")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Glycyrrhetinic acid and E.resveratroloside act as potential plant derived compounds against dopamine receptor D3 for Parkinson’s disease: a pharmacoinformatics study
Authors Mirza MU , Mirza AH , Ghori N, Ferdous S
Received 17 August 2014
Accepted for publication 23 September 2014
Published 18 December 2014 Volume 2015:9 Pages 187—198
DOI https://doi.org/10.2147/DDDT.S72794
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Muhammad Usman Mirza,1 A Hammad Mirza,2 Noor-Ul-Huda Ghori,3 Saba Ferdous4
1Centre for Research in Molecular Medicine, The University of Lahore, Lahore, Pakistan; 2Department of Bioscience, COMSATS Institute of Information Technology, Sahiwal, Pakistan; 3Atta-ur-Rehman School of Applied Biosciences, National University of Science and Technology, Islamabad, Pakistan; 4Institute of Structural and Molecular Biology, University College London, UK
Abstract: Parkinson’s disease (PD) is caused by loss in nigrostriatal dopaminergic neurons and is ranked as the second most common neurodegenerative disorder. Dopamine receptor D3 is considered as a potential target in drug development against PD because of its lesser side effects and higher degree of neuro-protection. One of the prominent therapies currently available for PD is the use of dopamine agonists which mimic the natural action of dopamine in the brain and stimulate dopamine receptors directly. Unfortunately, use of these pharmacological therapies such as bromocriptine, apomorphine, and ropinirole provides only temporary relief of the disease symptoms and is frequently linked with insomnia, anxiety, depression, and agitation. Thus, there is a need for an alternative treatment that not only hinders neurodegeneration, but also has few or no side effects. Since the past decade, much attention has been given to exploitation of phytochemicals and their use in alternative medicine research. This is because plants are a cheap, indispensable, and never ending resource of active compounds that are beneficial against various diseases. In the current study, 40 active phytochemicals against PD were selected through literature survey. These ligands were docked with dopamine receptor D3 using AutoDock and AutoDockVina. Binding energies were compared to docking results of drugs approved by the US Food and Drug Administration against PD. The compounds were further analyzed for their absorption, distribution, metabolism, and excretion-toxicity profile. From the study it is concluded that glycyrrhetinic acid and E.resveratroloside are potent compounds having high binding energies which should be considered as potential lead compounds for drug development against PD.
Keywords: AutoDock, AutoDockVina, molecular docking, parkinson’s disease, glycyrrhetinic acid, E.resveratroloside
Introduction
Parkinson’s disease (PD) is the most common form of Parkinsonism. It is a neurodegenerative disorder which notably causes rigidity, bradykinesia, tremor, stooped posture, dementia, and depression in PD patients. Due to unclear etiology this disease is also known as Idiopathic Parkinsonism. But certain factors ie, environmental chemicals, drinking water chemistry, pesticide exposure, and rural living are responsible for PD.1–3 Moreover, mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes are also found to be accompanied by PD.4
Degeneration of nigrostriatal pathway, noradrenergic locus ceruleus, motor vagal nucleus, the serotonergic raphe nuclei, cholinergic nucleus basalis of Meynert, pedunculopontine nucleus pars compacta, Edinger-Westphal nucleus, and many peptidergic brainstem nuclei are characteristic of PD.5 However, it is believed that the primary cause of the disease is loss in the nigrostriatal dopaminergic neurons, formation of intraneuronal, proteinaceous cytoplasmic inclusions called Lewy bodies, inability to produce dopamine, mitochondrial respiration defect, and oxidative stress. Consequently, a striatal dopamine-deficiency syndrome occurs being responsible for the classical motor symptoms of PD.6,7
PD is a progressive disorder, affecting one in every 100 people older than 65 years of age. The symptoms usually appear when approximately 60% of the dopamine producing neurons are lost.8,9 PD is ranked as the second most common neurodegenerative disorder after Alzheimer’s disease.10 Unfortunately, no therapeutic curative regimen has been devised yet.9 One of the most prominent treatments for PD is the use of dopamine agonists which mimic the natural action of dopamine in the brain and stimulate dopamine receptors directly.11 These dopamine agonists will meet the dopamine requirement of the brain and help in bringing about the restoration of normal functionality of the remaining dopaminergic neurons.
Biochemical and electrophysiological data suggest that dopamine acts on its very own special dopamine receptors. They show variance in their anatomical localization, functional substantiality, and pharmacological importance.12,13 This is why they are differentiated into five types of dopamine receptors (D1–D5). Dopamine receptor D3 (DRD3) serves as a potential target for drug development as several experimental data suggest that DRD3 agonists are involved in neuroprotection and alleviating motor dysfunctions.14 Furthermore, neuroprotection and neurorestoration are also seen in animal models by using DRD3 agonist.15
Dopamine carries out cell cycle modulation in both developing and adult brain and DRD3 receptor play a vital role in dopaminergic neuronal development. Activation of these receptor subtypes leads to propagation of neurogenesis in substantia nigra in animal models. Moreover, a persistent recovery in the locomotor functions was also seen.16
Drugs with DRD3 receptor proffering behavior, when acting on their receptors, causes an increase in production of dopamine neurotrophic factor in tissue culture. This factor can also be the autotrophic factor for the dopaminergic neurons. This protein is oxidant labile, therefore, the drugs having DRD3 receptor agonistic activity that results in the increase of this protein and also having the antioxidant profile will provide a unique therapeutic strategy.17
In early and moderate PD, the dopamine receptor agonists have shown effectiveness as symptomatic monotherapy.18 However, use of current pharmacological therapies such as bromocriptine, apomorphine, and ropinirole provides only temporary relief of the disease symptoms and is frequently linked with insomnia, anxiety, depression, and agitation.19 Moreover, dose failure or wearing-off effects are common and high doses are required to overcome the problem. Usually elderly patients are highly sensitive to wearing-off effects.9,20,21 Memory problems and confusion are associated with anticholinergics. Monoamine oxidase inhibitors precipitate many side effects when given in combination therapy. Psychiatric complications, pathological gambling, and depression is also reported with other anti-parkinsonian therapy.22,23 Thus, there is a need for an alternative treatment that not only hinders neurodegeneration but also has few or no side effects.
Since the past decade much interest has been seen in exploitation of phytochemicals. Phytochemicals are considered as a potential alternative cure for various neurodegenerative diseases including PD. This is because plants are a cheap, indispensable, and never ending resource of active compounds that are beneficial against various diseases. These phytochemicals belong to various classes of compounds such as phenolics, alkaloids, terpenoids, lignans etc. Several phytochemicals having beneficial affects against PD have been reported in literature. Many studies have also revealed promising results in decreasing levels of free radicals which is a major cause of neurodegeneracy.24–28 Therefore, the current study was designed to find potential lead compounds from different plants against PD that can be developed into commercial drugs.
In this investigation, various phytochemicals active against PD were identified through literature survey. They were screened through in silico docking analysis to find potential lead compounds for PD. The molecular docking tools, AutoDock and AutoDockVina (Scripps Research Institute, La Jolla, CA, USA) were used to dock 40 phytochemicals against DRD3. The ligands were also analyzed for their absorption, distribution, metabolism, and excretion-toxicity (ADMET) profile. ADMET profile determines the absorption, distribution, metabolism and excretion efficiency of the drug. It also predicts the risk of toxicity upon consumption of the compound.29 Lead compounds having impressive pharmacokinetics and pharmacodynamic properties have a better chance to be developed into commercial drugs. In vitro analysis of ADMET properties is usually carried out during the end stage of drug development due to which several potent compounds fail to achieve drug status. In silico ADMET analysis not only decreases the cost of drug designing but also skips the risk of rejection of a compound.30
From the results of the current study it is concluded that glycyrrhetinic acid, and E.resveratroloside showed high binding energy against dopamine DRD3 when docked with AutoDock and AutoDockVina. They also possess a good ADMET profile and should be considered potential lead compounds for drug development against PD.
Material and methods
Preparation of data set
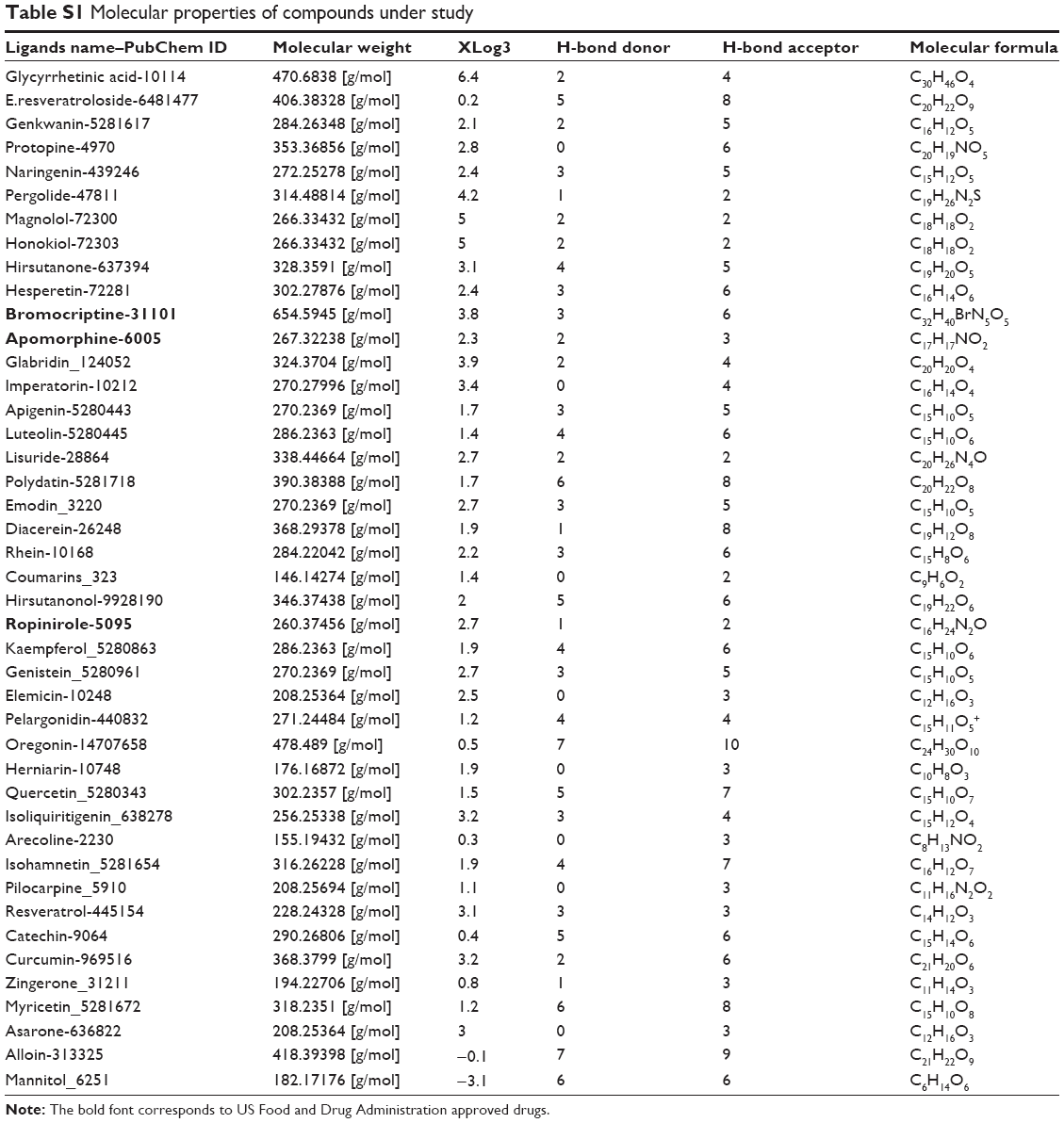
The three dimensional (3D) structure of DRD3 was retrieved from the protein data bank (PDB ID: 3PBL with a resolution [Å]: 2.89, R-Value: 0.245).31 This structure was complexed with dopamine 2/dopamine 3 selective antagonist eticlopride (3-chloro-5-ethyl-N-{[(2S)-1-ethylpyrrolidin-2-yl] methyl}-6-hydroxy-2-ethoxybenzamide). Phytochemicals for the study were collected after extensive literature survey. All phytochemicals included in this study have been reported to be active against PD after in vitro analysis in different studies.32–34 Their respective structures were obtained from the publicly available database, Pubchem.35 Selected phytochemicals were sorted according to Lipinski’s rule. Only those structures were selected for docking that strictly followed the Lipinski’s rule (details for all selected compounds are given in Table S1).
Protein and ligand optimization
In order to figure out the equilibrium configuration of biomolecules and solids, energy optimization methods were used. Crystal structure of DRD3 was further processed for molecular docking studies. For this, all the hetero-atoms and water molecules were removed from protein structure followed by energy minimization in order to remove all the bad steric clashes using the UCSF Chimera (Resource for Biocomputing, Visualization, and Informatics, University of California San Francisco, San Francisco, CA, USA) for 1,000 steepest descent steps at root-mean-square gradient of 0.02 with an update interval of 10 and using AMBER ff12SB force field.36,37 The structure-data file two dimensional structures of phytochemicals were converted into MOL 3D structure using Open babel. It was followed by energy minimization through Hyperchem’s (Gainesville, FL, USA) MM+ force field.
Determination of binding site
Binding sites of proteins are often located in the structural pockets and cavities which show high affinity for selective drugs. The binding site of DRD3 was revealed by knowledge based studies.31 The binding residues were further inspected by using Computed Atlas of Surface Topography of Proteins (CASTp) server and Q-Site Finder.38,39 CASTp server uses the weighted Delaunay triangulation and the alpha complex for shape measurements. It provides identification and measurements of surface accessible pockets as well as interior inaccessible cavities, for proteins and other molecules. It measures analytically the area and volume of each pocket and cavity, both in solvent accessible surface and molecular surface. Q-Site Finder uses the interaction energy between the protein and a simple van der Waals probe to locate energetically favorable binding sites. Pocket Finder is another tool used for analysis of binding sites of a protein and uses Ligsite algorithm. Ligsite algorithm is designed to heuristically search protein for probable interaction of ligand and generate profiles of best active sites.40
Computational docking strategy between DRD3 and phytochemicals
All computational docking studies were carried out using AutoDock 4.0 and AutoDockVina41 installed in a single machine running on an OptiPlex 980 (Dell, Round Rock, TX, USA), Corei3-550/3.2 card GT-203 (Intel, Santa Clara, CA, USA) with LINUX (Ubuntu, Canonical Group Limited, London, UK) as an operating system.42 Automated dockings were performed using the AutoDock 4.0 tool to locate the proper binding orientations and conformations of various ligands. For DRD3 binding precisely, ligands and receptors were subjected to polar hydrogen atoms and Gasteiger charges and all torsions were permissible to rotate while docking. Grid maps were created using the AutoGrid program (Scripps Research Institute). The dimensions of the grid were defined with points separated by 0.375A°. A random approach of starting positions, torsions, and orientations was applied for all ligands and default settings of the AutoDock program were used for translation, quaternion, and torsion steps. Minimization was done by using default parameters by Lamarckian genetic algorithm. The standard docking protocol consisted of 100 runs with an initial population of 150 randomly placed samples for rigid and flexible ligand docking with energy evaluations 2.5×105, mutation rate 0.02, crossover rate of 0.80, maximum number of 27,000 iterations, and an elitism value of 1 used as standard. Root-mean-square tolerance of 1.0A° was used for cluster analysis on the docked results. The cluster having a higher number of confirmation and a low binding energy was selected as the docked pose of the respective ligand; the binding energy of each cluster being the mean binding energy of sum of confirmations present in the cluster.
AutoDockVina was employed to find the most probable ligands for target protein, and ligands were docked at DRD3. For this purpose, blind docking was performed by covering the whole protein under the grid to detect if there is any other binding site available other than provided by PDB crystal structure. Gasteiger charges were added to convert protein and all ligand molecules into PDBQT files. Protein was prepared with identical steps used for AutoDock 4 docking experiment. All ligands were docked by AutoDockVina and results were further analyzed in conjunction with results by AutoDock tools using Chimera. Potential ligands were sorted on the basis of making highest interactions with binding pocket and minimum binding energy values. The comparison of results from both of the tools was considered based on the minimum binding energy.
Drug-likeness and ADMET analysis
In-silico analysis of drug likeness was performed in order to check the potential DRD3 ligands for their ability to follow Lipinski’s rule of five.43 This was done by uploading ligands to Mcule,44 Molsoft, and Molinspiration server (http://www.molinspiration.com/cgi-bin/properties) for calculation of their molecular properties (Table 1). The chemical structures of potential ligands were submitted to admetSAR (http://lmmd.ecust.edu.cn:8000/) server for in-silico prediction of ADME-Tox (absorption, distribution, metabolism, excretion and toxicity) properties.45 Furthermore, online server Osiris Property Explorer (http://www.organic-chemistry.org/prog/peo/) was used to predict the tumorigenic, reproductive, and mutagenic risks (Table 2).
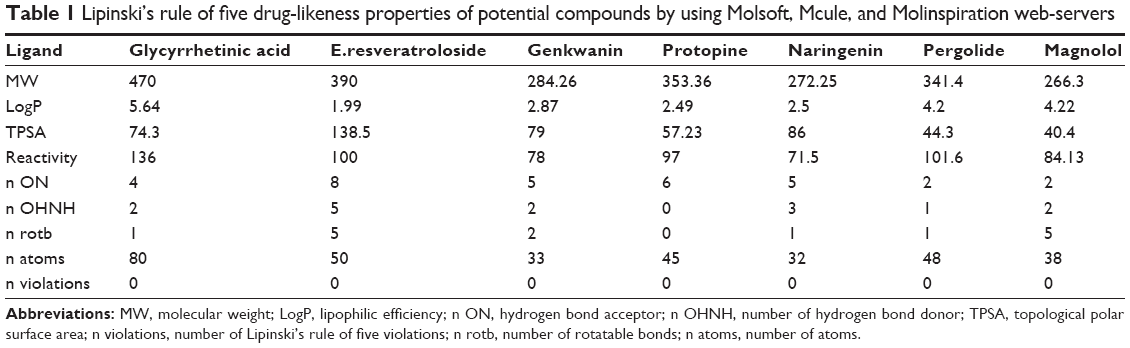 | Table 1 Lipinski’s rule of five drug-likeness properties of potential compounds by using Molsoft, Mcule, and Molinspiration web-servers |
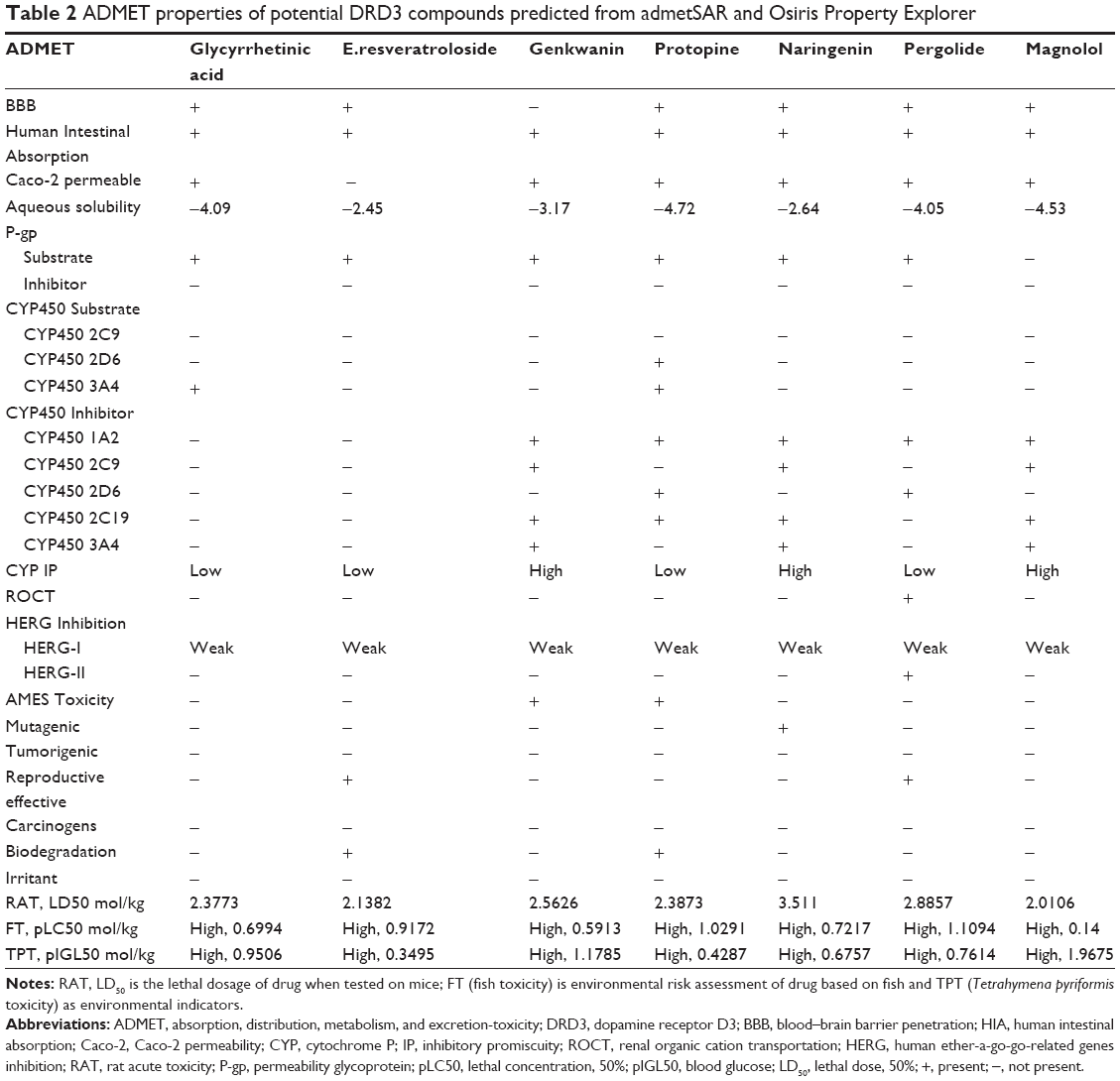 | Table 2 ADMET properties of potential DRD3 compounds predicted from admetSAR and Osiris Property Explorer |
Results and discussion
Molecular docking enables a scientist to virtually screen a number of candidate compounds based on their binding ability and binding orientation with a target molecule. It also allows one to select compounds with strong affinity for the target site. In the current study, phytochemicals have been docked in silico with DRD3 to find the best dopamine agonist candidates that can hinder neurodegeneration in PD. Saponins, flavonoids, alkaloids, lignin, and diarylheptanoids have shown high binding energy when docked with DRD3. Specifically, glycyrrhetinic acid (−7.11 and −12.7 kcal/mol) and E.resveratroloside (−7.03 and −11.7 kcal/mol) have shown stronger binding at the receptor’s binding site in experiments when analyzed using AutoDock and AutoDockVina respectively (Tables 3 and 4).
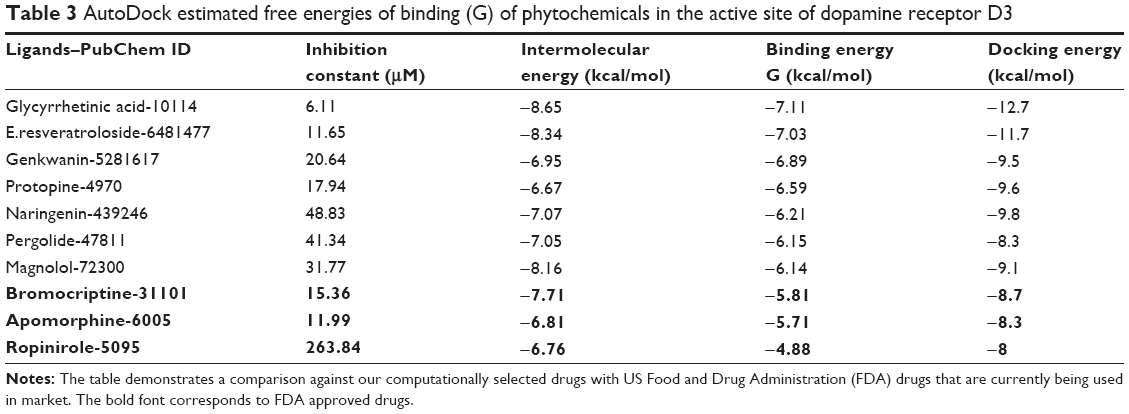 | Table 3 AutoDock estimated free energies of binding (G) of phytochemicals in the active site of dopamine receptor D3 |
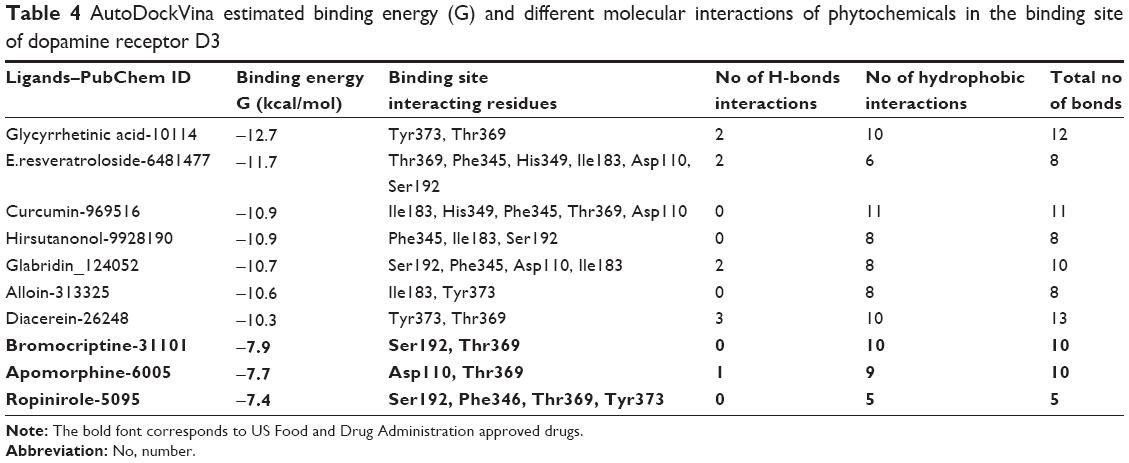 | Table 4 AutoDockVina estimated binding energy (G) and different molecular interactions of phytochemicals in the binding site of dopamine receptor D3 |
Binding residues’ analysis
We performed binding site analysis of our protein through CASTp and Pocket Finder. CASTp results were analyzed through special plugin of Chimera for better understanding of active site. It was observed that the pockets provided by CASTp were similar to those found in DRD3 crystal structure. It also provided other pockets with measurements. An in-depth analysis of pockets demonstrated that the pocket having residues similar to those found in DRD3 crystal structure could be the most potent active site. The results of CASTp were also compared with Pocket Finder results. The pockets generated through Pocket Finder were analyzed and compared with crystal structure of DRD3. It demonstrated residues already observed in crystal structure and also some additional potent active sites’ residues involved in the binding site of protein are shown in (Figure 1). The crystal structure and computational analysis of binding site support that Asp110, Ile183, Ser192, Phe346, His349, Thr369, Tyr373 act as active site residues in 3D crystal structure of human DRD3.31
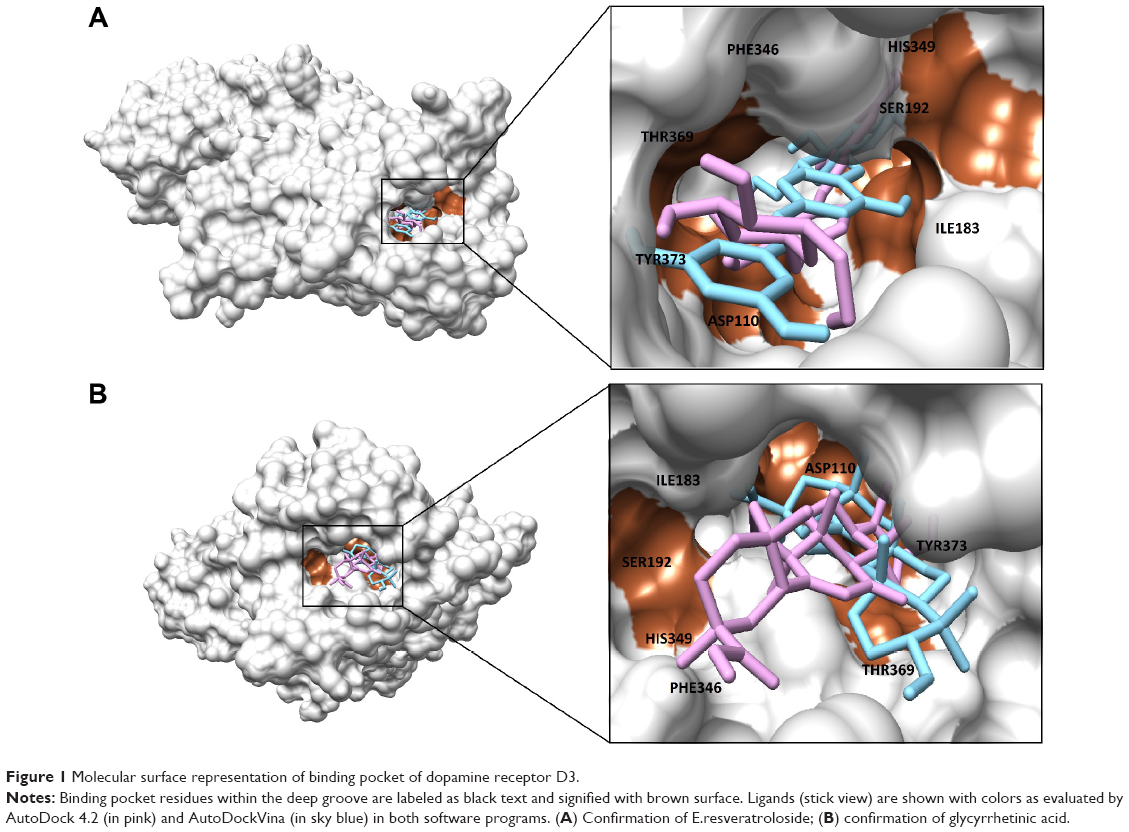 | Figure 1 Molecular surface representation of binding pocket of dopamine receptor D3. |
Molecular interactions
The location of drug binding is a crucial site for designing viable drug molecules against any disease. Therefore, the binding site was further confirmed with the help of CASTp and Pocket Finder. It was observed that DRD3 crystal structure shows valuable information of interacting residues, located in the binding pocket such as Asp110, Ile183, Ser192, Phe346, His349, Thr369, and Tyr373, respectively which play an important role in phytochemical binding. Our docking result with phytochemicals showed that many potential compounds were actively involved in hydrogen bonding with five polar residues such as, aspartic acid, serine, histidine, threonine, and tyrosine. Other crucial hydrophobic interactions were also found to exist that increase binding of phytochemicals with protein binding pocket (Table 5). In the docking results however, Thr369 was frequently seen to be a common residue amongst phytochemicals with high binding energies in the docking results. This is seen to be followed by Tyr373, Asp110, and Ile183 residues which also occur frequently. Thr369 and Tyr373 were also found to be common amongst the binding site of synthetic drugs used in the study namely bromocriptine, apomorphine, and ropinirole. Further, docking studies revealed the results with a binding score up to a maximum of −7.11 kcal/mol, which is higher than the binding energy range of US Food and Drug Administration (FDA) approved drugs (−4.88 to −5.81 kcal/mol). Analyses showed that the top nine compounds with considerable high affinity for DRD3 share a somewhat common active site. After careful observation of molecular interactions, it was recognized that the H-bond interactions with Thr369 and Tyr373 were reported in many phytochemicals that were showing binding energies greater than −6.00 kcal/mol, such as, glycyrrhetinic acid, E.resveratroloside, genkwanin, protopine, naringenin, pergolide, magnolol, honokiol, and hirsutanone. However, glycyrrhetinic acid and E.resveratroloside neatly fit into the binding pocket of DRD3 hence (Figure 1), justifying their high binding values as compared to other compounds.
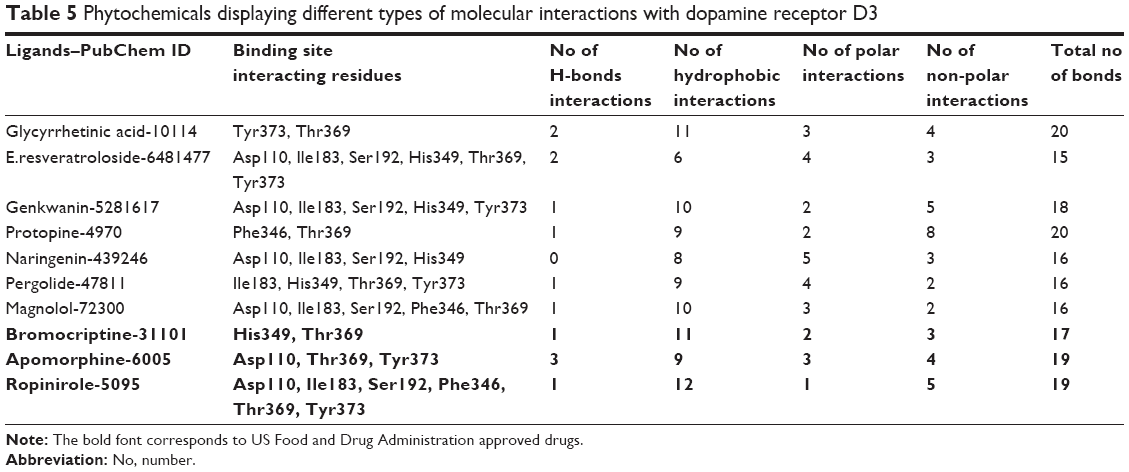 | Table 5 Phytochemicals displaying different types of molecular interactions with dopamine receptor D3 |
Comparative studies with AutoDockVina
The present comparative docking of phytochemicals with DRD3 using two different tools confirmed that AutoDockVina showed high binding energies as compared to AutoDock Tool. This is due to the fact that Vina uses a gradient optimization method in its local optimization procedure and has better accuracy of the binding mode prediction. Our results suggested that binding energy between ligand and target molecule has been enhanced in AutoDockVina. Binding energies calculated using AutoDock tool for glycyrrhetinic acid, E.resveratroloside, genkwanin, protopine, naringenin, pergolide, and magnolol were in the range (−6.14 to −7.11 kcal/mol). However, this range of binding energy increased (−8.3 to −12.7 kcal/mol) in the case of AutoDockVina and showed strong binding (Table 6). A varying trend of binding energies was seen amongst the phytochemicals docked in both tools. Flavonoids were observed to rank higher when docked in AutoDockVina as compared to AutoDock where a mixed trend of saponins, lignins, phenols, and alkaloids was seen to rank high.
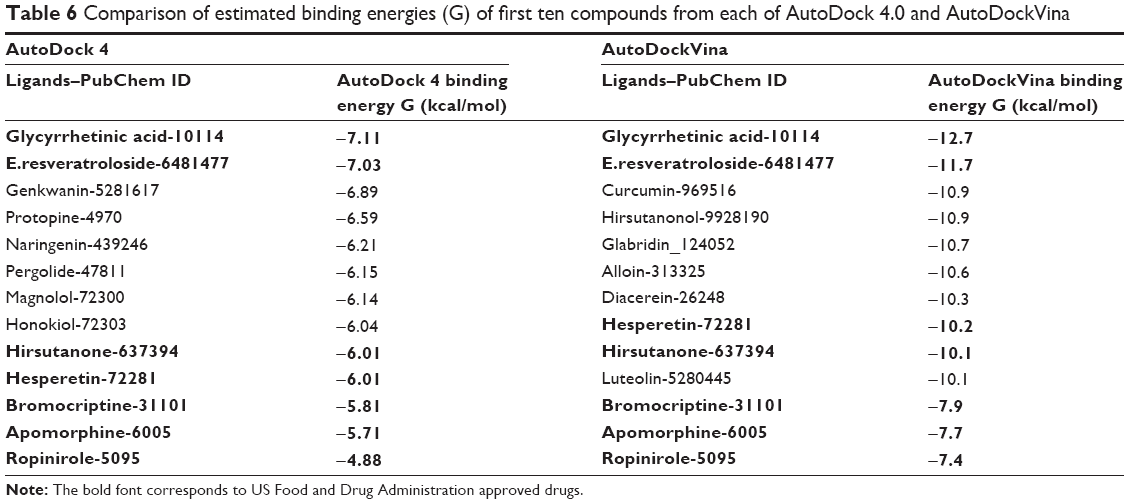 | Table 6 Comparison of estimated binding energies (G) of first ten compounds from each of AutoDock 4.0 and AutoDockVina |
Both AutoDock 4.0 and AutoDockVina use Monte-Carlo algorithm. In our experiment, we tried to find the most probable small molecules that may act against DRD3 to cure PD. Docking results were sorted and ranked on the basis of their binding energy values. The first ten compounds from each of AutoDock 4.0 and AutoDockVina results were chosen for further analysis. The comparison showed that four compounds were common that have higher binding energy values for DRD3. Except binding energy values, these four compounds were found to make more interactions with DRD3. These compounds were also compared with FDA approved drugs. The comparison with drugs showed that the most potent compounds, glycyrrhetinic acid and E.resveratroloside, showed the highest binding energy values when analyzed using both software and also presented strong interactions with DRD3 (Table 6).
Although differing in precision and binding affinities, glycyrrhetinic acid and E.resveratroloside were popularly recognized as lead compounds in both the software programs (Figure 1A, B). Both ligands were found to bind inside a deep groove surrounded by seven amino acid residues with considerably high binding energy values regardless of the tool being used. Figures 1 and 2 illustrate a detailed interaction of glycyrrhetinic acid and E.resveratroloside with DRD3 inside a deep groove.
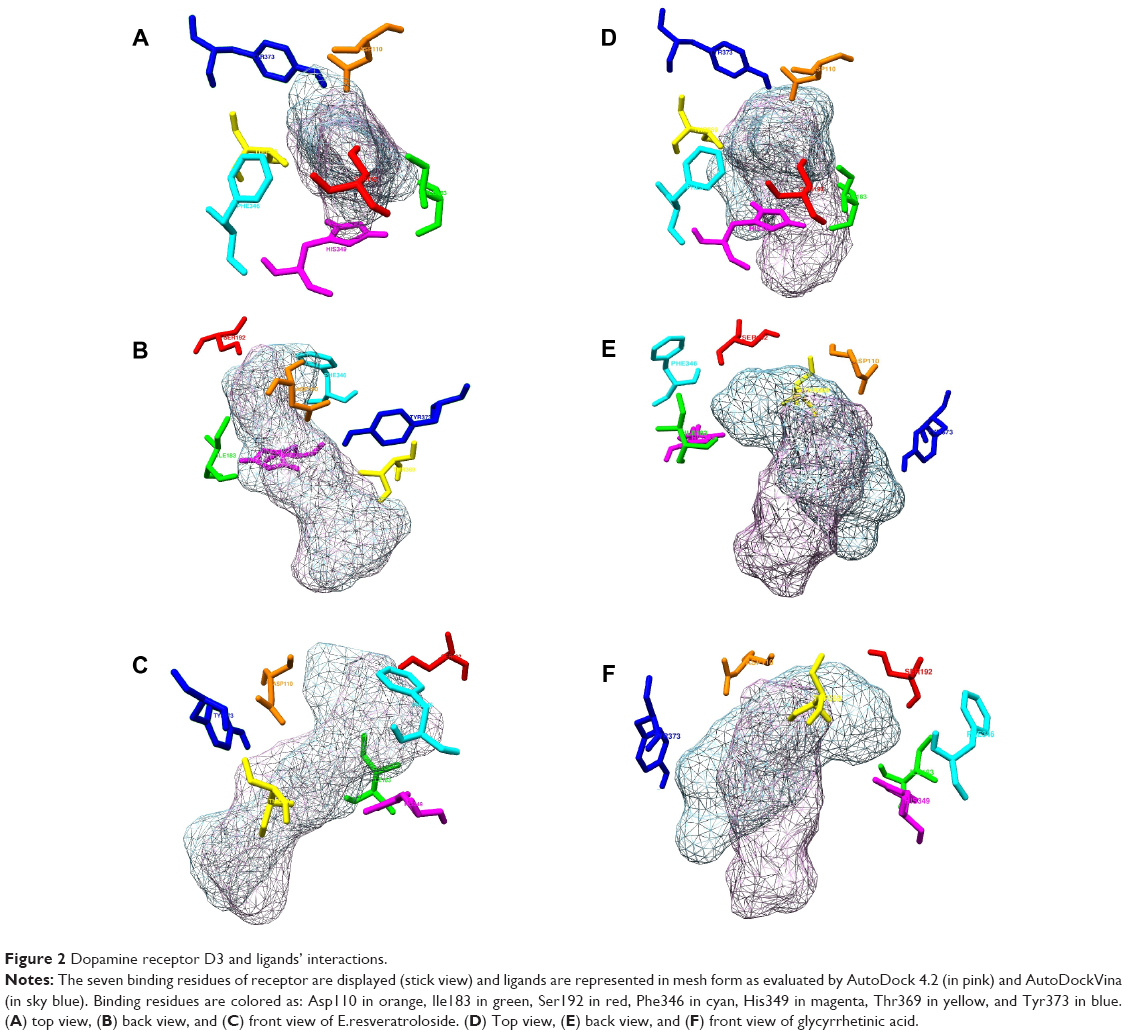 | Figure 2 Dopamine receptor D3 and ligands’ interactions. |
ADMET analysis
The molecular structures of potential ligands, glycyrrhetinic acid, E.resveratroloside, genkwanin, protopine, naringenin, pergolide, and magnolol were submitted to Mcule, Molsoft, Molinspiration, admetSAR, and Osiris property servers to determine their different properties including drug likeness and ADMET properties. All these potential compounds followed the Lipinski’s rule of five without any violation with respect to an octanol-water partition coefficient (LogP ≤5), molecular weight (≤500 KDa), number of H-bond donors (≤5), number of H-bond acceptors (≤10), molecular refractivity (40–130) as tabulated in Table 1. In ADMET assessment, different pharmacokinetic and pharmacodynamic parameters were considered such as aqueous solubility,46 human intestinal absorption,47 blood–brain barrier penetration, Caco-2 permeability, cytochrome P450 inhibition,48 cytochrome P (CYP) inhibitory promiscuity, renal organic cation transportation, human ether-a-go-go-related genes inhibition, rat acute toxicity, fish toxicity, Tetrahymena pyriformis toxicity, AMES toxicity, tumorigenic, reproductive, and mutagenic risks. The results have been summarized in Table 2. The bioavailability and toxicity risks of the potential compounds were predicted based on their ADMET properties. Interestingly, the analysis performed on admetSAR and Osiris property explorer revealed that only glycyrrhetinic acid, E.resveratroloside, and pergolide had no substantial ADMET properties that could cause adverse effects in humans. Whereas genkwanin, naringenin, and magnolol have the potential to show adverse effects in recipients. The analysis displays high CYP inhibitory promiscuity, as they inhibit most of the cytochrome P450 isoforms containing CYP450 1A2, 2C9, 2C19, and 3A4.49 The cytochrome P450 superfamily shows a significant role in metabolizing the drug and its clearance in the liver. Therefore, the inhibition of cytochrome P450 isoforms might affect the drug metabolism and elevate the toxicity level.50 The analysis also inferred that, except pergolide, none of the potential compounds were potential compounds of the human ether-a-go-go-related gene. In fact, other than pergolide, none of the compounds were carcinogenic, tumorigenic or irritant to humans. Most interestingly, it was noticed that both lead compounds, glycyrrhetinic acid and E.resveratroloside, indicated the most favorable ADMET properties. They were seen to show negative results for AMES toxicity, carcinogenicity, mutagenicity, and tumorigenicity. Both compounds also showed promising human intestinal absorption, blood–brain barrier ability and solubility, and potential non-inhibitor of CYP450 1A2, 2C9, 2D6, 2C19, 3A4, as these factors help in metabolizing and in flushing out the drugs from the body.49
Conclusion
The ascription of all the unique benefits to the DRD3 agonists is currently under investigation and there is a possibility of an increase in therapeutic index of regimes using these agonists. However, an increase in potency of drugs is evident using DRD3 agonists. Anti-parkinsonian efficacy has also dramatically increased using DRD3 agonists via the autotrophic factor pathway. A profound therapeutic leap seems to be promising using these agonists.
In the current study 40 phytochemicals were retrieved from a literature survey and docked using AutoDock and AutoDockVina against DRD3 to find potent lead compounds for PD treatment. The compounds were also assessed for their ADMET properties. From this study it can be concluded that Thr369, Tyr373, Asp110, and Ile183 are likely target sites for designing drugs against PD. Also, glycyrrhetinic acid and E.resveratroloside have been identified as potential compounds that should be scrutinized for use against PD as potent drugs. These two compounds have shown strong binding affinity for DRD3. The ligand-receptor complex of glycyrrhetinic acid and E.resveratroloside shows strong hydrogen bonding as well as van der Waals forces between the two. It is apparent that this bonding firmly grips the ligand into the receptor’s binding pocket. We performed an in-depth analysis of protein binding site and comparative docking to ascertain best docked poses of ligands. The best ligands we found were based on making high interactions with protein residues and had minimum energy values. The ligand docking results were compared with docking results of FDA approved drugs. Interestingly, a wide difference in affinity values was observed between glycyrrhetinic acid, E.resveratroloside, and FDA approved drugs. A consensus of result was observed when docking results of AutoDock and AutoDockVina were evaluated. Further, glycyrrhetinic acid and E.resveratroloside also exhibited good ADMET profiles and can be considered safe for development into a commercial drug. These results are certainly enlightening that glycyrrhetinic acid and E.resveratroloside can be considered as a template for future drug designing against PD.
Acknowledgment
We thank Mr Sohaib Mushtaq for excellent pharmacological assistance in reviewing the pharmacology of DRD3 with special reference to PD.
Disclosure
The authors declare that they have no conflict of interest.
References
Tanner CM. The role of environmental toxins in the etiology of Parkinson’s disease. Trends in Neurosciences. 1989;12(2):49–54. | ||
Rajput AH, Uitti RJ, Stern W, et al. Geography, drinking water chemistry, pesticides and herbicides and the etiology of Parkinson’s disease. Can J Neurol Sci. 1989;14(3 Suppl):414–418. | ||
Hubble JP, Cao T, Hassanein RES, Neuberger JS, Roller WC. Risk factors for Parkinson’s disease. Neurology. 1993;43(9):1693–1693. | ||
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat. 2010;31(7):763–780. | ||
Jellinger KA. Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol. 1991;14(3):153–197. | ||
Lloyd KG, Davidson L, Hornykiewicz O. The neurochemistry of Parkinson’s disease: effect of L-dopa therapy. J Pharmacol Exp Ther. 1975;195(3):453–464. | ||
Jellinger K, Paulus W, Grundke-Iqbal I, Riederer P, Youdim MB. Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J Neural Transm Park Dis Dement Sect. 1990;2(4):327–340. | ||
Dauer W, Przedborski S. Parkinson’s disease: Mechanisms and Models. Neuron. 2003;39(6):889–909. | ||
Singh N, Pillay V, Choonara YE. Advances in the treatment of Parkinson’s disease. Prog Neurobiol. 2007;81(1):29–44. | ||
De Rijk MC, Launer LJ, Berger K, et al. Prevalence of Parkinson’s disease in Europe: a collaborative study of population based cohorts. Neurology. 2000;54(11 Suppl 5):21–23. | ||
Pahwa R, Lyons KE. Handbook of Parkinson’s disease. 4th ed. New York: Informa Healthcare Publishers; 2007. | ||
Kebabian JW, Calne DB. Multiple receptors for dopamine. Nature. 1979;277(5692):93–69. | ||
Stoof JC, Kebabian JW. Two dopamine receptors: biochemistry, physiology and pharmacology. Life Sci. 1984;35(23):2281–2296. | ||
Jayadeepa RM, Niveditha MS. Computational approaches to screen candidate ligands with anti-Parkinson’s activity using R programming. Curr Top Med Chem. 2012;12(16):1807–1814. | ||
Joyce JN, Millan MJ. Dopamine D3 receptor agonists for protection and repair in Parkinson’s disease. Curr Opin Pharmacol. 2007;7(1):100–105. | ||
Van Kampen JM, Eckman CB. Dopamine D3 receptor agonist delivery to a model of Parkinson’s disease restores the nigrostriatal pathway and improves locomotor behavior. J Neurosci. 2006;26(27):7272–7280. | ||
Carvey PM, McGuire SO, Ling ZD. Neuroprotective effects of D3 dopamine receptor agonists. Parkinsonism Relat Disord. 2001;7(3):213–223. | ||
Rezak M. Current Pharmacotherapeutic treatment options in Parkinson’s disease. Dis Mon. 2007;53(4):214–222. | ||
Katzung BG. Pharmacological management of parkinsonism and other movement disorders. In: Katzung BH, editor. Basic and Clinical Pharmacology 8th ed. New York: Lange Medical Books/McGraw Hill, Inc; 2001. | ||
Jankovic J. Motor fluctuations and dyskinesias in Parkinson’s disease: clinical manifestations. Mov Disord. 2005;20 Suppl 11:S11–S16. | ||
Olanow CW, Agid Y, Mizuno Y. Levodopa in the treatment of Parkinson’s disease: current controversies. Mov Disord. 2004;19(9):997–1005. | ||
Saint-Cyr JA, Taylor AE, Lang AE. Neuropsychological and psychiatric side effects in the treatment of Parkinson’s disease. Neurology. 1993;43(12 Suppl 6):S47–S52. | ||
Dodd ML, Klos KJ, Bower JH, Geda YE, Josephs KA, Ahlskog JE. Pathological gambling caused by drugs used to treat Parkinson disease. Arch Neurol. 2005;62(9):1377–1381. | ||
Sudha K, Rao AV, Rao S, Rao A. Free radical toxicity and antioxidants in Parkinson’s disease. Neurol India. 2003;51(1):60–62. | ||
Roghani M, Behzadi G. Neuroprotective effect of Vitamin E on the early model of Parkinson’s disease in rat: behavioral and histochemical evidence. Brain Res. 2001;892(1):211–217. | ||
Nicoletti G, Crescibene L, Scornaienchi M, et al. Plasma levels of Vitamin E in Parkinson’s disease. Arch Gerontol Geriatr. 2001;33(1):7–12. | ||
Etminan M, Gill SS, Samii A. Intake of Vitamin E, Vitamin C, and carotenoids and the risk of Parkinson’s disease: a meta-analysis. Lancet Neurol. 2005;4(6):362–365. | ||
Paraskevas GP, Kapaki E, Petropoulou O, Anagnostouli M, Vagenas V, Papageorgiou C. Plasma levels of antioxidant vitamins C and E are decreased in vascular parkinsonism. J Neurol Sci. 2003;21(1–2):51–55. | ||
Alavijeh MS, Chishty M, Qaiser MZ, Palmer AM. Drug Metabolism and Pharmacokinetics, the Blood–Brain Barrier, and Central Nervous System Drug Discovery. NeuroRx. 2005;2(4):554–571. | ||
Rosen DM, Grimes K, editors. Discovery and Preclinical Work. A Practical Guide to Drug Development in Academia. Springer International Publishing; 2014. | ||
Chien EY, Liu W, Zhao Q, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330(6007):1091–1095. | ||
Kita T, Asanuma M, Miyazaki I, Takeshima M. Protective Effects of Phytochemical Antioxidants Against Neurotoxin–Induced Degeneration of Dopaminergic Neurons. J Pharmacol Sci. 2014;124(3):313–319. | ||
Ebrahimi A, Schluesener H. Natural polyphenols against neurodegenerative disorders: Potentials and pitfalls. Ageing Res Rev. 2012;11(2):329–345. | ||
Siddique YH, Naz F, Jyoti S. Effect of Curcumin on Lifespan, Activity Pattern, Oxidative Stress, and Apoptosis in the Brains of Transgenic Drosophila Model of Parkinson’s Disease. Biomed Res Int. 2014;2014:606928. | ||
Li Q, Cheng T, Wang Y, Bryant SH. PubChem as a public resource for drug discovery. Drug Discov Today. 2010;15(23–24):1052–1057. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. | ||
Case DA, Darden TA, Cheatham III TE, et al. AMBER 9. University of California, San Francisco. 2006. Available from: http://ambermd.org/. Accessed November 24, 2014. | ||
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server issue):W116–W118. | ||
Laurie AT, Jackson RM. Q–SiteFinder: an energy-based method for the prediction of protein–ligand binding sites. Bioinformatics. 2005;21(9):1908–1916. | ||
Hendlich M, Rippmann F, Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model. 1997;15(6):359–363. | ||
Trott O, Olson AJ. AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. | ||
Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1(4):337–341. | ||
Kiss R, Sandor M, Szalai, FA. http://Mcule.com: a public web service for drug. J Cheminform. 2012;4(Suppl 1):17. | ||
Cheng F, Li W, Zhou Y, et al. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099–3105. | ||
Cheng A, Merz KM Jr. Prediction of aqueous solubility of a diverse set of compounds using quantitative structure-property relationships. J Med Chem. 2003;46(17):3572–3580. | ||
Egan WJ, Merz KM Jr, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43(21):3867–3877. | ||
Susnow RG, Dixon SL. Use of robust classification techniques for the prediction of human cytochrome P450 2D6 inhibition. J Chem Inf Comput Sci. 2003;43(4):1308–1315. | ||
Vasanthanathan P, Taboureau O, Oostenbrink C, Vermeulen NP, Olsen L, Jørgensen FS. Classification of cytochrome P450 1A2 inhibitors and noninhibitors by machine learning techniques. Drug Metab Dispos. 2009;37(3):658–664. | ||
Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76(3):391–396. |
Supplementary material
 | Table S1 Molecular properties of compounds under study |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.