")
Back to Archived Journals » Research and Reports in Biology » Volume 6
Hedgehog-Gli signaling in basal cell carcinoma and other skin cancers: prospects for therapy
Authors Pandolfi S, Stecca B
Received 3 December 2014
Accepted for publication 6 March 2015
Published 8 May 2015 Volume 2015:6 Pages 55—71
DOI https://doi.org/10.2147/RRB.S60262
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Zvi Kelman
Silvia Pandolfi,1 Barbara Stecca1,2
1Laboratory of Tumor Cell Biology, Core Research Laboratory, Istituto Toscano Tumori, 2Department of Oncology, Azienda Ospedaliero- Universitaria Careggi, Florence, Italy
Abstract: The Hedgehog (Hh) signaling pathway is of critical importance during embryonic development, where it directs a number of cellular processes, including cell proliferation, differentiation, and patterning. In normal adult tissues, Hh signaling is mostly involved in stem cell maintenance, tissue repair, and regeneration. Over the last two decades, aberrant activation of Hh signaling has been linked to several types of cancer, including those of the skin. In particular, the critical role of Hh signaling in the development of basal cell carcinoma has been demonstrated by several mouse models and genetic mutation analyses. In addition, several clinical trials using Hh signaling inhibitors have been shown to be effective treatments in basal cell carcinoma. Recent evidence indicates that activation of the Hh pathway plays an important role in other types of human skin cancer, such as melanoma and cutaneous squamous cell carcinoma. In this review, we provide an overview of the roles of Hh pathway in skin cancers, including basal cell carcinoma, melanoma, and squamous cell carcinoma. Finally, we discuss the rapid development of drugs that target the Hh pathway and the implications for skin cancer therapy.
Keywords: Hedgehog, Gli transcription factors, basal cell carcinoma, melanoma, squamous cell carcinoma, small-molecule inhibitors
Introduction
Major progress has been made in our understanding of how the Hedgehog (Hh) pathway operates since the discovery in 1980 of Hh as a segment polarity gene in Drosophila.1 Several years passed before the finding that inactivation of the Sonic Hedgehog gene is responsible for the hereditary developmental disorder holoprosencephaly.2,3 Since then, Hh signaling has been shown to play a critical role not only in embryonic development, but also in other normal processes, such as regulation of stem cells and maintenance of tissue homeostasis.4 The initial association between the Hh pathway and human cancers was made as a result of the discovery that loss-of-function mutations in human Ptch1 are associated with Gorlin syndrome, a rare and hereditary disorder. Patients with Gorlin syndrome show a broad spectrum of developmental defects and have a predisposition to develop basal cell carcinoma (BCC) and medulloblastoma.5 During the past 15 years, numerous studies have revealed aberrant activation of Hh signaling not only in BCC, but also in medulloblastoma, glioblastoma, leukemia, and gastrointestinal, lung, ovarian, breast, liver, pancreatic, and prostate cancers.6,7 Recent evidence indicates that activation of Hh signaling plays an important role not only in BCC but also in other types of skin cancer, such as melanoma and squamous cell carcinoma (SCC). Here, we focus on the role of Hh signaling in the development of skin cancer and review what has been learned from experimental mouse models carrying genetic modifications and from preclinical studies using specific Hh inhibitors. Lastly, we discuss the rapid development of drugs that target the Hh pathway and the implications for skin cancer therapy.
Hedgehog signaling in vertebrates
Hh signaling is highly conserved from Drosophila to humans,8 although there are significant differences between vertebrates and invertebrates. Ptch and SMO proteins are conserved and maintain their function in mammals, whereas the Hh ligand has diversified into Sonic (Shh), Indian (Ihh), and Desert (Dhh) Hedgehog, and the function of the downstream transcription factor Cubitus interruptus (Ci) has multiplied into three Gli proteins (Gli1, Gli2, and Gli3). Figure 1 shows a simplified view of the Hh signaling pathway in vertebrates.
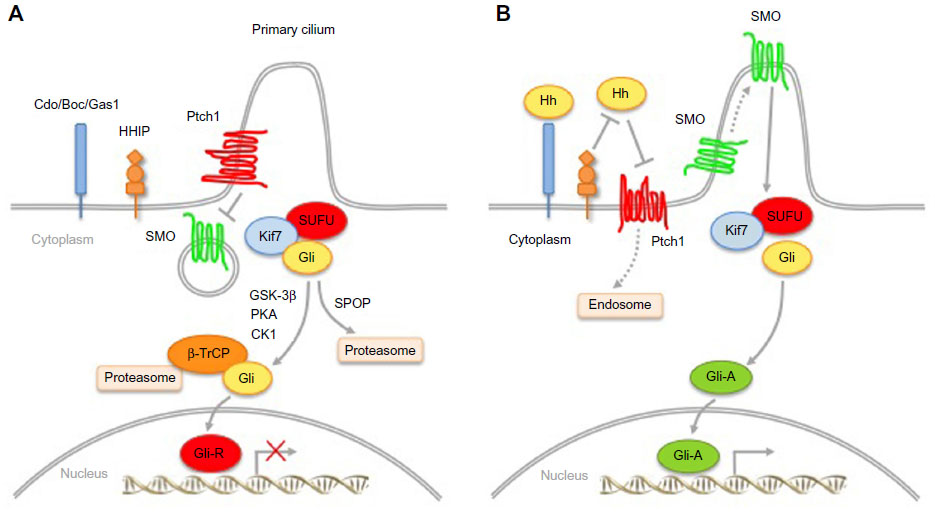 | Figure 1 Simplified Hedgehog (Hh) signaling pathway in mammals. |
Signal transduction depends on the secretion of Hh ligands from the producing cell. Hh ligands are synthesized as precursors, which undergo autoproteolytic cleavage to form amino-terminal protein fragments. This cleavage is essential for the covalent attachment of a cholesterol molecule at the carboxyl terminus9 and palmitic acid at the amino terminus10 by Skinny, the Hh acyltransferase.11 These lipid modifications are required for the correct movement and reception of the ligands.12,13 Secretion of mature Hh ligands is mediated by Dispatched (Disp), a 12-transmembrane protein with structural homology to Ptch.14,15 In addition to Disp, several other proteins are involved in this process, including the proteoglycans Dally and Dally-like,16–18 Tout-velu, and Sulfateless.19,20
The signaling cascade of the Hh pathway is initiated by binding of Hh ligands to the 12-pass transmembrane protein receptor Ptch, which resides in the primary cilium, a non-motile structure that plays an important role in the transduction of Hh signaling.21,22 Several factors are involved in the binding of Hh ligands to Ptch. The Hh-interacting protein (HHIP) can compete with Ptch for Hh binding, thus acting as a negative regulator of Hh signaling.23 Conversely, Cdo, Boc, Gas1, and glypican-3 act as coreceptors of Hh.24–29 Binding of Hh ligands to Ptch leads to relocalization of the ligand/receptor complex from the primary cilium to the endosomal vesicles and induces a conformational change of SMO.21 Activated SMO then translocates into the cilium30 and triggers a series of intracellular events that promote the formation of Gli activator forms (Gli-A). Gli2/3-A translocate into the nucleus and induce Hh target genes31,32 (Figure 1A). In the absence of ligands, Ptch inhibits activation of the pathway by preventing SMO from entering the cilium. This results in phosphorylation and proteasome-mediated carboxyl cleavage of Gli3 and, to a lesser extent, of Gli2 to repressor forms (Gli2/3-R);33,34 Gli1 is transcriptionally repressed, with consequent silencing of the pathway (Figure 1).
The final effectors downstream of SMO in the mammalian Hh signaling pathway are the Gli transcription factors (Gli1, Gli2, Gli3), all members of the Kruppel family. They share five conserved C2-H2 zinc-finger DNA binding domains and a consensus histidine/cysteine linker sequence between zinc fingers. The Gli factors recognize the consensus sequence 5′-GACCACCCA-3′ in the promoter of their target genes,35 although they can bind to variant Gli binding sites with lower affinity, still leading to strong transcriptional activation.36
Gli1 is a potent transcriptional activator and a direct target of Gli2.37 Being directly regulated by Hh signaling, Gli1 represents the best read-out of Hh pathway activation.38 Gli2 has a N-terminal repressor domain and a C-terminal activator domain. Gli2 can act as an activator or, in its C-terminal deleted form, as a repressor. Gli3 acts mostly as a repressor in its C-terminal cleaved form, although it can also have positive effects.39 The activity of the Gli transcription factors is regulated by a number of post-translational modifications, including cytoplasmic-nuclear shuttling, phosphorylation, acetylation, ubiquitination, and protein degradation.
Suppressor of Fused (SUFU) is the main negative regulator of Hh signaling, and controls Gli nuclear localization and transcriptional activity;40,41 in turn, Hh signaling regulates SUFU activity by inducing its turnover via the ubiquitin-proteasome system.42 Protein kinase A (PKA) can retain Gli1 in the cytoplasm, inhibiting its transcriptional activity.43 Gli2 and Gli3 processing is triggered by PKA-dependent phosphorylations, which are required for subsequent casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3beta) phosphorylations and recruitment of the β-transducin repeat-containing protein (β-TrCP) ubiquitin ligase.33,34,44,45 In this context, Kif7 plays a regulatory role in controlling the efficient relocalization of Gli3 to the cilium in response to Shh and its processing to Gli3-R.46 The dual specificity Yak-1-related kinases 1 (Dyrk1) and 2 (Dyrk2) modulate the Hh pathway in opposite ways. Dyrk1 increases Gli1 nuclear retention and transcriptional activity,47 whereas Dyrk2 directly phosphorylates Gli2 and induces its degradation by the ubiquitin-proteasome system.48 The serine/threonine unc-51-like kinase 3 enhances Gli1 (and Gli2) transcriptional activity.49 Atypical protein kinase C (aPKC) ι/λ50 and the downstream effector of the mammalian target of rapamycin (mTOR) pathway ribosomal protein S6 kinase 1 activate Gli1.51 Deacetylation of Gli1 and Gli2 by histone deacetylase 1 increases their transcriptional activity.52
Degradation is also important for Gli1; two degradation sequences, degron N and degron C, mediate recognition by the β-TrCP E3 ubiquitin ligase to allow ubiquitination and degradation by the proteasome.53 Gli1 is also targeted for proteolysis by Itch, another E3 ubiquitin ligase.54 Similarly, Ci/Gli can be degraded through the ubiquitin E3 ligase adaptors Roadkill and HIB/SPOP in an Hh-dependent manner,55,56 the latter being mediated by multiple Ser/Thr-rich degrons.57 Upon genotoxic stress, p53 induces the acetyltransferase p300/CREB-binding protein (CBP)-associated factor (PCAF), identified as a novel E3 ubiquitin ligase targeting Gli1 for proteasomal degradation.58
Skin cancers linked to aberrant Hedgehog pathway activity
Skin cancer is by far the most frequent cancer worldwide and its incidence is increasing every year. Most of the insights into the role of Hh signaling in human cancers came from studies on BCC. However, recent evidence indicates that activation of the Hh pathway plays an important role in other types of human skin cancer, including melanoma and SCC.
Basal cell carcinoma
BCC is the most frequent form of human cancer. The incidence of BCC is strongly associated with exposure to ultraviolet radiation, and it often develops in elderly people with fair skin phototypes, especially on the head and neck. Additional risk factors include ionizing radiation, arsenic exposure, smoking, and immunosuppression.59,60 Although disfiguring if allowed to grow, BCCs are benign tumors that rarely metastasize beyond the primary tumor site. The most common histological subtypes are nodular and superficial, both showing a less aggressive clinical course. On the other hand, micronodular, infiltrative, and mixed morphological variants are more aggressive. Clinically, BCCs appear as a pearly papule or nodule with overlying telangiectasias and rolled borders, with or without ulceration, and may be pigmented.59,60
The first link between Hh signaling and cancer came from the finding that loss-of-function mutations in the Ptch1 gene on chromosome 9q22 were the cause of the nevoid BCC syndrome (NBCCS) or Gorlin syndrome.61–63 NBCCS is an autosomal dominant disorder strongly predisposing to the development of BCC at a young age.5 Moreover, these patients show a broad spectrum of developmental defects and a high incidence of other neoplasms, particularly medulloblastoma, meningioma, ovarian and heart fibroma, fetal rhabdomyoma, and rhabdomyosarcoma.64 In BCC from NBCCS patients, it was found that one allele of the Ptch1 gene is mutated and the other allele is deleted, showing for the first time that Ptch1 behaves as a classical tumor suppressor according to Knudson’s two-hit model.65 More recently, a truncating germline mutation in SUFU was found in a family with features of Gorlin syndrome, presenting medulloblastoma but not BCC. Of note, no Ptch1 mutations were detected in this family, suggesting that mutations in SUFU might be another cause of Gorlin syndrome.66
Later, it was found that sporadic BCCs also have a high frequency of loss-of-function mutations in Ptch1 and, to a lesser extent, activating mutations in SMO (Table 1). Notably, the downstream effectors of the Hh pathway (Gli1/2/3) are rarely found to be mutated in sporadic BCCs (Table 1). Inactivating mutations in Ptch1 occur in about 70%–80% of BCCs, and they mostly produce a truncated protein or are frameshift or missense mutations.62,63,67–71 A nonsense mutation of Ptch1 (Q688X) produces a truncated form of the protein, which enhances Gli1 activity independent of stimulation with Shh.72 Recently, three different splice site mutations have been described in sporadic BCCs.71 In another fraction of BCCs, activation of Hh signaling results from gain-of-function mutations in SMO (about 6%–21%),70,73–75 or less frequently from inactivating mutations of SUFU.70 Mutational analysis identified recurrent mutations in SMO, such as the G-to-A transition at base pair 1,685 of exon 10, which produces the amino acid change of Arg to Gln at codon 562 (activating mutation M1)73 or G-to-T transversion at base pair 1,604 of exon 9, which changes codon 535 from Trp to Leu (activating mutation M2).73,75 Both hot spot mutations lead to constitutive activation of Hh signaling. Recently, a translocation in Shh has been identified in a sporadic case of BCC. The translocation occurs between chromosomes 7 and Y, and fuses the middle of the Shh promoter with Y chromosome sequences, leaving 140 kb of regulatory sequences upstream of the Shh transcription start site. The authors demonstrated that the mutant promoter drives high expression of Shh protein in the skin,76 in contrast with the absence of expression of Shh in sporadic BCCs with Ptch1 and SMO mutations.77
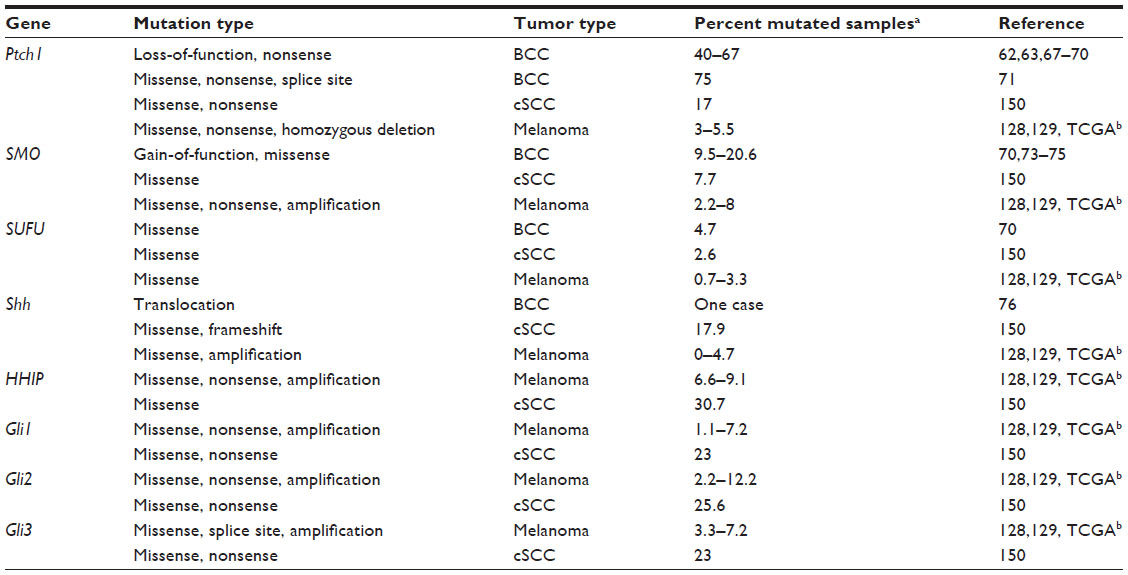 | Table 1 Genetic aberrations in the Hedgehog pathway identified in skin cancers |
Mouse models of BCC
Genetic data in patients have established the role of the Hh pathway in BCC; nevertheless, in the last few years, several mouse models using tissue-specific activation of the Hh pathway have provided a powerful tool to investigate the mechanisms of Hh-mediated development of BCC and to identify the BCC cell of origin. All current BCC mouse models target different components of Hh signaling, from the Shh ligand, to the transmembrane proteins Ptch and SMO, SUFU, and the Gli transcription factors, and are summarized in Table 2.
 | Table 2 Mouse models of BCC with genetically modified Hedgehog pathway components |
Ptch1+/− mice kept under normal conditions rarely develop full-blown BCCs; rather, they show skin proliferation similar to human basaloid follicular hamartoma.78 BCCs occur only after exposure to ultraviolet ionizing radiation78,79 similar to development of BCC in NBCCS patients, suggesting that ultraviolet exposure is a very important risk factor for BCC. Because homozygous Ptch1 knockdown leads to embryonic lethality due to heart and neural tube closure defects,80–82 skin-specific knockout of Ptch1 has been generated. Combining conditional Ptch1 knockout driven by K6a-Cre, an inducible K6a promoter, Ptch1-/- mice developed BCC-like lesions.83 Similarly, the use of other skin-specific Cre strains to drive homozygous Ptch1 ablation, such as K14-cre and Mx1-Cre, or Ptchflox/flox mice crossed with Rosa26CreERT2 mice (R26-CreERT2+/−) produced BCC lesions.84,85 Interestingly, in the latter model, 100% of animals develop BCCs with features of the nodular subtype, are noninvasive, and are characterized by strong Hh pathway activation, as witnessed by abundant expression of Gli1 and Ptch1.85 In a similar model, Ptchflox/flox mice were crossed with K5-CreERT mice, which express the transgene in cells of the basal layer of the skin. All mice develop BCC lesions, but the K5-CreERT is highly leaky, resulting in formation of BCCs even without Cre activation.86 In addition to Ptch1 inactivation, overexpression of Gli transcription factors leads to BCC. Ectopic expression of Gli1 in the embryonic frog epidermis leads to formation of epidermal tumors87 and its overexpression in the mouse epidermis drives formation of BCC-like tumors.88 Likewise, overexpression of Gli2 in mouse skin leads to development of BCC,89 and sustained Hh signaling appears to be required for growth of BCC in a mouse model allowing conditional Gli2 expression, because transgene inactivation leads to BCC regression.90 Similarly, K14-Shh and K5-SMO-M2 transgenic mice develop BCC-like tumors at an early age.73,91 However, expression of mutant human SMO under the control of a truncated Keratin 5 (K5) promoter (ΔK5-SMO-M2) is not sufficient for development of full-blown BCCs.92 Skin-specific knockin SMO-M2 mice (CAGGS-Cre; R26-SMO-M2 or K14-CreERT; R26-SMO-M2) develop multiple BCCs.93,94 Sufu+/− mice develop basaloid follicular hamartoma lesions similar to those seen in Ptch1+/− mice, but more frequently,95 and even compound Ptch1+/−; SUFU+/− mice lack signs of full-blown BCC lesions.96 This suggests that the combined reduction in gene dosage of these two negative regulators is still insufficient to reach the threshold of Gli activity required for development of BCC. The current view is that increasing levels of Hh pathway activation determines various stages in a spectrum ranging from benign hamartomas to malignant BCC lesions. For instance, comparing two different BCC models, Grachtchouk et al found that K5-Gli2 mice with strong Hh signaling developed full-blown BCCs,89 while the weaker Hh signal in ΔK5-SMO-M2 mice results in follicular hamartomas.92
As mentioned earlier, primary cilia are an important organelle for Hh signal transduction in mammals. Primary cilia contain multiple components of the Hh pathway, including Shh, Ptch1, SMO, and Gli, and are present in the majority of human BCCs. The question of whether primary cilia are required for Hh-driven development of BCC has been addressed using a tissue-specific knockout. Genetic disruption of cilia formation by conditional ablation of Kif3a and of Ift88, both of which required for ciliogenesis, decreases formation of BCC in K14-CreERT; R26-SMO-M2 mice, but increases tumor formation in a Gli2 conditional transgenic model.94 This apparently paradoxical effect suggests that cilia play a dual role in Hh-mediated tumorigenesis, which could be explained by the fact that cilia are required for formation of both repressor and activator forms of Gli.97–99
Cell of origin of BCC
The cellular origin of BCC has been debated for a long time. It has been proposed that BCCs may arise from the outer root sheet or bulge of the hair follicle, and stem and progenitor cells are thought to be the probable source of initiation of BCC due to their long life and self-renewal ability. However, recent studies using Cre-mediated cell-specific targeting both by lineage tracing and specific activation of Hh signaling in distinct skin cell populations gave unexpected results (Table 3). The first of these studies, using cell-specific Cre to activate expression of R26-SMO-M2 in mice, localized the cell of origin of BCC in the long-term resident progenitor cells of the interfollicular epidermis (IFE) and the upper infundibulum.100 However, K15-Cre and K14-Cre; R26-SMO-M2 mice, where SMO-M2 is mostly targeted in stem cells and transient amplifying progenitors of the hair follicle (HF), show only basaloid lesions and never BCCs.100 In contrast, using cell fate tracking in Ptch1+/− mice exposed to X-ray, Wang et al101 demonstrated the BCC cell of origin in keratin 15-positive stem cells of the HF bulge. Interestingly, loss of p53 in these mice was associated with enhanced BCC formation, not only from the HF bulge but also from the IFE.101 Using conditional expression of SMO-M2 in keratin 15-positive cells, another group showed that wounding recruits HF SMO-M2-expressing cells to the wound site (IFE), where they give rise to superficial BCC-like tumors.102 The same type of tumor was observed upon conditional expression of SMO-M2 in keratin 14-positive cells, even in the absence of wounding.102 Conditional overexpression of Gli1 or homozygous deletion of Ptch1 under the control of the K5 promoter results in BCC formation, preferentially in the IFE but also in the HF.103 Lineage tracing of Lgr5+ stem cells showed that HF-associated and IFE-associated lesions had distinct cells of origin and that Lgr5-labeled HF cells were able to give rise to BCCs in the IFE upon wounding,103 in agreement with Wong and Reiter.102 Another study revealed that low levels of Gli2ΔN expression in the basal compartment do not lead to nodular BCCs in the HF, but to slow-growing basaloid follicular hamartomas resembling tumors found in ΔK5-SMO-M2 mice.104 These studies suggest that BCCs can arise from cells competent to receive Hh signal and able to activate Gli transcription factors.
 | Table 3 Mouse models used to identify the BCC cell of origin |
Hh targets and interaction with other pathways in BCC
Both Gli1 and Gli2 are highly expressed in human BCCs.87,105 In particular, Gli2 directly regulates the expression of Gli1, further activating the Hh pathway.37 Gli2 silencing reduces growth of BCC xenografts by decreasing vascularization and increasing apoptosis.106 Activation of the Hh pathway might exert its mitogenic effect on keratinocytes by activation of several targets. For instance, Gli1 can induce expression of platelet-derived growth factor receptor alpha107 and FOXM1,108 and Gli2 activates FOXE1.109 Gli2 has been shown to activate the antiapoptotic factor Bcl-2110 and to counteract death ligand-mediated apoptosis by inducing expression of the caspase-8 inhibitor c-FLIP,111 suggesting an antiapoptotic role in BCC. Hh signaling enhances ribosomal RNA transcription in BCC by increasing basonuclin gene expression.112 In addition, insulin-like growth factor binding protein 2 is upregulated in both murine and human BCCs and has been shown to play a role in Hh-mediated expansion of epidermal progenitor cells in K14-Cre; Ptch1fl/fl skin explants.113
Hh signaling has been shown to cross-talk with other signaling pathways. The Wnt pathway plays a critical role in development of the HF, and both human and murine BCCs show increased levels of β-catenin.114 Conditional overexpression of the Wnt pathway antagonist Dkk1 results in inhibition of Hh-driven benign hamartomas.115 The epidermal growth factor receptor/MEK/ERK pathway has been shown to modulate Gli-dependent transcription in human keratinocytes116 and to synergize with Hh signaling in inducing oncogenic transformation of human keratinocytes through activation of c-Jun.117 In accordance with these studies, an increase of c-Jun upon Hh or epidermal growth factor receptor activation inhibits the tumor suppressor miR-203, which in turn represses c-Jun, creating a negative regulatory loop.118 Interestingly, Hh and epidermal growth factor receptor signaling synergistically activate a number of cooperation response genes, including SOX2, SOX9, JUN, CXCR4, and FGF19, which are required for growth of BCC in vivo.119 In addition, loss of p53 has been shown to accelerate tumorigenesis of BCC in Ptch1+/− mice,101 likely through Gli1 activation.120
Melanoma
Melanoma is the most aggressive form of skin cancer and originates from the malignant transformation of melanocytes or neural crest-derived precursors.121 For the purpose of this review, we focus on malignant melanoma affecting the skin, ie, cutaneous melanoma (herein referred to as melanoma). While melanoma accounts for less than 10% of all skin cancers, it is responsible for more than 75% of skin cancer-related deaths.122 The overall 5-year survival rate for patients with localized melanoma is about 98%, but it falls to 62% and 16% for patients with lymph nodes and distant metastases, respectively.123 Cutaneous melanomas harbor mutually exclusive activating mutations in BRAF and NRAS, occurring respectively in 50% and 15%–20% of cases, and deletion of the CDKN2A locus.124–126 Recent exome sequencing studies have identified a variety of additional alterations, including mutations in GRIN2A, PPP6C, and KIT, and amplification of TERT, CCND1, KIT, and MITF-M.127–129
The importance of Hh signaling in melanoma tumorigenesis was not revealed until recently (Table 4). Our group was the first to demonstrate that human melanomas express Hh pathway components, and they require active Hh signaling for growth and proliferation. We showed that melanoma cells, but not the surrounding stroma, express Shh, Gli1, and Ptch1 and that melanoma cells in vitro respond to inhibition of the Hh pathway through cyclopamine (a SMO antagonist) or silencing of Gli1 and Gli2 by drastically decreasing their proliferation. Importantly, treatment with cyclopamine or silencing of SMO reduced tumor growth in an orthotopic xenograft model and abolished tumor recurrence, and systemic treatment with cyclopamine prevented metastatic growth in the lungs of mice.130 These effects appear to be specific, because Gli1 epistatically rescues the inhibitory effect of cyclopamine on cell proliferation, and the latter mimics inhibition of SMO via RNA interference.130 In addition, we showed that Hh signaling is required also in tumors induced by oncogenic NRAS in a Tyrosinase-NRASQ61K; Ink4a−/− mouse model. Our study provided the first evidence that endogenous Ras-MEK and AKT signaling regulate the nuclear localization and transcriptional activity of Gli1 in melanoma cells.130
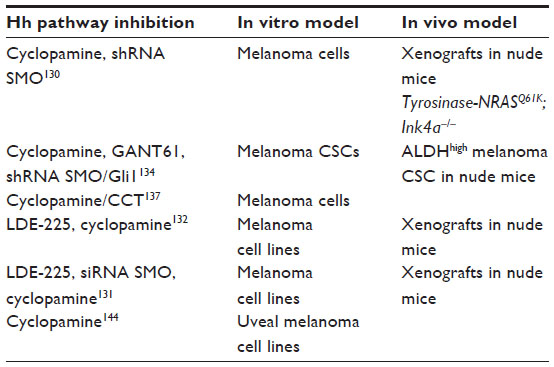 | Table 4 Experimental in vitro and in vivo models in which interference with the Hedgehog pathway reduces growth of melanoma cells |
Two recent studies confirmed and extended our previous findings (Table 4). In one study, it was shown that Hh pathway members are highly expressed in a subset (50%) of melanoma cell lines; in particular SMO, Gli2, and Ptch1 are upregulated, while the negative regulators of Hh signaling, PKA and DYRK2, are downregulated when compared with melanocytes.131 Interestingly, high Hh pathway activity is associated with decreased post-recurrence survival in patients with metastatic melanoma.131 The second study found that expression of Gli1 is higher in human primary melanoma harboring BRAF(V600E) mutation than in those with wild-type BRAF.132 These studies also showed that LDE-225, a well tolerated oral SMO antagonist currently in Phase II clinical trials for advanced BCC (see later), reduces proliferation of human melanoma cell lines by inducing apoptosis, and decreases human melanoma xenograft growth in nude mice.131,132 Interestingly, a study reported by O’Reilly et al131 showed a more dramatic proliferative effect in BRAF mutant cell lines than in wild-type BRAF cells and a modest but significant effect of combining BRAF and Hh inhibitors, suggesting that combined therapy targeting mutant BRAF and Hh could be beneficial in patients with mutated BRAF and activated Hh signaling. Interestingly, inhibition of Hh signaling with LDE-225 leads to upregulation of the programmed cell death mediator XAF1 and apoptosis in vitro and in vivo.131
Multiple lines of evidence indicate that Hh signaling regulates cancer stem cells (CSCs) in several types of cancer,133 including melanoma. Putative melanoma CSCs with high aldehyde dehydrogenase activity have been shown to require Hh signaling, because pharmacological inhibition of the Hh pathway with cyclopamine or GANT61 (a Gli1/2 inhibitor) and genetic silencing of SMO and Gli1 drastically reduces self-renewal of melanoma CSCs and tumorigenicity in vivo.134 The critical role of Hh in CSCs is elicited through the subverted regulation of stemness genes, such as Nanog and SOX2,135,136 which are overexpressed in certain types of cancer. For instance, it was recently shown by our group that both Gli1 and Gli2 bind to the SOX2 promoter in melanoma cells and that SOX2 function is required for Hh pathway-mediated self-renewal of melanoma CSCs.136
Activation of Hh signaling in melanoma can be mediated by the oncogenic phosphatase WIP1, which increases the stability and transcriptional activity of Gli1 in patient-derived primary melanoma cells.137 Ectopic expression of Gli1 or of an active form of Gli2 (ΔNGli2) in N/TERT human keratinocytes increases their resistance to apoptosis induced by ultraviolet B and DNA-alkylating agents and induces epithelial-to-mesenchymal transition.138 The Hh pathway is also involved in resistance to treatment with BRAF inhibitors, because it mediates upregulation of platelet-derived growth factor receptor alpha observed in BRAF inhibitor-resistant cells, and inhibition of the Hh pathway restores the sensitivity of the cell to BRAF inhibitors.139
Hh signaling also plays a role in the progression of melanoma by contributing to the acquisition of invasive behavior. Melanoma cells with high Gli2 expression are characterized by an invasive and metastatic phenotype associated with loss of E-cadherin and secretion of metalloproteases, and metastasize to bone more quickly than cells with low Gli2 expression.140 Gli2 represses MITF-M by direct binding to its promoter and contributes to the loss of melanocytic differentiation markers.141,142 Gli1 directly induces osteopontin, and their high expression levels correlate with tumor progression and metastasis in human melanoma.143 Likewise, inhibition of Hh signaling in uveal melanoma cell lines decreases cell viability, epithelial-mesenchymal transition, and angiogenesis.144
Hh pathway mutations in melanoma have been identified in two whole-exome sequencing studies128,129 and in The Cancer Genome Atlas Research Network (http://cancergenome.nih.gov/), where 91, 121, and 278 melanoma samples were respectively analyzed. Interestingly, data from The Cancer Genome Atlas indicate that 35% of patients show one alteration (mutation or copy number variation) in at least one component of the Hh pathway, with co-occurrence of amplifications in SMO and Shh (P<0.001), co-occurrence of mutations in Gli1 and Gli2 (P<0.005), and mutations in Ptch1 and HHIP (P=0.014).145,146 These alterations are predominantly missense mutations and amplifications, and all occur in the main components of the Hh pathway, from Ptch1 and SMO to the downstream effectors Gli1/2/3, in contrast with BCC mutations, which are mostly found in Ptch1 and SMO (Table 1). The relevance of these mutations in melanoma remains to be determined, and only further functional studies will shed light on their impact on the development and progression of melanoma. Unlike in BCC, no genetic mouse models have been established thus far for Hh pathway-mediated development of melanoma. Nevertheless, K5-Gli2 transgenic mice form hyperpigmented BCC-like tumors,89 and ΔK5-SMO-M2 transgenic mice92 and aging mice lacking one Sufu allele95 develop skin pigmentation. In addition, injection of Gli1 mRNA into the epidermis/neural crest of Xenopus embryos induces formation of pigmented epithelial tumors expressing high levels of the melanoma marker Mitf.130
Squamous cell carcinoma
SCC originates from the squamous epithelium of the skin, but can occur also in the lungs, oral mucous membranes, esophagus, cervix, bladder, and genitals. SCCs usually appear in areas exposed to the sun and look like scaly red patches, open sores, elevated growths with a central depression, or warts. SCC is the second most common cancer of the skin after BCC. However, SCC is more aggressive than BCC, because it grows faster and has higher rates of metastasis and mortality.147
Although the link between aberrant activation of the Hh pathway and SCC is not as strong as that in BCC, a number of studies have shown a potential role of the Hh pathway in different types of SCC. However, for the purpose of this review, the focus is on skin SCC. Ping et al148 provided the first evidence that Ptch is mutated in a subset of skin SCC from individuals with a history of multiple BCC. Later, a high prevalence of allelic loss at 9q22.3, including Ptch, was demonstrated in skin SCC.149 A recent exome sequencing analysis in 39 cases of aggressive cutaneous SCCs identified missense mutations in most of the components of Hh signaling, including the Gli transcription factors150 (Table 1). However, these mutations so far are of unknown significance. Evidence of activation of Hh signaling in skin SCC derives mainly from immunochemistry studies, which reveal high expression of major components of Hh signaling in the tumor compared with control tissues.151,152
While the role of Ptch1 in BCC tumor suppression is clear, mouse models suggest an opposing function of Ptch1 in SCC. Wakabayashi et al153 reported that a single polymorphism at the Ptch1 C-terminus (T1267N) confers increased susceptibility to Ras-induced tumor formation in FVB mice. Overexpression of the Ptch1FVB allele driven by the K14 promoter (K14-Ptch1FVB) is sufficient to drive Ras-induced formation of early post-natal SCC in mice with C57BL/6 background, but is not required for tumor maintenance. A second study from the same group showed that chemically induced development of SCC increased and tumor latency decreased in adult K14-Ptch1FVB mice compared with their wild-type counterparts, without aberrant activation of the Hh pathway.154 These authors proposed that Ptch1FVB promotes SCC formation through regulation of Ras-induced apoptosis; high levels of active Ras and inhibition of the tumor suppressor Tid1 by Ptch1FVB results in SCC tumor formation, whereas in the C57BL/6 background, SCC tumor formation is suppressed, because Ptch1 is unable to inhibit Tid1, resulting in an increase in Ras-induced apoptosis.153,154
Inhibitors of the Hh pathway and their application in skin cancer
Three major targeting sites for Hh signaling inhibitors have been identified, ie, SMO protein, Gli transcription factors, and other agents that directly or indirectly modulate the Hh pathway. Table 5 lists Hh signaling inhibitors that are in use or have potential clinical use for the treatment of BCC.
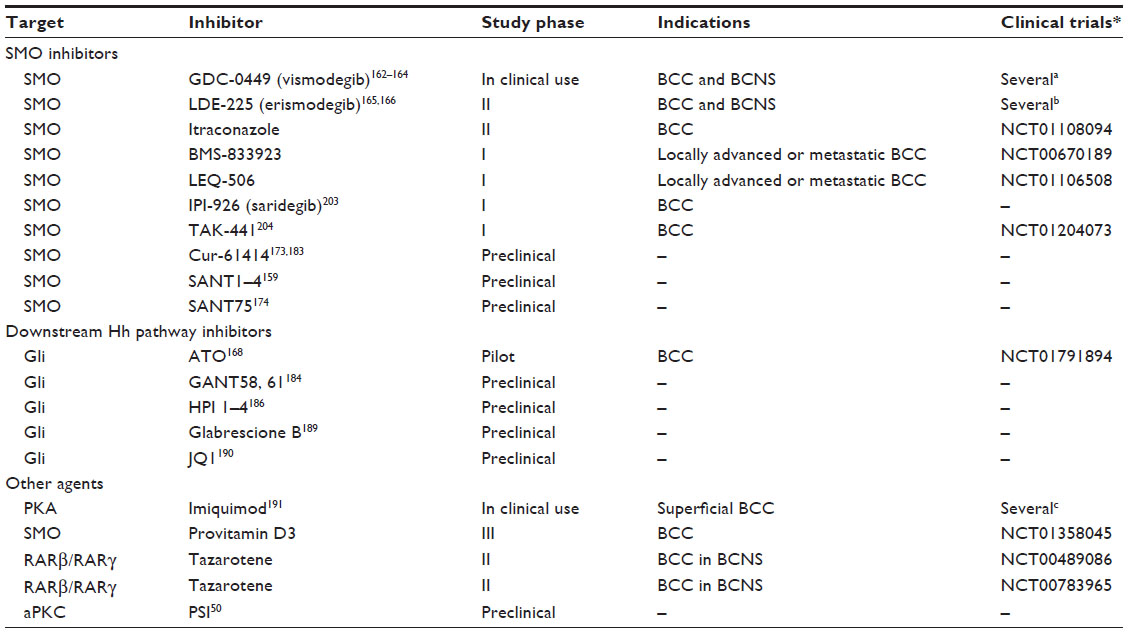 | Table 5 Hedgehog pathway antagonists in use or with potential clinical use for the treatment of BCC |
SMO inhibitors
The idea of targeting the Hh pathway for treating cancers came from the finding that ingestion of corn lilies (Veratrum californicum) by pregnant sheep induced birth defects in their offspring (cyclopia)155 similar to those observed in mice lacking Shh.156 The active compound, cyclopamine, was later purified and shown to inhibit Hh signaling157,158 and to bind SMO.159 Initial studies showed that oral administration of cyclopamine drastically reduced growth and development of BCCs in Ptch+/− mice exposed to ultraviolet light,160 and topical application of cyclopamine can induce regression of human BCC.161 Several groups have confirmed that cyclopamine also decreases growth of many human cancer cell lines in xenotransplantation, including melanoma.6 However, cyclopamine is not suitable for clinical development because of its poor oral solubility. Subsequent work led to the discovery of a number of new SMO antagonists, including GDC-0449 (vismodegib), the first SMO inhibitor approved by the US Food and Drug Administration for locally advanced and metastatic BCC.162–164 Other current SMO inhibitors that are in Phase I or Phase II clinical trials to treat locally advanced or metastatic BCC are LDE-225 (erismodegib),165,166 itraconazole,167,168 BMS-833923,169 and LEQ-506.170 Two SMO inhibitors, IPI-926 (saridegib) and TAK-441, have been discontinued for lack of efficacy.171,172 A number of additional SMO antagonists have been used in preclinical studies, including Cur-61414 (HhAntag),173 Sant1-4,159 and Sant75174 (Table 5).
Clinical trials showed a heterogeneous response to vismodegib depending on the type of BCC. The most sensitive patients are those with NBCCS, who showed a 100% response rate without signs of resistance during treatment, as described for medulloblastoma.163 In contrast, in sporadic cases, only 57% of patients with late advanced or metastatic BCC showed tumor regression in Phase I clinical trials,162,175 and only 30% of metastatic and 43% of late advanced BCCs responded in Phase II clinical trials.164 These results suggest that tumors with a low mutation rate, such as in patients with NBCCS, are predicted to respond well to SMO inhibition, whereas metastatic BCCs with a high mutation rate have a greater likelihood of developing acquired resistance during treatment.176
Little is known about resistance mechanisms in BCC, but studies in other Hh-driven cancers suggest that BCCs can bypass SMO inhibition through Hh-specific genetic alterations or compensatory adaptation.176 Studies in mice and humans harboring SMO inhibitor-resistant medulloblastoma have shed light on the mechanisms of acquired resistance, which could derive from mutations in human SMO (D473H) and the matching mutation in the mouse (D477G),177 amplification of downstream Hh target genes, such as Gli2 and CCND1,178,179 and upregulation of other oncogenic pathways, such as that for phosphatidylinositide 3-kinase-AKT.178
Treatment with vismodegib and other systemic Hh pathway inhibitors is associated with mild to moderate side effects, which include muscle cramps, nausea, diarrhea, alteration in taste perception, weight loss, and alopecia. Most of these side effects cease after patients stopped taking the drug. It is likely that hair loss, altered taste, and diarrhea are related directly to inhibition of the intended target SMO, since Hh signaling is known to be active in the HF, taste buds, and the gastrointestinal tract.180–182 Therefore, such effects are unlikely to be avoided by modifying the molecular structure of these agents. The location of BCCs makes them ideal for topical use of these inhibitors, thus limiting the side effects associated with systemic treatment. One study using topical LDE-225 for 4 weeks documented an effective reduction in tumor size or clinical clearing, correlated with inhibition of Hh signaling.165 In another study, the treatment was ineffective, probably because of lack of Hh pathway blockade, despite the fact that the drug (Cur-61414) showed efficacy in mice.183
Itraconazole, an antifungal agent approved by the US Food and Drug Administration, has been identified as a potent inhibitor of the Hh pathway by preventing cilial translocation of SMO.167 Systemic administration of itraconazole has been shown to delay development of BCC in Ptch1+/− mice, and it is also active against drug-resistant mutant SMO D477G and Gli2 overexpression.168 At present, itraconazole is under Phase II investigation as a treatment for BCC (Table 5).
Gli antagonists
The development of molecules able to directly target the Gli transcription factors, the final effectors of the Hh signaling, would be effective in skin tumors with mutations that are downstream of SMO and perhaps overcome anti-SMO drug resistance. Unfortunately, so far, only few molecules acting on Gli proteins have been identified and their use is limited to preclinical studies. Cell-based screening for inhibitors of Gli1-mediated transcription identified two structurally different compounds, GANT61 and GANT58. Both are capable of interfering with Gli1-mediated and Gli2-mediated transcription and inhibit tumor cell growth in a Gli-dependent manner.184 Screening of natural products identified physalins F and B as inhibitors of Gli-mediated transcriptional activity.185 More recently, HPI-1/4 were described to act at or downstream of SUFU through various mechanisms, such as interfering with processing or activation of Gli. In particular, HPI-1 and HPI-4 have been shown to increase the proteolytic cleavage of Gli2 to its repressor form, whereas HPI-4 also decreases Gli1 stability.186
One example of a drug found to target the Gli transcription factors is arsenic trioxide,187 a therapeutic agent already approved for acute promyelocytic leukemia. Mechanistically, arsenic trioxide binds directly to Gli1 protein and inhibits its transcriptional activity188 and blocks Hh-induced ciliary accumulation of Gli2.187 The in vivo efficacy of arsenic trioxide was demonstrated in mouse models of medulloblastoma,187,188 and a pilot clinical study of arsenic trioxide in BCC is ongoing (Table 5).
Recently, the small molecule Glabrescione B was shown to interfere with the interaction between Gli1 and target DNA.189 Glabrescione B is an isoflavone naturally present in the seeds of Derris glabrescens. Remarkably, as a consequence of its strong inhibition of Gli1 activity, Glabrescione B inhibits growth of Hh-dependent BCC and medulloblastoma tumor cells in vitro and in vivo.189
Inhibition of BET bromodomain proteins has recently emerged as a novel strategy for epigenetic targeting of the transcriptional output of the Hh pathway.190 The BET bromodomain protein BRD4 is a critical regulator of Gli1 and Gli2 transcription via direct occupancy of their promoter. Interestingly, occupancy of Gli1 and Gli2 promoters by BRD4 and transcriptional activation at cancer-specific Gli promoter-binding sites are markedly inhibited by the BET inhibitor JQ1. In Ptch-deficient BCC and medulloblastoma mouse models and in patient-derived tumors with constitutive Hh pathway activation, JQ1 decreases proliferation and viability of tumor cells in vitro and in vivo.190
Other agents
Other compounds might inhibit Hh signaling by targeting proteins and/or pathways that modulate Gli transcription factors. For instance, imiquimod, a nucleoside analog of the imidazoquinoline family and approved by the US Food and Drug Administration for the treatment of BCC,191 has been shown to directly repress Hh signaling by inducing PKA-mediated Gli phosphorylation with consequent reduction of Gli activator levels in BCC and medulloblastoma.192
Vitamin D3 has been shown to efficiently block Hh signaling in vitro and to mimic the SMO loss-of-function phenotype in a zebrafish model system.193 Vitamin D3 also inhibits proliferation and Hh signaling in BCC cell lines. These effects seem to be independent of the vitamin D receptor, because its genetic silencing does not abrogate the inhibitory effect of vitamin D3.194 In addition, it was shown by a different group that application of calcitriol, the physiologically active form of vitamin D3, inhibits proliferation and growth of BCC in Ptch-deficient mice in vitro and in vivo, and stimulates differentiation of BCC.195 Of note, in the same paper, the authors showed that calcitriol inhibits Hh signaling at the level of SMO in a vitamin D receptor-independent manner. At present, a Phase III trial combining application of topical vitamin D3 and treatment with anti-inflammatory agents is ongoing (Table 5).
Topical treatment with the RARβ/RARγ-selective retinoid tazarotene has been shown to reduce the number and size of BCC in irradiated Ptch1+/− mice.196 Tazarotene also inhibited in vitro growth of a murine BCC keratinocyte cell line and downregulated Gli1.197 Its efficacy in BCC patients is under investigation in Phase II clinical trials (Table 5).
Another potentially interesting therapeutic target recently identified as a novel activator of Gli1 transcriptional activity is aPKC. Loss of aPKC or inhibition with a specific peptide inhibitor suppressed Hh signaling and growth of a mouse BCC cell line. Of note, aPKC activity is increased in sensitive and resistant human BCCs when compared with normal skin, and topical treatment with the specific peptide inhibitor led to reduction of tumor growth in mouse BCC allografts.50
Prospects for melanoma and SCC
Recent studies have started to elucidate the role of Hh signaling in melanoma, demonstrating its requirement for melanoma cell growth. In fact, inhibition of the Hh pathway with the SMO antagonist cyclopamine or LDE-225 or with the Gli1/2 inhibitor GANT61, or through genetic silencing of SMO and Gli1 drastically reduces proliferation of human melanoma cells in vitro and in mouse xenografts in vivo.130–132,134 Interestingly, BRAF mutant melanoma cell lines are more sensitive to SMO inhibition with LDE-225 than melanoma cells with wild-type BRAF.131 Moreover, combination of LDE-225 with the mutant BRAF inhibitor vemurafenib reduced the proliferation of melanoma cells more efficiently than the single agent alone, suggesting the attractive possibility of a combined therapy targeting both mutant BRAF and Hh signaling in patients with melanoma.131 From a therapeutic point of view, it would also be interesting to test whether SMO inhibition might be effective in vemurafenib-resistant melanomas with mutant BRAF. In support of this, a recent report showed that upregulation of Hh-mediated platelet-derived growth factor receptor alpha leads to BRAF inhibitor resistance and suggested that blockade of the Hh pathway restores sensitivity of melanoma cells to BRAF inhibitors.139 These data represent a rationale for translation of combinatorial treatment using Hh and BRAF inhibitors for melanoma to the clinical setting.
The role of Hh signaling in SCC is less clear. Our knowledge of the involvement of Hh signaling in SCC of the human skin derives mainly from immunochemistry studies, which reveal expression of Hh pathway components in cancer cells, but functional studies in human SCCs are lacking. More importantly, mouse models suggest that Ptch1 mainly promotes development of murine SCC.153,154 Therefore, inhibition of Hh might have completely opposite effects in BCC and SCC, ie, Hh inhibitors may be therapeutic agents for BCC, but might promote SCC. In support of this notion, recent reports described cases of patients developing SCC soon after initiation of vismodegib for BCC.198–200 This could be due to appearance of a squamous component within a metatypical BCC or to squamous differentiation of stem cells present in the deep epidermal layer or in the follicular bulge through inhibition of the Hh pathway.198 Therefore, further studies are needed to investigate the molecular mechanisms by which Hh signaling mediates development of SCC before advocating use of Hh inhibitors in SCC patients.
Conclusion
Recent advances have shed light on the role of Hh signaling in skin cancers, including BCC, melanoma, and SCC. What are the prospects for skin cancer therapy? While recent clinical trials using Hh inhibitors have identified effective treatments for BCC, further basic understanding of the molecular mechanisms by which Hh signaling mediates tumor development in melanoma and especially in SCC is required. In particular, animal models of Hh-mediated tumorigenesis for melanoma have not been established as yet. Another challenging question is the nature of the genetic and molecular events that cooperate with active Hh signaling in melanoma tumorigenesis. Only a clear understanding of the mechanisms leading to Gli activation will allow for selection of the appropriate Hh pathway inhibitors and, in cases of a crosstalk between Hh and other oncogenic pathways, the optimal combinatorial partner. This is particularly relevant for melanomas, a cancer characterized by a non-canonical activation of the Hh pathway.
Another complication is that tumors, in particular melanomas, are heterogeneous, with multiple clones in the same lesion. It is possible that during treatment, drug-resistant clones that are initially present in low numbers become dominant as they gain a growth advantage and sensitive clones die. This suggests that initially targeting multiple pathways at the same time may be more effective that targeting a single one. In addition, given the opposite roles of Ptch1 in BCC and SCC, inhibitors of Hh signaling may be therapeutic agents for BCCs, but might have additional effects in promoting SCCs. Therefore, it will be equally important to monitor the appearance of SCC in BCC patients treated with Hh inhibitors. Future studies will certainly provide answers to these questions, and hopefully what has been learned from treating BCC with Hh pathway inhibitors will open new prospects for the treatment of melanoma, SCC, and possibly other tumors that depend on active Hh signaling.
Acknowledgments
Support for this work was provided by grants from the Associazione Italiana per la Ricerca sul Cancro (IG-9566 and IG-14184) and the Tuscany Health Research Program 2009.
Disclosure
The authors report no conflicts of interest in this work.
References
Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in drosophila. Nature. 1980;287(5785):795–801. | |
Roessler E, Belloni E, Gaudenz K, et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14(3):357–360. | |
Belloni E, Muenke M, Roessler E, et al. Identification of Sonic Hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet. 1996;14(3):353–356. | |
Petrova R, Joyner AL. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development. 2014;141(18):3445–3457. | |
Gorlin RJ. Nevoid basal cell carcinoma syndrome. Dermatol Clin. 1995; 13(1):113–125. | |
Teglund S, Toftgard R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010;1805(2):181–208. | |
Stecca B, Ruiz i Altaba A. Context-dependent regulation of the GLI code in cancer by Hedgehog and non-Hedgehog signals. J Mol Cell Biol. 2010;2(2):84–95. | |
Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15(23):3059–3087. | |
Porter JA, Young KE, Beachy PA. Cholesterol modification of Hedgehog signaling proteins in animal development. Science. 1996;274(5285):255–259. | |
Pepinsky RB, Zeng C, Wen D, et al. Identification of a palmitic acid-modified form of human Sonic Hedgehog. J Biol Chem. 1998;273(22):14037–14045. | |
Chamoun Z, Mann RK, Nellen D, et al. Skinny Hedgehog, an acyltransferase required for palmitoylation and activity of the Hedgehog signal. Science. 2001;293(5537):2080–2084. | |
Lewis PM, Dunn MP, McMahon JA, et al. Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by ptc1. Cell. 2001;105(5):599–612. | |
Chen MH, Li YJ, Kawakami T, Xu SM, Chuang PT. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 2004;18(6):641–659. | |
Caspary T, Garcia-Garcia MJ, Huangfu D, et al. Mouse Dispatched homolog1 is required for long-range, but not juxtacrine, Hh signaling. Curr Biol. 2002;12(18):1628–1632. | |
Ma Y, Erkner A, Gong R, et al. Hedgehog-mediated patterning of the mammalian embryo requires transporter-like function of Dispatched. Cell. 2002;111(1):63–75. | |
Ayers KL, Gallet A, Staccini-Lavenant L, Therond PP. The long-range activity of Hedgehog is regulated in the apical extracellular space by the glypican dally and the hydrolase notum. Dev Cell. 2010;18(4):605–620. | |
Beckett K, Franch-Marro X, Vincent JP. Glypican-mediated endocytosis of Hedgehog has opposite effects in flies and mice. Trends Cell Biol. 2008;18(8):360–363. | |
Lum L, Yao S, Mozer B, et al. Identification of Hedgehog pathway components by RNAi in drosophila cultured cells. Science. 2003;299(5615):2039–2045. | |
Bellaiche Y, The I, Perrimon N. Tout-velu is a Drosophila homologue of the putative tumour suppressor ext-1 and is needed for hh diffusion. Nature. 1998;394(6688):85–88. | |
Toyoda H, Kinoshita-Toyoda A, Fox B, Selleck SB. Structural analysis of glycosaminoglycans in animals bearing mutations in sugarless, sulfateless, and tout-velu. Drosophila homologues of vertebrate genes encoding glycosaminoglycan biosynthetic enzymes. J Biol Chem. 2000;275(29):21856–21861. | |
Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates Hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–376. | |
Eggenschwiler JT, Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007;23:345–373. | |
Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999;397(6720):617–621. | |
Zhang W, Kang JS, Cole F, Yi MJ, Krauss RS. Cdo functions at multiple points in the Sonic Hedgehog pathway, and Cdo-deficient mice accurately model human holoprosencephaly. Dev Cell. 2006;10(5):657–665. | |
Tenzen T, Allen BL, Cole F, et al. The cell surface membrane proteins Cdo and Boc are components and targets of the Hedgehog signaling pathway and feedback network in mice. Dev Cell. 2006;10(5):647–656. | |
Okada A, Charron F, Morin S, et al. Boc is a receptor for Sonic Hedgehog in the guidance of commissural axons. Nature. 2006;444(7117):369–373. | |
Allen BL, Tenzen T, McMahon AP. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007;21(10):1244–1257. | |
Martinelli DC, Fan CM. Gas1 extends the range of Hedgehog action by facilitating its signaling. Genes Dev. 2007;21(10):1231–1243. | |
Capurro MI, Xu P, Shi W, et al. Glypican-3 inhibits Hedgehog signaling during development by competing with Patched for Hedgehog binding. Dev Cell. 2008;14(5):700–711. | |
Corbit KC, Aanstad P, Singla V, et al. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–1021. | |
Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15(6):801–812. | |
Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22(18):2454–2472. | |
Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100(4):423–434. | |
Pan Y, Bai CB, Joyner AL, Wang B. Sonic Hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26(9):3365–3377. | |
Kinzler KW, Vogelstein B. The Gli gene encodes a nuclear protein which binds specific sequences in the human genome. Mol Cell Biol. 1990;10(2):634–642. | |
Winklmayr M, Schmid C, Laner-Plamberger S, et al. Non-consensus Gli binding sites in Hedgehog target gene regulation. BMC Mol Biol. 2010;11:2. | |
Ikram MS, Neill GW, Regl G, et al. Gli2 is expressed in normal human epidermis and BCC and induces Gli1 expression by binding to its promoter. J Invest Dermatol. 2004;122(6):1503–1509. | |
Lee J, Platt KA, Censullo P, Ruiz i Altaba A. Gli1 is a target of Sonic Hedgehog that induces ventral neural tube development. Development. 1997;124(13):2537–2552. | |
Ruiz i Altaba A. Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development. 1999;126(14):3205–3216. | |
Kogerman P, Grimm T, Kogerman L, et al. Mammalian Suppressor-of-Fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1(5):312–319. | |
Ding Q, Fukami S, Meng X, et al. Mouse Suppressor of Fused is a negative regulator of Sonic Hedgehog signaling and alters the subcellular distribution of Gli1. Curr Biol. 1999;9(19):1119–1122. | |
Yue S, Chen Y, Cheng SY. Hedgehog signaling promotes the degradation of tumor suppressor SUFU through the ubiquitin-proteasome pathway. Oncogene. 2009;28(4):492–499. | |
Sheng T, Chi S, Zhang X, Xie J. Regulation of Gli1 localization by the cAMP/protein kinase A signaling axis through a site near the nuclear localization signal. J Biol Chem. 2006;281(1):9–12. | |
Tempe D, Casas M, Karaz S, Blanchet-Tournier MF, Concordet JP. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by beta-TrCP. Mol Cell Biol. 2006;26(11):4316–4326. | |
Niewiadomski P, Kong JH, Ahrends R, et al. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014;6(1):168–181. | |
Endoh-Yamagami S, Evangelista M, Wilson D, et al. The mammalian cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr Biol. 2009;19(15):1320–1326. | |
Mao J, Maye P, Kogerman P, et al. Regulation of Gli1 transcriptional activity in the nucleus by DYRK1. J Biol Chem. 2002;277(38):35156–35161. | |
Varjosalo M, Bjorklund M, Cheng F, et al. Application of active and kinase-deficient kinome collection for identification of kinases regulating Hedgehog signaling. Cell. 2008;133(3):537–548. | |
Maloverjan A, Piirsoo M, Michelson P, Kogerman P, Osterlund T. Identification of a novel serine/threonine kinase ULK3 as a positive regulator of Hedgehog pathway. Exp Cell Res. 2010;316(4):627–637. | |
Atwood SX, Li M, Lee A, Tang JY, Oro AE. Gli activation by atypical protein kinase C i/l regulates the growth of basal cell carcinomas. Nature. 2013;494(7438):484–488. | |
Wang Y, Ding Q, Yen CJ, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 2012;21(3):374–387. | |
Canettieri G, Di Marcotullio L, Greco A, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12(2):132–142. | |
Huntzicker EG, Estay IS, Zhen H, et al. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006;20(3):276–281. | |
Di Marcotullio L, Ferretti E, Greco A, et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for ITCH-dependent ubiquitination. Nat Cell Biol. 2006;8(12):1415–1423. | |
Kent D, Bush EW, Hooper JE. Roadkill attenuates Hedgehog responses through degradation of cubitus interruptus. Development. 2006;133(10):2001–2010. | |
Zhang Q, Zhang L, Wang B, et al. A Hedgehog-induced BTB protein modulates Hedgehog signaling by degrading Ci/Gli transcription factor. Dev Cell. 2006;10(6):719–729. | |
Zhang Q, Shi Q, Chen Y, et al. Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-Hib/SPOP E3 ubiquitin ligase. Proc Natl Acad Sci U S A. 2009;106(50):21191–21196. | |
Mazza D, Infante P, Colicchia V, et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013;20(12):1688–1697. | |
Rubin AI, Chen EH, Ratner D. Basal-cell carcinoma. N Engl J Med. 2005;353(21):2262–2269. | |
Epstein EH. Basal cell carcinomas: attack of the Hedgehog. Nat Rev Cancer. 2008;8(10):743–754. | |
Gailani MR, Bale SJ, Leffell DJ, et al. Developmental defects in Gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell. 1992;69(1):111–117. | |
Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85(6):841–851. | |
Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996; 272(5268):1668–1671. | |
Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6(6):530–539. | |
Levanat S, Gorlin RJ, Fallet S, et al. A two-hit model for developmental defects in Gorlin syndrome. Nat Genet. 1996;12(1):85–87. | |
Pastorino L, Ghiorzo P, Nasti S, et al. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A. 2009;149A(7):1539–1543. | |
Unden AB, Holmberg E, Lundh-Rozell B, et al. Mutations in the human homologue of drosophila patched (ptch) in basal cell carcinomas and the Gorlin syndrome: different in vivo mechanisms of Ptch inactivation. Cancer Res. 1996;56(20):4562–4565. | |
Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G. Mutations in the human homologue of the drosophila segment polarity gene patched (ptch) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1997;57(13):2581–2585. | |
Kim MY, Park HJ, Baek SC, Byun DG, Houh D. Mutations of the p53 and PTCH gene in basal cell carcinomas: UV mutation signature and strand bias. J Dermatol Sci. 2002;29(1):1–9. | |
Reifenberger J, Wolter M, Knobbe CB, et al. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br J Dermatol. 2005;152(1):43–51. | |
Jayaraman SS, Rayhan DJ, Hazany S, Kolodney MS. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J Invest Dermatol. 2014;134(1):213–220. | |
Barnes EA, Heidtman KJ, Donoghue DJ. Constitutive activation of the SHh-PTC1 pathway by a Patched1 mutation identified in BCC. Oncogene. 2005;24(5):902–915. | |
Xie J, Murone M, Luoh SM, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–92. | |
Reifenberger J, Wolter M, Weber RG, et al. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998;58(9):1798–1803. | |
Lam CW, Xie J, To KF, et al. A frequent activated Smoothened mutation in sporadic basal cell carcinomas. Oncogene. 1999;18(3):833–836. | |
Gomez-Ospina N, Chang AL, Qu K, Oro AE. Translocation affecting Sonic Hedgehog genes in basal-cell carcinoma. N Engl J Med. 2012;366(23):2233–2234. | |
Kallassy M, Toftgard R, Ueda M, et al. Patched (PTCH)-associated preferential expression of smoothened (SMOH) in human basal cell carcinoma of the skin. Cancer Res. 1997;57(21):4731–4735. | |
Aszterbaum M, Beech J, Epstein EH Jr. Ultraviolet radiation mutagenesis of Hedgehog pathway genes in basal cell carcinomas. J Investig Dermatol Symp Proc. 1999;4(1):41–45. | |
Mancuso M, Pazzaglia S, Tanori M, et al. Basal cell carcinoma and its development: insights from radiation-induced tumors in Ptch1-deficient mice. Cancer Res. 2004;64(3):934–941. | |
Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse Patched mutants. Science. 1997;277(5329):1109–1113. | |
Hahn H, Wojnowski L, Zimmer AM, et al. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. 1998;4(5):619–622. | |
Ellis T, Smyth I, Riley E, et al. Patched 1 conditional null allele in mice. Genesis. 2003;36(3):158–161. | |
Adolphe C, Hetherington R, Ellis T, Wainwright B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006;66(4):2081–2088. | |
Siggins SL, Nguyen NY, McCormack MP, et al. The Hedgehog receptor Patched1 regulates myeloid and lymphoid progenitors by distinct cell-extrinsic mechanisms. Blood. 2009;114(5):995–1004. | |
Zibat A, Uhmann A, Nitzki F, et al. Time-point and dosage of gene inactivation determine the tumor spectrum in conditional Ptch knockouts. Carcinogenesis. 2009;30(6):918–926. | |
Nitzki F, Becker M, Frommhold A, Schulz-Schaeffer W, Hahn H. Patched knockout mouse models of basal cell carcinoma. J Skin Cancer. 2012;2012:907543. | |
Dahmane N, Lee J, Robins P, Heller P, Ruiz i Altaba A. Activation of the transcription factor Gli1 and the Sonic Hedgehog signalling pathway in skin tumours. Nature. 1997;389(6653):876–881. | |
Nilsson M, Unden AB, Krause D, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing Gli-1. Proc Natl Acad Sci U S A. 2000;97(7):3438–3443. | |
Grachtchouk M, Mo R, Yu S, et al. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat Genet. 2000;24(3):216–217. | |
Hutchin ME, Kariapper MS, Grachtchouk M, et al. Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev. 2005;19(2):214–223. | |
Oro AE, Higgins KM, Hu Z, et al. Basal cell carcinomas in mice overexpressing Sonic Hedgehog. Science. 1997;276(5313):817–821. | |
Grachtchouk V, Grachtchouk M, Lowe L, et al. The magnitude of Hedgehog signaling activity defines skin tumor phenotype. EMBO J. 2003;22(11):2741–2751. | |
Mao J, Ligon KL, Rakhlin EY, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66(20):10171–10178. | |
Wong SY, Seol AD, So PL, et al. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med. 2009;15(9):1055–1061. | |
Svard J, Heby-Henricson K, Persson-Lek M, et al. Genetic elimination of Suppressor of Fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10(2):187–197. | |
Svard J, Rozell B, Toftgard R, Teglund S. Tumor suppressor gene co-operativity in compound Patched1 and Suppressor of Fused heterozygous mutant mice. Mol Carcinog. 2009;48(5):408–419. | |
Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102(32):11325–11330. | |
Haycraft CJ, Banizs B, Aydin-Son Y, et al. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1(4):e53. | |
Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of gli transcription factors. Development. 2005;132(13):3103–3111. | |
Youssef KK, Van Keymeulen A, Lapouge G, et al. Identification of the cell lineage at the origin of basal cell carcinoma. Nat Cell Biol. 2010;12(3):299–305. | |
Wang GY, Wang J, Mancianti ML, Epstein EH Jr. Basal cell carcinomas arise from hair follicle stem cells in Ptch1(+/−) mice. Cancer Cell. 2011;19(1):114–124. | |
Wong SY, Reiter JF. Wounding mobilizes hair follicle stem cells to form tumors. Proc Natl Acad Sci U S A. 2011;108(10):4093–4098. | |
Kasper M, Jaks V, Are A, et al. Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes. Proc Natl Acad Sci U S A. 2011;108(10):4099–4104. | |
Grachtchouk M, Pero J, Yang SH, et al. Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. J Clin Invest. 2011;121(5):1768–1781. | |
Regl G, Neill GW, Eichberger T, et al. Human Gli2 and Gli1 are part of a positive feedback mechanism in basal cell carcinoma. Oncogene. 2002;21(36):5529–5539. | |
Ji J, Kump E, Wernli M, Erb P. Gene silencing of transcription factor Gli2 inhibits basal cell carcinomalike tumor growth in vivo. Int J Cancer. 2008;122(1):50–56. | |
Xie J, Aszterbaum M, Zhang X, et al. A role of PDGFRalpha in basal cell carcinoma proliferation. Proc Natl Acad Sci U S A. 2001;98(16):9255–9259. | |
Teh MT, Wong ST, Neill GW, et al. Foxm1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002;62(16):4773–4780. | |
Eichberger T, Regl G, Ikram MS, et al. FoxE1, a new transcriptional target of Gli2 is expressed in human epidermis and basal cell carcinoma. J Invest Dermatol. 2004;122(5):1180–1187. | |
Regl G, Kasper M, Schnidar H, et al. Activation of the Bcl2 promoter in response to Hedgehog/Gli signal transduction is predominantly mediated by Gli2. Cancer Res. 2004;64(21):7724–7731. | |
Kump E, Ji J, Wernli M, Hausermann P, Erb P. Gli2 upregulates cFLIP and renders basal cell carcinoma cells resistant to death ligand-mediated apoptosis. Oncogene. 2008;27(27):3856–3864. | |
Cui C, Elsam T, Tian Q, et al. Gli proteins up-regulate the expression of Basonuclin in basal cell carcinoma. Cancer Res. 2004;64(16):5651–5658. | |
Villani RM, Adolphe C, Palmer J, Waters MJ, Wainwright BJ. Patched1 inhibits epidermal progenitor cell expansion and basal cell carcinoma formation by limiting IGFBP2 activity. Cancer Prev Res (Phila). 2010;3(10):1222–1234. | |
El-Bahrawy M, El-Masry N, Alison M, Poulsom R, Fallowfield M. Expression of beta-Catenin in basal cell carcinoma. Br J Dermatol. 2003;148(5):964–970. | |
Yang SH, Andl T, Grachtchouk V, et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical WNT/beta3-Catenin signaling. Nat Genet. 2008;40(9):1130–1135. | |
Kasper M, Schnidar H, Neill GW, et al. Selective modulation of Hedgehog/Gli target gene expression by epidermal growth factor signaling in human keratinocytes. Mol Cell Biol. 2006;26(16):6283–6298. | |
Schnidar H, Eberl M, Klingler S, et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/Gli in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009;69(4):1284–1292. | |
Sonkoly E, Loven J, Xu N, et al. MicroRNA-203 functions as a tumor suppressor in basal cell carcinoma. Oncogenesis. 2012;1:e3. | |
Eberl M, Klingler S, Mangelberger D, et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol Med. 2012;4(3):218–233. | |
Stecca B, Ruiz i Altaba A. A Gli1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009;28(6):663–676. | |
Tsao H, Chin L, Garraway LA, Fisher DE. Melanoma: from mutations to medicine. Genes Dev. 2012;26(11):1131–1155. | |
Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351(10):998–1012. | |
Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–6206. | |
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | |
Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20(16):2149–2182. | |
Goldstein AM, Chan M, Harland M, et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across genoMEL. Cancer Res. 2006;66(20):9818–9828. | |
Wei X, Walia V, Lin JC, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43(5):442–446. | |
Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44(9):1006–1014. | |
Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–263. | |
Stecca B, Mas C, Clement V, et al. Melanomas require Hedgehog-Gli signaling regulated by interactions between Gli1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A. 2007;104(14):5895–5900. | |
O’Reilly KE, de Miera EV, Segura MF, et al. Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals (Basel). 2013;6(11):1429–1450. | |
Jalili A, Mertz KD, Romanov J, et al. NVP-LDE225, a potent and selective smoothened antagonist reduces melanoma growth in vitro and in vivo. PLoS One. 2013;8(7):e69064. | |
Coni S, Infante P, Gulino A. Control of stem cells and cancer stem cells by Hedgehog signaling: pharmacologic clues from pathway dissection. Biochem Pharmacol. 2013;85(5):623–628. | |
Santini R, Vinci MC, Pandolfi S, et al. Hedgehog-Gli signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells. 2012;30(9):1808–1818. | |
Po A, Ferretti E, Miele E, et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 2010;29(15):2646–2658. | |
Santini R, Pietrobono S, Pandolfi S, et al. Sox2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene. 2014;33(38):4697–4708. | |
Pandolfi S, Montagnani V, Penachioni JY, et al. WIP1 phosphatase modulates the Hedgehog signaling by enhancing GLI1 function. Oncogene. 2013;32(40):4737–4747. | |
Harrison W, Cochrane B, Neill G, Philpott M. The oncogenic Gli transcription factors facilitate keratinocyte survival and transformation upon exposure to genotoxic agents. Oncogene. 2014;33(19):2432–2440. | |
Sabbatino F, Wang Y, Wang X, et al. PDGFRalpha up-regulation mediated by Sonic Hedgehog pathway activation leads to BRAF inhibitor resistance in melanoma cells with BRAF mutation. Oncotarget. 2014;5(7):1926–1941. | |
Alexaki VI, Javelaud D, Van Kempen LC, et al. Gli2-mediated melanoma invasion and metastasis. J Natl Cancer Inst. 2010;102(15):1148–1159. | |
Javelaud D, Alexaki VI, Pierrat MJ, et al. Gli2 and M-MITF transcription factors control exclusive gene expression programs and inversely regulate invasion in human melanoma cells. Pigment Cell Melanoma Res. 2011;24(5):932–943. | |
Pierrat MJ, Marsaud V, Mauviel A, Javelaud D. Expression of microphthalmia-associated transcription factor (MITF), which is critical for melanoma progression, is inhibited by both transcription factor Gli2 and transforming growth factor-beta. J Biol Chem. 2012;287(22):17996–18004. | |
Das S, Harris LG, Metge BJ, et al. The Hedgehog pathway transcription factor Gli1 promotes malignant behavior of cancer cells by up-regulating osteopontin. J Biol Chem. 2009;284(34):22888–22897. | |
Duan F, Lin M, Li C, et al. Effects of inhibition of Hedgehog signaling on cell growth and migration of uveal melanoma cells. Cancer Biol Ther. 2014;15(5):544–559. | |
Gao L, Hou X, Wu L, et al. Drosophila miR-960 negatively regulates Hedgehog signaling by suppressing Smoothened, Costal-2 and Fused. Cell Signal. 2013;25(5):1301–1309. | |
Cerami E, Gao J, Dogrusoz U, et al. The cBIO cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. | |
Preston DS, Stern RS. Nonmelanoma cancers of the skin. N Engl J Med. 1992;327(23):1649–1662. | |
Ping XL, Ratner D, Zhang H, et al. Ptch mutations in squamous cell carcinoma of the skin. J Invest Dermatol. 2001;116(4):614–616. | |
Danaee H, Karagas MR, Kelsey KT, Perry AE, Nelson HH. Allelic loss at drosophila patched gene is highly prevalent in basal and squamous cell carcinomas of the skin. J Invest Dermatol. 2006;126(5):1152–1158. | |
Pickering CR, Zhou JH, Lee JJ, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. 2014; 20(24):6582–6592. | |
Schneider S, Thurnher D, Kloimstein P, et al. Expression of the sonic Hedgehog pathway in squamous cell carcinoma of the skin and the mucosa of the head and neck. Head Neck. 2011;33(2):244–250. | |
Bakry OA, Samaka RM, Shoeib MA, Megahed DM. Immunolocalization of glioma-associated oncogene homolog 1 in non melanoma skin cancer. Ultrastruct Pathol. 2015;39(2):135–146. | |
Wakabayashi Y, Mao JH, Brown K, Girardi M, Balmain A. Promotion of hRAS-induced squamous carcinomas by a polymorphic variant of the Patched gene in FVB mice. Nature. 2007;445(7129):761–765. | |
Kang HC, Wakabayashi Y, Jen KY, et al. Ptch1 overexpression drives skin carcinogenesis and developmental defects in k14ptch(fvb) mice. J Invest Dermatol. 2013;133(5):1311–1320. | |
Keeler RF, Binns W. Teratogenic compounds of veratrum californicum (durand). II. Production of ovine fetal cyclopia by fractions and alkaloid preparations. Can J Biochem. 1966;44(6):829–838. | |
Chiang C, Litingtung Y, Lee E, et al. Cyclopia and defective axial patterning in mice lacking sonic Hedgehog gene function. Nature. 1996;383(6599):407–413. | |
Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic veratrum alkaloid cyclopamine inhibits sonic Hedgehog signal transduction. Development. 1998;125(18):3553–3562. | |
Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280(5369):1603–1607. | |
Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci U S A. 2002; 99(22):14071–14076. | |
Athar M, Li C, Tang X, et al. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of Fas expression and apoptosis. Cancer Res. 2004;64(20):7545–7552. | |
Tabs S, Avci O. Induction of the differentiation and apoptosis of tumor cells in vivo with efficiency and selectivity. Eur J Dermatol. 2004;14(2):96–102. | |
Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the Hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361(12):1164–1172. | |
Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the Hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med. 2012;366(23):2180–2188. | |
Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366(23):2171–2179. | |
Skvara H, Kalthoff F, Meingassner JG, et al. Topical treatment of basal cell carcinomas in nevoid basal cell carcinoma syndrome with a Smoothened inhibitor. J Invest Dermatol. 2011;131(8):1735–1744. | |
Rodon J, Tawbi HA, Thomas AL, et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral Smoothened inhibitor sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res. 2014;20(7):1900–1909. | |
Kim J, Tang JY, Gong R, et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17(4):388–399. | |
Kim J, Aftab BT, Tang JY, et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23(1):23–34. | |
Siu LL, Papadopoulos K, Alberts SR, et al. A first-in-human, phase I study of an oral Hedgehog (Hh) pathway antagonist, BMS-833923 (XL139), in subjects with advanced or metastatic solid tumors. J Clin Oncol 2010; 28:15 Suppl:Abstr 2501. | |
Peukert S, He F, Dai M, et al. Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem. 2013;8(8):1261–1265. | |
Williams R. Discontinued in 2012: oncology drugs. Expert Opin Investig Drugs. 2013;22(12):1627–1644. | |
Williams R. Discontinued in 2013: oncology drugs. Expert Opin Investig Drugs. 2015;24(1):95–110. | |
Williams JA, Guicherit OM, Zaharian BI, et al. Identification of a small molecule inhibitor of the Hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proc Natl Acad Sci U S A. 2003;100(8):4616–4621. | |
Yang H, Xiang J, Wang N, et al. Converse conformational control of Smoothened activity by structurally related small molecules. J Biol Chem. 2009;284(31):20876–20884. | |
LoRusso PM, Rudin CM, Reddy JC, et al. Phase I trial of Hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011; 17(8):2502–2511. | |
Atwood SX, Whitson RJ, Oro AE. Advanced treatment for basal cell carcinomas. Cold Spring Harb Perspect Med. 2014;4(7):a013581. | |
Yauch RL, Dijkgraaf GJ, Alicke B, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326(5952):572–574. | |
Buonamici S, Williams J, Morrissey M, et al. Interfering with resistance to Smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2(51):51ra70. | |
Dijkgraaf GJ, Alicke B, Weinmann L, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71(2):435–444. | |
Hall JM, Bell ML, Finger TE. Disruption of Sonic Hedgehog signaling alters growth and patterning of lingual taste papillae. Dev Biol. 2003; 255(2):263–277. | |
St-Jacques B, Dassule HR, Karavanova I, et al. Sonic Hedgehog signaling is essential for hair development. Curr Biol. 1998;8(19):1058–1068. | |
Ramalho-Santos M, Melton DA, McMahon AP. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development. 2000;127(12):2763–2772. | |
Tang T, Tang JY, Li D, et al. Targeting superficial or nodular basal cell carcinoma with topically formulated small molecule inhibitor of Smoothened. Clin Cancer Res. 2011;17(10):3378–3387. | |
Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104(20):8455–8460. | |
Hosoya T, Arai MA, Koyano T, Kowithayakorn T, Ishibashi M. Naturally occurring small-molecule inhibitors of Hedgehog/Gli-mediated transcription. Chembiochem. 2008;9(7):1082–1092. | |
Hyman JM, Firestone AJ, Heine VM, et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc Natl Acad Sci U S A. 2009;106(33):14132–14137. | |
Kim J, Lee JJ, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A. 2010;107(30):13432–13437. | |
Beauchamp EM, Ringer L, Bulut G, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/Gli pathway. J Clin Invest. 2011;121(1):148–160. | |
Infante P, Canettieri G, Gulino A, Di Marcotullio L. Yin-yang strands of PCAF/Hedgehog axis in cancer control. Trends Mol Med. 2014;20(8):416–418. | |
Tang Y, Gholamin S, Schubert S, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014;20(7):732–740. | |
Lacarrubba F, Potenza MC, Gurgone S, Micali G. Successful treatment and management of large superficial basal cell carcinomas with topical imiquimod 5% cream: a case series and review. J Dermatolog Treat. 2011;22(6):353–358. | |
Wolff F, Loipetzberger A, Gruber W, et al. Imiquimod directly inhibits Hedgehog signalling by stimulating adenosine receptor/protein kinase A-mediated GLI phosphorylation. Oncogene. 2013;32(50):5574–5581. | |
Bijlsma MF, Spek CA, Zivkovic D, et al. Repression of Smoothened by Patched-dependent (pro-) vitamin D3 secretion. PLoS Biol. 2006; 4(8):e232. | |
Tang JY, Xiao TZ, Oda Y, et al. Vitamin D3 inhibits Hedgehog signaling and proliferation in murine basal cell carcinomas. Cancer Prev Res (Phila). 2011;4(5):744–751. | |
Uhmann A, Niemann H, Lammering B, et al. Antitumoral effects of calcitriol in basal cell carcinomas involve inhibition of Hedgehog signaling and induction of vitamin D receptor signaling and differentiation. Mol Cancer Ther. 2011;10(11):2179–2188. | |
So PL, Lee K, Hebert J, et al. Topical tazarotene chemoprevention reduces basal cell carcinoma number and size in Ptch1+/− mice exposed to ultraviolet or ionizing radiation. Cancer Res. 2004;64(13):4385–4389. | |
So PL, Fujimoto MA, Epstein EH Jr. Pharmacologic retinoid signaling and physiologic retinoic acid receptor signaling inhibit basal cell carcinoma tumorigenesis. Mol Cancer Ther. 2008;7(5):1275–1284. | |
Iarrobino A, Messina JL, Kudchadkar R, Sondak VK. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69(1):e33–e34. | |
Orouji A, Goerdt S, Utikal J, Leverkus M. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171(2):431–433. | |
Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. July 1, 2014. [Epub ahead of print.] | |
Sheng H, Goich S, Wang A, et al. Dissecting the oncogenic potential of Gli2: deletion of an NH(2)-terminal fragment alters skin tumor phenotype. Cancer Res. 2002;62(18):5308–5316. | |
Uhmann A, Hess I, Frommhold A, et al. DMBA/TPA treatment is necessary for BCC formation from patched deficient epidermal cells in Ptch(flox/flox)CD4Cre(+/−) mice. J Invest Dermatol. 2014;134(10):2620–2629. | |
Jimeno A, Weiss GJ, Miller WH Jr, et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin Cancer Res. 2013;19(10):2766–2774. | |
Ishii T, Shimizu Y, Nakashima K, et al. Inhibition mechanism exploration of investigational drug TAK-441 as inhibitor against vismodegib-resistant Smoothened mutant. Eur J Pharmacol. 2014;723:305–313. | |
National Cancer Institute [homepage on the Internet]. The Cancer Genome Atlas. Available from: http://cancergenome.nih.gov/. Accessed May 1, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.