14–9.07±4.86 pg/mL [P=0.63] in the atorvastatin 20 mg group). We observed the same finding for ox-LDL (87.25±34.44–49.08±14.01 ng/mL [P<0.0001] in the higher-dosage group and 76.23±35.04–65.14±20.96 ng/mL [P=0.07] in the moderate-dosage group).
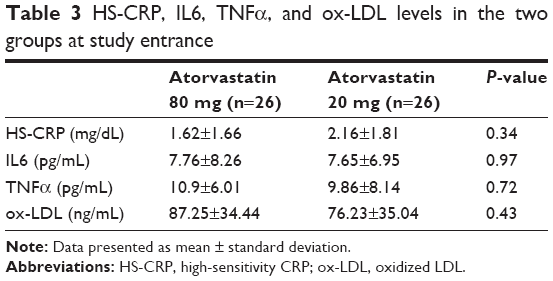 | Table 3 HS-CRP, IL6, TNFα, and ox-LDL levels in the two groups at study entrance |
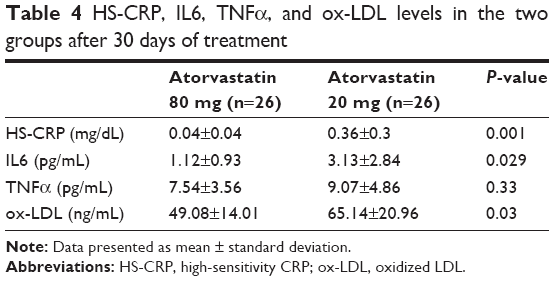 | Table 4 HS-CRP, IL6, TNFα, and ox-LDL levels in the two groups after 30 days of treatment |
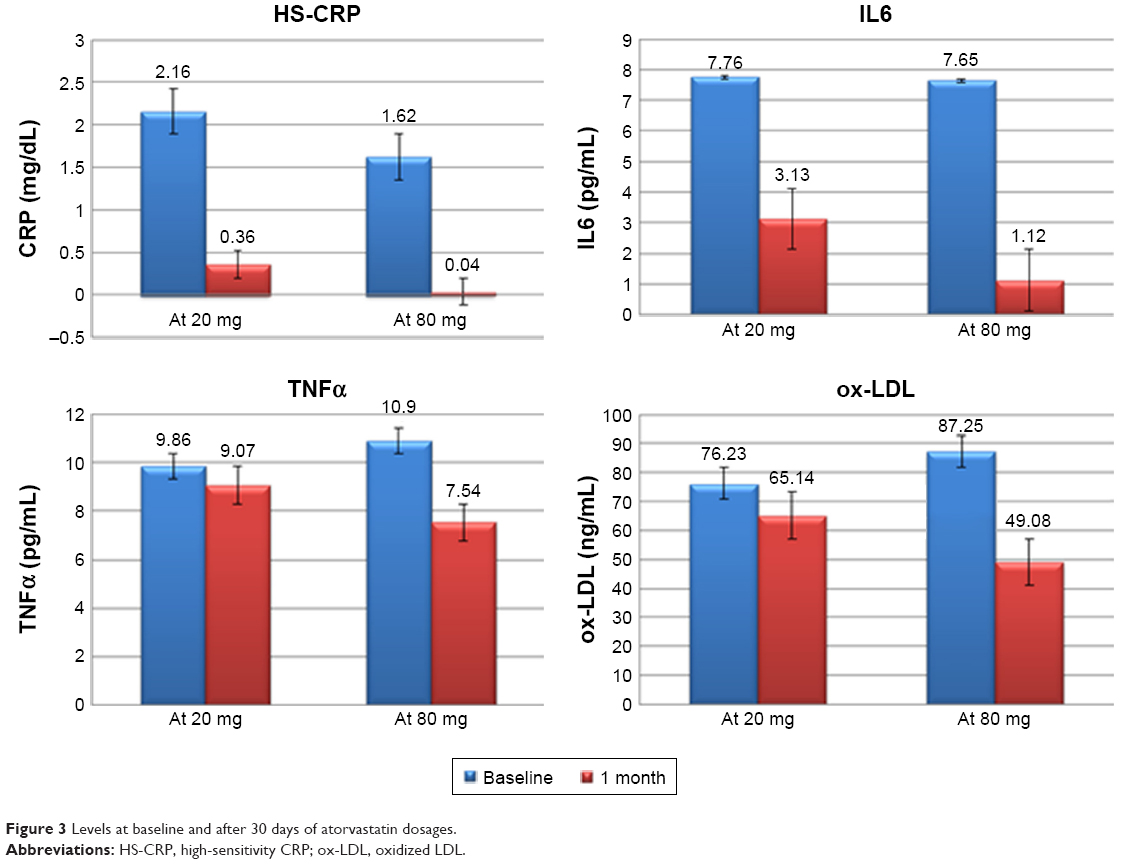 | Figure 3 Levels at baseline and after 30 days of atorvastatin dosages. |
Endothelial function
The mean value of the RHI at baseline was 1.55±0.31 and did not differ between the two groups (Table 5). After 30 days of therapy, the RHI had improved in the whole population, reaching a value of 1.83±0.21 (P<0.0001). In the group treated with the higher dosage, compared with the lower dosage there was a significant increase in the RH-PAT index (1.96±0.16 vs 1.72±0.19, P=0.002) (Table 5 and Figure 4). In patients treated with higher dose of atorvastatin, we observed a significant direct correlation between the RH-PAT index and HS-CRP obtained after 30 days of treatment in the whole population (r=0.5, P=0.01) (Figure 5), while there was no correlation with TNFα, IL6, or ox-LDL values at 30 days.
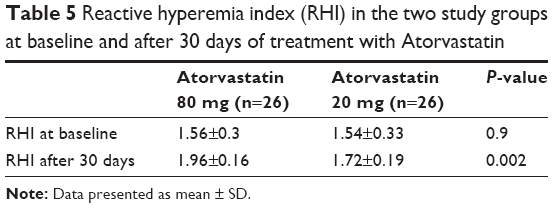 | Table 5 Reactive hyperemia index (RHI) in the two study groups at baseline and after 30 days of treatment with Atorvastatin |
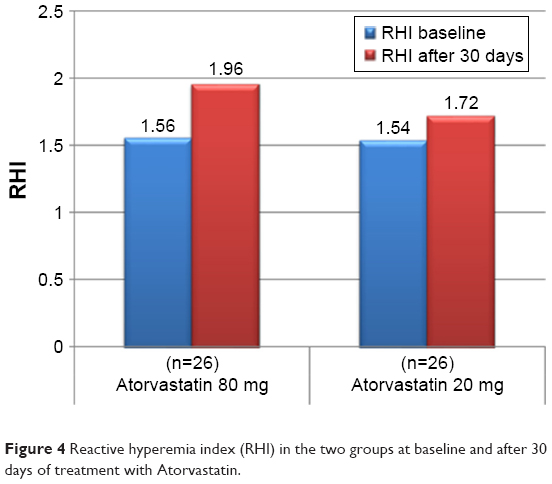 | Figure 4 Reactive hyperemia index (RHI) in the two groups at baseline and after 30 days of treatment with Atorvastatin. |
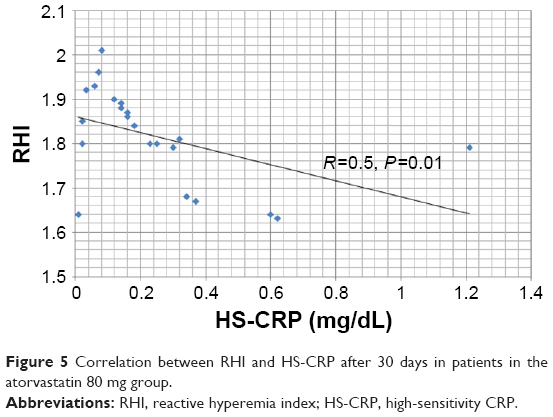 | Figure 5 Correlation between RHI and HS-CRP after 30 days in patients in the atorvastatin 80 mg group. |
Discussion
The present study evaluated the short-term effects of early intensive treatment with atorvastatin compared to moderate-dose atorvastatin on vascular endothelial function and inflammatory markers in patients with STEMI undergoing primary PCI. In our study, 53.8% of patients had endothelial dysfunction at randomization (expressed as RH-PAT index value <1.67). In all patients, we observed an improvement in endothelial function after 1 month, as expected for the period after acute coronary event and coronary stenting, but in the group treated with intensive statin therapy, higher RHI values were reached (1.56±0.3–1.96±0.16 [P<0.0001] in the atorvastatin 80 mg daily group vs 1.54±0.33–1.72±0.19 in the atorvastatin 20 mg daily group [P=0.03]).
Endothelial function provides cardiovascular homeostasis in both primary and secondary prevention.25 Several studies have shown the relationship between noninvasive evaluation of endothelial dysfunction and rate of cardiovascular events in primary prevention26–28 and secondary prevention.29,30 Wang et al found a relationship between endothelial function (measured as flow-mediated dilatation) and cardiovascular events in patients with STEMI undergoing primary PCI,31 affirming the role of evaluation of endothelial function in patients with ACS. The PROSPECT study32 demonstrated that in patients who presented with ACS and underwent PCI, cardiovascular events occurring during follow-up were attributable to both recurrence at the site of the first event (culprit lesion) and to other sites that become vulnerable in the first phase after the coronary event. Demonstration of endothelial dysfunction, even in nonculprit coronary arteries (by angiography), revealed that during the acute phase after ACS, there could exist an injury on the entire coronary tree, and one of the possible mechanisms is impairment of endothelial function.
In this acute vulnerable phase, improvement in endothelial function within some weeks of an event correlates with a lower number of future events.33 A small study found endothelial dysfunction (assessed by acetylcholine test) in 83% of patients with non-STEMI,34 and after a few days of intensive statin therapy (80 mg for 7 days), a trend of improvement in endothelial coronary function, without statistical significance. The authors of this study hypothesized that this failing of significance could have depended on the shortness of the treatment (7 days) in a clinical setting for ACS, in which, as is known, there is a general exacerbation of inflammation. Several studies have confirmed vascular inflammation plays a protagonist role in the development of atherosclerosis and its complications (as in triggering ACS).18–21
Plasmatic levels of markers of systemic inflammation (ie, HS-CRP, IL6, TNFα) have been found to predict adverse cardiovascular events and increase global and cardiovascular mortality.35–37 CRP is an acute-phase reactant that plays an active role in the progression of atherosclerosis through its direct proatherogenic effects on the vasculature.38 This protein is produced chiefly by the liver in response to proinflammatory cytokines and is secreted into systemic circulation.39 Serum CRP is a strong independent predictor of future cardiovascular events. High CRP levels predict an increase in risk that goes beyond classical risk factors.40 Similarly, serum CRP levels are elevated in patients with unstable coronary artery disease, as opposed to those with stable levels.41 It has been reported that in endothelial cells, CRP increases the expression of cell-adhesion molecules and decreases endothelial nitric oxide synthase expression and activity.42,43 Moreover, it promotes the production of reactive oxygen species.44 It should also be considered that compared with non-STEMI, STEMI is characterized by greater inflammatory response, which is exacerbated by mechanical injury due to primary angioplasty and/or stent implantation.15
IL6 is a major inducer of CRP production in the liver, and its elevation is associated with worse short- and long-term prognosis in patients with unstable angina.45 TNFα is a multifunctional circulating cytokine derived from endothelial and smooth-muscle cells and macrophages associated with coronary atheroma.46 It has been demonstrated that plasma concentrations of this factor are persistently elevated among post-MI patients47 and that it is upregulated in the myocardium in response to both transient myocardial ischemia and reperfusion, moreover, persistent overexpression of TNFα after ischemia leads to adverse coronary outcomes.48 ox-LDL can induce atherosclerosis by stimulating monocyte infiltration and smooth-muscle-cell migration and proliferation. Furthermore, it contributes to atherothrombosis by inducing endothelial-cell apoptosis and subsequent plaque erosion, impairing the anticoagulant balance in the endothelium.49
Clinical trials have demonstrated the efficacy of statin therapy in reducing inflammation after ACS.50,51 In our study, both atorvastatin 80 mg daily 20 mg daily reduced inflammatory biomarkers, with better effects reached for each inflammatory biomarker in the higher-dose group. In addition, we observed an interesting inverse correlation between HS-CRP and RHI values at the end of the month of treatment with high-dose statins. As demonstrated in previous work, systemic inflammation and endothelial dysfunction are closely related during acute coronary events,52 resulting in a highly vulnerable state that predisposes to other future plaque ruptures.53 Statins (especially hydrophobic and efficacy molecules, such as atorvastatin)21 have been demonstrated to inhibit the endothelial expression and action of HMG CoA reductase (not only in the liver), so increasing the bioavailability of NO and reducing plasma proinflammatory cytokines (TNFα, IL6) that are responsible for the synthesis and release of HS-CRP by the liver.10 HS-CRP itself plays an active role in perpetuating this pathological circuit, because it reduces eNOS expression,44 among other properties.54 Acute vascular effects of statin therapy could be more important in patients with STEMI, given the presence of microvascular dysfunction and the no-reflow phenomenon, for which one mechanism hypothesized is acute endothelial dysfunction.
Trials with nonstatin lipid-lowering therapies1,55 have been published recently. Both these trials enrolled patients after ACS that were already on statin therapy, and demonstrated that further reduction of LDL-C (less than 50 mg/dL) led to further reduction of coronary events and ischemic stroke without improving total or cardiovascular mortality. Ours and other previous studies confirm the need to treat patients optimally up to the target and even far below (even lower is better) in the setting of acute coronary event and in the immediate post-discharge vulnerable period, titrating dose of statin therapy, as first.
Limitations
Our study did not have the statistical power to evaluate hard end points (cardiovascular events). The small number of patients did not allow more complex statistical analysis, such as multivariate analysis of end points, according to baseline patient characteristics.
Conclusion
In our study, atorvastatin led to a reduction in HS-CRP, IL6, TNFα and improvement in endothelial function in a dose-dependent manner. This could be related to improvements in lipid profile and simultaneously to vascular direct effects that are early and dose-dependent. This finding reaffirms the central role of therapeutic targeting (even lower, even better) in patients presenting with acute coronary events.
Disclosure
The authors report no conflicts of interest in this work.
References
Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–2397. | ||
Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. | ||
de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004;292:1307–1316. | ||
Schwartz GG, Oliver MF, Ezekowitz MD, et al. Rationale and design of the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACLE) study that evaluates atorvastatin in unstable angina pectoris and in non-Q-wave acute myocardial infarction. Am J Cardiol. 1998;81:578–581. | ||
Briel M, Schwartz GG, Thompson PL, et al. Effects of early treatment with statins on short-term clinical outcomes in acute coronary syndromes: a meta-analysis of randomized controlled trials. JAMA. 2006;295:2046–2056. | ||
Patti G, Pasceri V, Colonna G, et al. Atorvastatin pretreatment improves outcomes in patients with acute coronary syndromes undergoing early percutaneous coronary intervention: results of the ARMYDA-ACS randomized trial. J Am Coll Cardiol. 2007;49:1272–1278. | ||
Briguori C, Visconti G, Focaccio A, et al. Novel approaches for preventing or limiting events (Naples) II trial: impact of a single high loading dose of atorvastatin on periprocedural myocardial infarction. J Am Coll Cardiol. 2009;54:2157–2163. | ||
Di Sciascio G, Patti G, Pasceri V, Gaspardone A, Colonna G, Montinaro A. Efficacy of atorvastatin reload in patients on chronic statin therapy undergoing percutaneous coronary intervention: results of the ARMYDA-RECAPTURE (Atorvastatin for Reduction of Myocardial Damage During Angioplasty) randomized trial. J Am Coll Cardiol. 2009;54:558–565. | ||
Sposito AC, Chapman MJ. Statin therapy in acute coronary syndromes: mechanistic insight into clinical benefit. Arterioscler Thromb Vasc Biol. 2002;22:1524–1534. | ||
Ray KK, Cannon CP. The potential relevance of the multiple lipid-independent (pleiotropic) effects of statins in the management of acute coronary syndromes. J Am Coll Cardiol. 2005;46:1425–1433. | ||
Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol. 2005;96:24F–33F. | ||
Marzilli M. Pleiotropic effects of statins: evidence for benefits beyond LDL-cholesterol lowering. Am J Cardiovasc Drugs. 2010;10:3–9. | ||
Schönbeck U, Libby P. Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation. 2004;109:II18–II26. | ||
Dilaveris P, Giannopoulos G, Riga M, Synetos A, Stefanadis C. Beneficial effects of statins on endothelial dysfunction and vascular stiffness. Curr Vasc Pharmacol. 2007;5:227–237. | ||
Di Stefano R, Di Bello V, Barsotti MC, et al. Inflammatory markers and cardiac function in acute coronary syndrome: difference in ST-segment elevation myocardial infarction (STEMI) and in non-STEMI models. Biomed Pharmacother. 2009;63(10):773–780. | ||
Dibra A, Mehilli J, Braun S, et al. Inflammatory response after intervention assessed by serial C-reactive protein measurements correlates with restenosis in patients treated with coronary stenting. Am Heart J. 2005;150:344–350. | ||
Durante A, Camici PG. Novel insights into an “old” phenomenon: the no reflow. Int J Cardiol. 2015;187:273–280. | ||
Kim JS, Kim J, Choi D, et al. Efficacy of high-dose atorvastatin loading before primary percutaneous coronary intervention in ST-segment elevation myocardial infarction: the STATIN STEMI trial. JACC Cardiovasc Interv. 2010;3:332–339. | ||
Aydin MU, Aygul N, Altunkeser BB, Unlu A, Taner A. Comparative effects of high-dose atorvastatin versus moderate-dose rosuvastatin on lipid parameters, oxidized-LDL and inflammatory markers in ST elevation myocardial infarction. Atherosclerosis. 2015;239:439–443. | ||
Matetzky S, Fefer P, Shenkman B, et al. Statins have an early antiplatelet effect in patients with acute myocardial infarction. Platelets. 2011;22:103–110. | ||
Chitose T, Sugiyama S, Sakamoto K, et al. Effect of a hydrophilic and a hydrophobic statin on cardiac salvage after ST-elevated acute myocardial infarction: a pilot study. Atherosclerosis. 2014;237:251–258. | ||
Van de Werf F, Bax J, Betriu A, et al. Management of acute myocardial infarction in patients presenting with persistent ST-segment elevation. Eur Heart J. 2008;29:2909–2945. | ||
Smith SC Jr, Feldman TE, Hirshfeld JW Jr, et al. ACC/AHA/SCAI 2005 guideline update for percutaneous coronary intervention: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/SCAI Writing Committee to Update 2001 Guidelines for Percutaneous Coronary Intervention). Circulation. 2006;113:e166–e286. | ||
Bonetti PO, Pumper GM, Higano ST, Holmes DR Jr, Kuvin JT, Lerman A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J Am Coll Cardiol. 2004;44:2137–2141. | ||
Lüscher TF, Barton M. Biology of the endothelium. Clin Cardiol. 1997;20:II3–II10. | ||
Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23:168–175. | ||
Kuvin JT, Patel AR, Sliney KA, et al. Peripheral vascular endothelial function testing as a noninvasive indicator of coronary artery disease. J Am Coll Cardiol. 2001;38:1843–1849. | ||
Celermajer DS, Sorensen KE, Bull C, Robinson J, Deanfield JE. Endothelium-dependent dilation in the systemic arteries of asymptomatic subjects relates to coronary risk factors and their interaction. J Am Coll Cardiol. 1994;24:1468–1474. | ||
Kuvin JT, Patel AR, Sliney KA, et al. Assessment of peripheral vascular endothelial function with finger arterial pulse wave amplitude. Am Heart J. 2003;146:168–174. | ||
Kitta Y, Obata JE, Nakamura T, et al. Persistent impairment of endothelial vasomotor function has a negative impact on outcome in patients with coronary artery disease. J Am Coll Cardiol. 2009;53:323–330. | ||
Wang X, Guo F, Li G, Cao Y, Fu H. Prognostic role of brachial reactivity in patients with ST myocardial infarction after percutaneous coronary intervention. Coron Artery Dis. 2009;20:467–472. | ||
Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364:226–235. | ||
Elbaz M, Carrié D, Baudeux JL, et al. High frequency of endothelial vasomotor dysfunction after acute coronary syndromes in non-culprit and angiographically normal coronary arteries: a reversible phenomenon. Atherosclerosis. 2005;181:311–319. | ||
Chyrchel M, Dudek D, Rzeszutko L, et al. Effects of short-term anti-inflammatory therapy on endothelial function in patients with non-ST-segment elevation acute coronary syndrome. Cardiovasc Revasc Med. 2011;12:2–9. | ||
Blake GJ, Ridker PM. C-reactive protein and other inflammatory risk markers in acute coronary syndromes. J Am Coll Cardiol. 2003;41:37S–42S. | ||
Suleiman M, Khatib R, Agmon Y, et al. Early inflammation and risk of long-term development of heart failure and mortality in survivors of acute myocardial infarction predictive role of C-reactive protein. J Am Coll Cardiol. 2006;47:962–968. | ||
He LP, Tang XY, Ling WH, Chen WQ, Chen YM. Early C-reactive protein in the prediction of long-term outcomes after acute coronary syndromes: a meta-analysis of longitudinal studies. Heart. 2010;96:339–346. | ||
Scirica BM, Morrow DA. Is C-reactive protein an innocent bystander or proatherogenic culprit? The verdict is still out. Circulation. 2006;113:2128–2134. | ||
Arnaud C, Burger F, Steffens S, et al. Statins reduce interleukin-6-induced C-reactive protein in human hepatocytes: new evidence for direct antiinflammatory effects of statins. Arterioscler Thromb Vasc Biol. 2005;25:1231–1236. | ||
Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation. 2001;103:1813–1818. | ||
Berk BC, Weintraub WS, Alexander RW. Elevation of C-reactive protein in “active” coronary artery disease. Am J Cardiol. 1990;65:168–172. | ||
Venugopal SK, Devaraj S, Yuhanna I, Shaul P, Jialal I. Demonstration that C-reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106:1439–1441. | ||
Zhong Y, Li SH, Liu SM, et al. C-reactive protein upregulates receptor for advanced glycation end products expression in human endothelial cells. Hypertension. 2006;48:504–511. | ||
Huang X, Zhang J, Liu J, et al. C-reactive protein promotes adhesion of monocytes to endothelial cells via NADPH oxidase-mediated oxidative stress. J Cell Biochem. 2012;113:857–867. | ||
Biasucci LM, Vitelli A, Liuzzo G, et al. Elevated levels of interleukin-6 in unstable angina. Circulation. 1996;94:874–877. | ||
Kukeilka GL, Smith CW, Manning AM, Youker KA, Michael KH, Entman ML. Induction of interleukin synthesis in the myocardium: potential role in postreperfusion inflammatory injury. Circulation. 1995;92:1866–1875. | ||
Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol. 1995;146:419–428. | ||
Ridker PM, Rifai N, Pfeffer M, et al. Elevation of tumor necrosis factor-a and increased risk of recurrent coronary events after myocardial infarction. Circulation. 2000;101:2149–2153. | ||
Ho E, Galougahi KK, Liu CC, et al. Biological markers of oxidative stress: applications to cardiovascular research and practice. Redox Biol. 2013;1:483–491. | ||
Ray KK, Morrow DA, Shui A, Rifai N, Cannon CP. Relation between soluble intercellular adhesion molecule-1, statin therapy, and long-term risk of clinical cardiovascular events in patients with previous acute coronary syndrome (from PROVE IT-TIMI 22). Am J Cardiol. 2006;98:861–865. | ||
Kinlay S, Schwartz GG, Olsson AG, et al. Effect of atorvastatin on risk of recurrent cardiovascular events after an acute coronary syndrome associated with high soluble CD40 ligand in the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) study. Circulation. 2004;110:386–391. | ||
Cleland SJ, Sattar N, Petrie JR, Forouhi NG, Elliott HL, Connell JM. Endothelial dysfunction as a possible link between C-reactive protein levels and cardiovascular disease. Clin Sci (Lond). 2000;98:531–535. | ||
Peng HB, Libby P, Liao JK. Induction and stabilization of IκBα by nitric oxide mediates inhibition of NF-κB. J Biol Chem. 1995;270:14214–14219. | ||
Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000;102:2165–2168. | ||
Sabatine M, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.