")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Glutathione S-transferase π: a potential role in antitumor therapy
Authors Dong S, Sha H, Xu X, Hu T, Lou R, Li H, Wu J, Dan C, Feng J
Received 31 March 2018
Accepted for publication 27 June 2018
Published 23 October 2018 Volume 2018:12 Pages 3535—3547
DOI https://doi.org/10.2147/DDDT.S169833
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Shu-Chen Dong,1,* Huan-Huan Sha,1,* Xiao-Yue Xu,1 Tian-Mu Hu,2 Rui Lou,1 Huizi Li,1 Jian-Zhong Wu,1 Chen Dan,1 Jifeng Feng1
1Jiangsu Cancer Hospital and Jiangsu Institute of Cancer Research and Nanjing Medical University Affiliated Cancer Hospital, Nanjing 210009, China; 2Department of Biological Science, Purdue University, West Lafayette, IN, USA
*These authors contributed equally to this work
Abstract: Glutathione S-transferase π (GSTπ) is a Phase II metabolic enzyme that is an important facilitator of cellular detoxification. Traditional dogma asserts that GSTπ functions to catalyze glutathione (GSH)-substrate conjunction to preserve the macromolecule upon exposure to oxidative stress, thus defending cells against various toxic compounds. Over the past 20 years, abnormal GSTπ expression has been linked to the occurrence of tumor resistance to chemotherapy drugs, demonstrating that this enzyme possesses functions beyond metabolism. This revelation reveals exciting possibilities in the realm of drug discovery, as GSTπ inhibitors and its prodrugs offer a feasible strategy in designing anticancer drugs with the primary purpose of reversing tumor resistance. In connection with the authors’ current research, we provide a review on the biological function of GSTπ and current developments in GSTπ-targeting drugs, as well as the prospects of future strategies.
Keywords: tumor resistance, glutathione S-transferase pi, drug treatment
Introduction
In addition to combating a variety of noxious substances from the external environment, cell-detoxification mechanisms are capable of resisting the deleterious effects of certain endogenous substances (eg, ROS, a product generated from normal cellular metabolism), in order to maintain physiological homeostasis. Drug metabolism represents an important component of cellular detoxification and involves two enzymes: Phase I and Phase II drug-metabolism enzymes. The Glutathione S-transferase π (GST) family of enzymes is a group of typical Phase II detoxification enzymes and is found in many prokaryotes and eukaryotes.1 Of these enzymes, GSTπ catalyzes the conjunction between GSH and its electrophilic substrates upon exposure to damaging free radicals. Besides metabolite detoxification, GSTπ also exhibits ligand-binding properties that initiate cellular apoptosis when triggered by cellular stress.2 Further research has also demonstrated that GSTπ is expressed abundantly in tumor cells and associated closely to carcinogenesis, tumor formation, and chemotherapy resistance.3,4 Moreover, experiments involving drug-resistant cell lines have also demonstrated increased GSTπ expression.5,6 In multidrug-resistant HL60/VCR acute myelogenous leukemia cells, GSTπ is found to be expressed at higher levels than HL60.7 GSTπ is involved in facilitating tumor resistance and suppressing apoptosis in tumor cells via two mechanisms. First, GSTπ weakens the efficacy of chemotherapy drugs by promoting their in vitro extrusion. Second, GSTπ also functions as an MAPK-pathway inhibitor to prevent tumor-cell apoptosis.
A variety of anticancer drugs based on these principles have been synthesized in efforts to improve their therapeutic indices and reverse tumor resistance. Drugs that work through the GST system include GSTπ inhibitors and their respective prodrugs. The former work by exerting high GSTπ-inhibitory activity, while the latter comprise inactive compounds designed to target tumor tissue locally by undergoing GSTπ catalysis in the tumor to release cytotoxic metabolites.8 Development of therapies targeting GSTπ is a major field of research. As such, we believe that there is a need for more detailed studies outlining the diverse biological functions of GSTπ, in order to assist drug discovery and unlock more exciting possibilities in the realm of tumor treatments.
Structure
GSTs consist of the following three superfamilies: cytoplasmic (cGSTs), mitochondrial (κGSTs), and microsomal (membrane-associated proteins in eicosanoid and glutathione metabolism [MAPEG]). Among these families, cGSTs are the most complex and most closely linked to the development of human diseases.9,10 The cGSTs are divided into seven subtypes according to similarities in amino-acid sequence, different structure of genes, and immunological cross-reactivity. These subtypes are α, π, μ, θ, ω, σ, and δ.11 Among them, GSTα is highly expressed in many normal cells.12 However, recent studies have shown that GSTα also takes part in promoting multidrug resistance in p53-mutated lung cancer cells.13 GSTμ has been found to be able to act synergistically with MRP1 to decrease the effects of vincristine treatment.14 GSTπ is widespread in tumor cells, and is intricately involved with cellular carcinogenesis, tumor formation, and tumor-drug resistance.15 Current evidence supports the role of cGSTs in facilitating multidrug resistance across different types of tumors.16,17
GSTπ is the most frequently and extensively studied of all the GSTs. Its encoding gene is located on chromosome 11 and is composed of seven exons. In humans, GSTπ often consists of two identical dimer subunits, with each subunit consisting of 210 amino acids and two binding sites, the G-site and the H-site (Figure 1). Different G- and H-site locations in the amino-acid residue of different GSTs exert different functions. GSTπ is able specifically to bind to GSH or GSH analogs via the G-site, which catalyzes the interaction between GST amino-acid residues with GSH sulfhydryl and conventional electrophilic substances at the H-sites to promote catalytic action.18 Therefore, G-site modification often guides the development of specific GSTπ inhibitors.
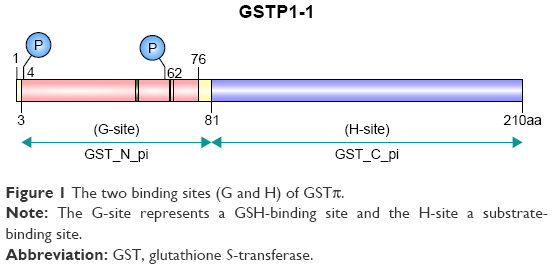 | Figure 1 The two binding sites (G and H) of GSTπ. |
Biological function
GSTπ in metabolite detoxification and antioxidation
The classical view holds that as a dimeric isoenzyme, GSTπ can conjugate GSH with substrate molecules in efforts to promote clearance of active ionic substances.19 However, tumor cells also utilize GSTπ to form a GSH–X complex between antitumor drugs and GSH, proceeding to excrete the complex out of the cell by Pgp and MRP. Pgp, encoded by MDR1, is often found to be highly expressed in tumor cells.6,20 What is more, GSTπ and Pgp or MRPs (eg, MRP2) are synergistic in driving the development of multidrug resistance in tumor cells21 (Figure 2). Studies have documented high GSTπ expression in various tumor cells, such as cancers of the gastrointestinal tract,6 pancreas,22 breast,23 liver,24 lymphoma,25 as well as melanoma.26
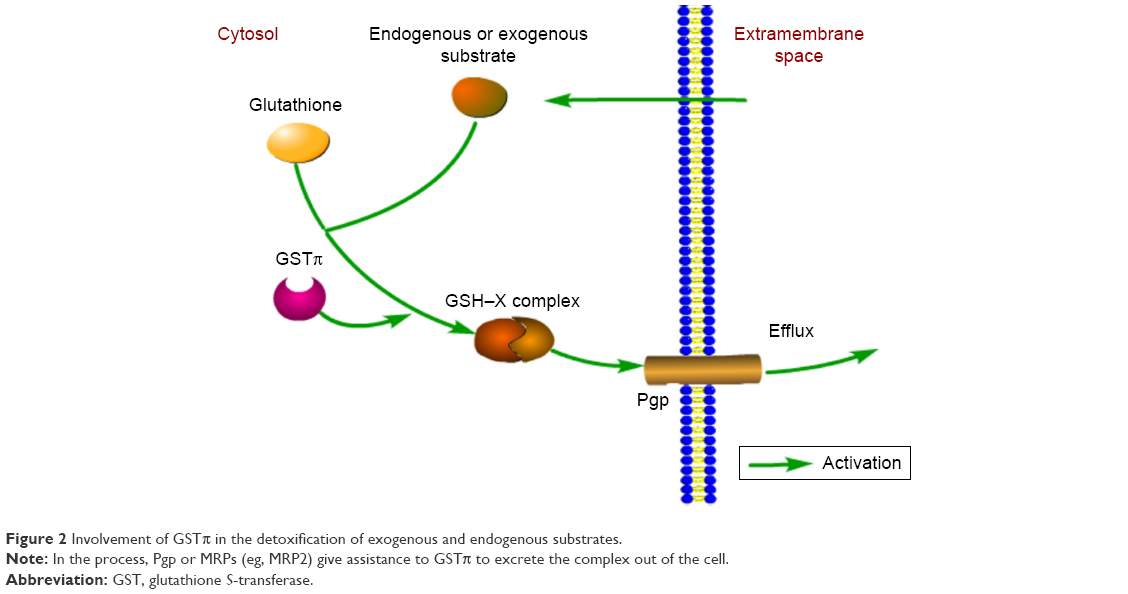 | Figure 2 Involvement of GSTπ in the detoxification of exogenous and endogenous substrates. |
Recent literature has characterized GSH and other related metabolic enzymes as vital in protecting cells from ROS27 through oxidation and reduction (redox) mechanisms.28 GSH carries a cysteine residue with an active thiol group and is responsible for maintaining thiol equilibrium. Meanwhile, it can also modulate the activities of many signaling molecules and redox-sensitive transcription factors through S-glutathionylation, a form of posttranslational modification that combines cysteine residues with GSH.29 GSTπ serves as a general S-glutathionylase enzyme and promotes S-glutathionylation. Its enzymatic function is based on two aspects: its catalytic activity and the auto-S-glutathionylation of GSTπ by itself on Cys47 and Cys10, both of which disturb the subsequent interaction with c-Jun NH2-terminal kinase (JNK), resulting in the formation of a GSTπ multimer30 (Figure 3). Besides GSTπ, other members of the GSH-redox system, such as glutamate cysteine ligase,31 glutathione peroxidase, and glutathione reductase, also play significant roles in this process.32
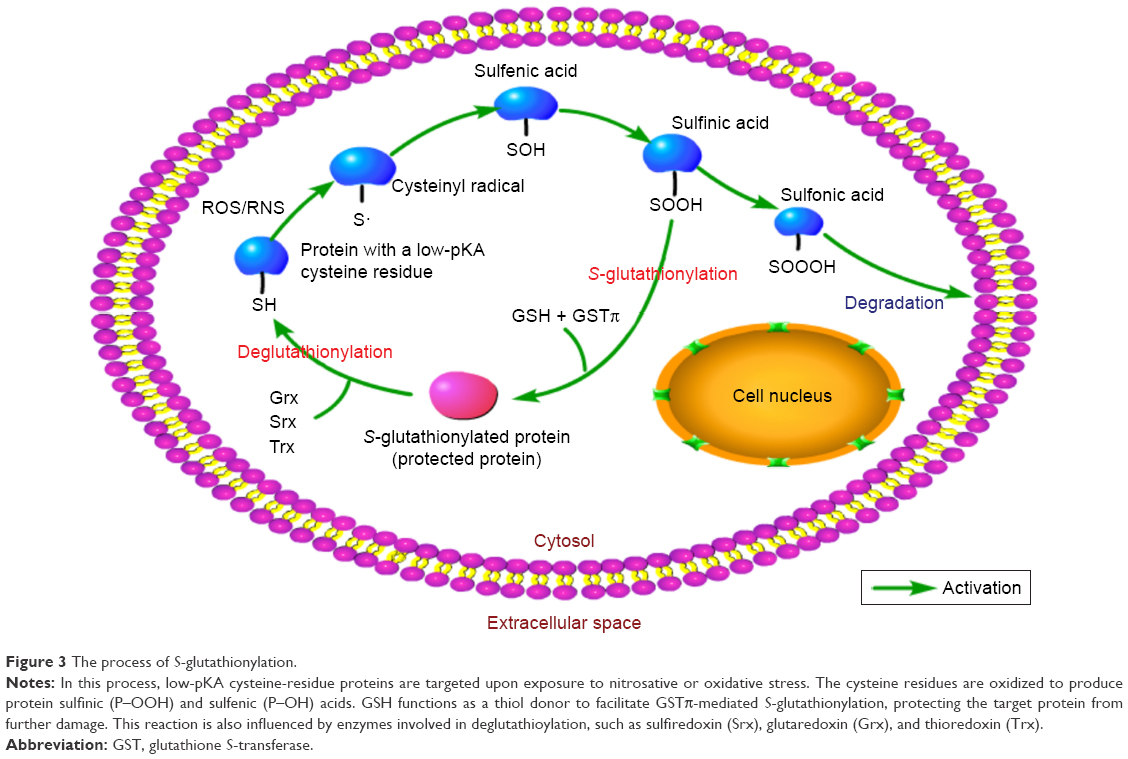 | Figure 3 The process of S-glutathionylation. |
GSTπ in regulation of MAPK pathway
Besides metabolite detoxification, GSTπ also exhibits ligand-binding properties that allow the enzyme to interact covalently and noncovalently with compounds, resulting in inhibition of conjugation activity.18 GSTπ can induce cellular apoptosis in the setting of cellular stress by activating MAPK, MKK4, downstream JNK-signal components, and p38 kinase. Normal cells have low basal JNK activity to maintain optimal cellular growth conditions. However, in the presence of oxidative or nitrosative stress, GSTπ can form homodimers that alter the reduced states of cysteine residues in its structure, resulting in JNK dissociation from the GSTπ–JNK heterocomplex and causing subsequent activation of the c-Jun protein. Ultimately, these series of reactions will activate the apoptotic pathways33,34 (Figure 4).
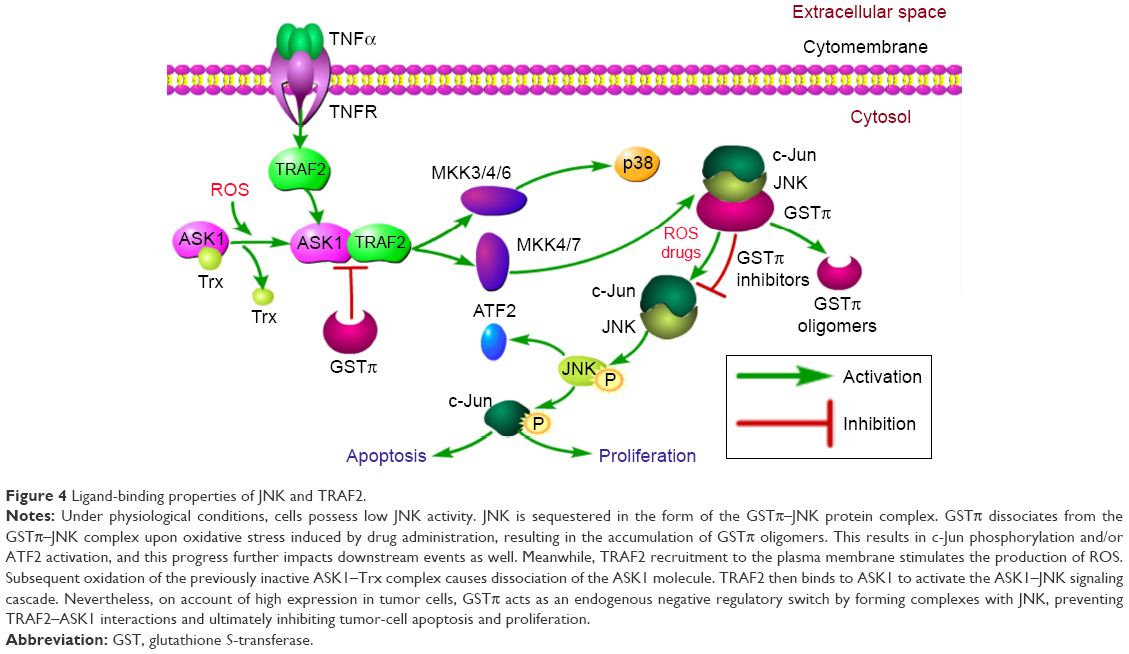 | Figure 4 Ligand-binding properties of JNK and TRAF2. |
Further research indicates that GSTπ can influence the MAPK pathway both through JNK and TRAF2 modulation. In-depth analysis of biological information tools has revealed that the TRAF family is strongly linked to GSTπ. Of all the TRAF members, TRAF2 is expressed most abundantly and has been subjected to intense research.35 Following the activation of TNFR though TNFα binding, TRAF2 is recruited to the plasma membrane, resulting in the production of ROS. ROS generation leads to oxidation of the ASK1 inhibitor thioredoxin, separating and activating ASK1 from the inactive ASK1–thioredoxin complex. ASK1 goes on to bind to TRAF2, which in turn activates downstream-signaling cascades, including the MKK3/4/6–p38 and MKK4/7–JNK signaling pathways36,37 (Figure 4). Further evidence from steady-state fluorescence analysis confirms that direct binding between TRAF2 and GSTπ also exists.38–42 In tumor cells, GSTπ can suppress JNK activity and block the interaction between TRAF2 and ASK1 to inhibit tumor-cell apoptosis. Consequently, regarding the formation of multidrug resistance in tumor cells, besides acting as a detoxification enzyme though excretion of drugs to decrease pharmacological efficacy, GSTπ can also act as an MAPK-pathway inhibitor to improve tumor-cell survival. GSTπ also represents a scaffold protein to unite different members across signaling pathways.
Other functions
GSTπ also functions as a chaperone protein that regulates common but significant cellular functions. It interacts with several key cellular proteins, including TGM243 and FANCC.44 Research has found STAT3 to be an active factor in signal transduction and transcription. STAT3 overexpression is a key molecule that drives the progression of hepatocellular carcinoma, and its activation may be critical in initiating oncogenesis.33,45,46 There are studies illustrating that GSTπ interaction with STAT3 can inhibit the STAT3-signaling pathway, curb aberrant cell-cycle progression, and decrease cell proliferation.36 Furthermore, GSTπ can participate in nonhomologous end-joining DNA repair by inhibiting DNA-dependent protein kinase.47 In conclusion, these findings mentioned show that seemingly disparate functions of GSTπ in fact work on several aspects of tumor carcinogenesis, making it an ideal molecule for further antitumor therapy research.
Gene variants and polymorphisms
The study of pharmacogenomics in recent decades has proved that genetic polymorphism of drug-metabolizing enzymes is an important mediating factor in determining individual drug responses. Genetic polymorphism is due to a single-nucleotide mutation in the genomic sequence that fundamentally changes how a person responds to chemotherapeutic drugs. Polymorphisms affecting GSTπ have been documented to exert significant modulatory effects on the biological cascade of carcinogenesis and have been discussed vigorously in the literature.48
The human GSTP1 gene exists as two functionally different variants, with both variants having documented nucleotide transitions from isoleucine to valine at codon 105 (A→G) and alanine to Val at 114 (C→T). This results in four alleles of GSTP1: wild-type GSTP1*A (Ile105 + Ala114), GSTP1*B (Val105 + Ala114), GSTP1*C (Val105 + Val114), and GSTP1*D (Ile105 + Val114).49 Besides GSTP1, cytosolic GSTs demonstrate clinically significant gene polymorphism (Table 1). These changes occur in active sites, leading to a decrease in encoded protein activity, decreased excretion of foreign substances, and suboptimal catalytic efficiency. On the other hand, reducing the rate of drug excretion has the benefit of improving an individual’s sensitivity to chemotherapeutic drugs, enhancing their curative effects.50–52
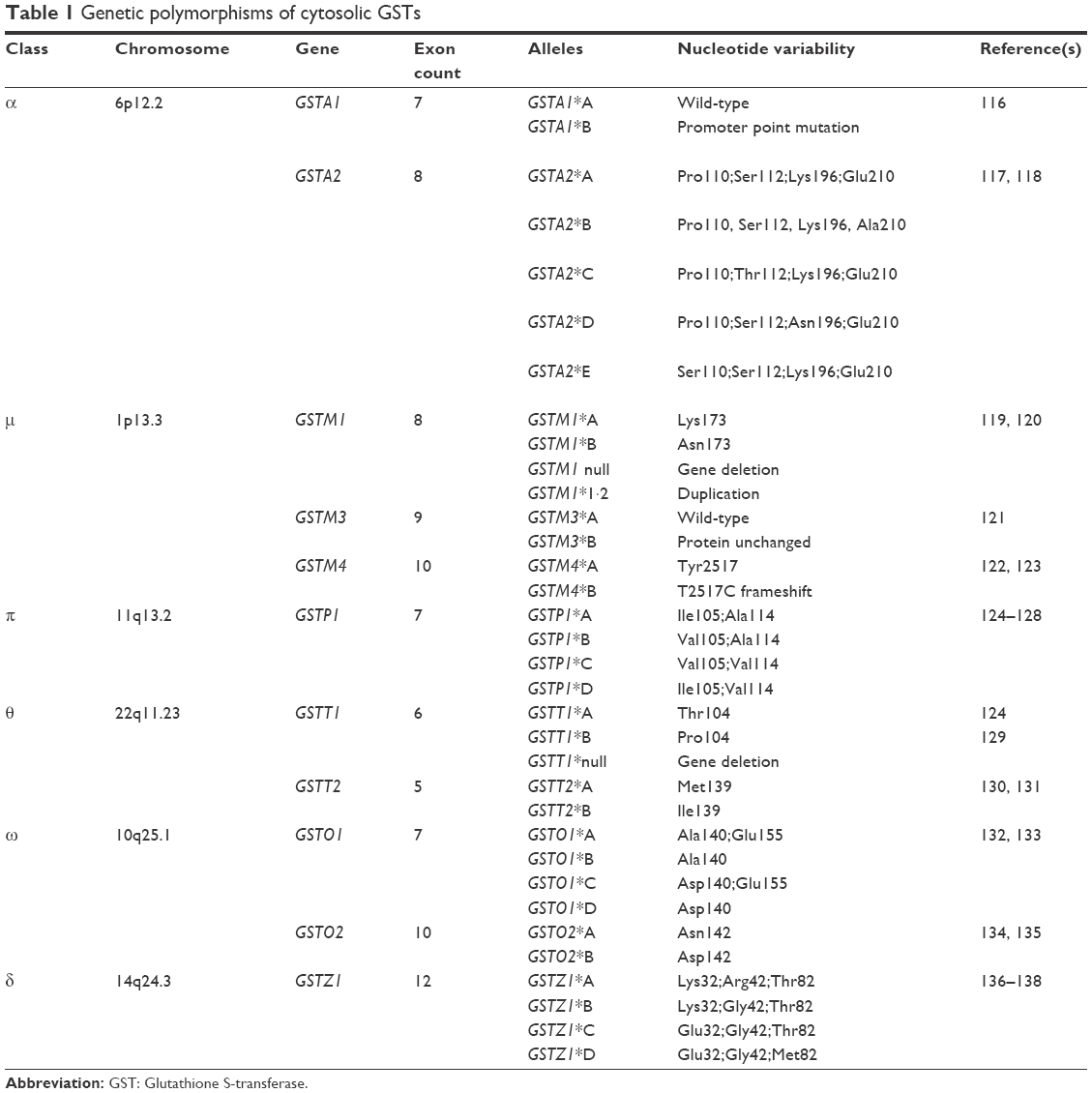 | Table 1 Genetic polymorphisms of cytosolic GSTs |
There are a great number of reviews that have summarized the associations between individual GSTπ variability and the drug sensitivity of malignant tumors.53–55 However, data are scarce regarding the relationship between GSTπ and clinical response to chemotherapy. Khrunin et al showed that 104 patients with ovarian cancer with mutant-type GSTπ possessed longer progression-free survival compared to wild-type GSTπ.56 These results highlight the possibility that genetic variation may have significant effects on susceptibility toward cancer. In future, precise genetic polymorphic screening tests may play a more central role in determining chemotherapeutic treatment regimens for patients.
GSTπ inhibitors
GSTπ inhibitors reverse tumor resistance by means of suppressing GSTπ activity and improving the chemotherapeutic drug sensitivity of tumor cells. Ethacrynic acid (EA) is a classic GSTπ inhibitor.57 However, due its aspecific pharmacological properties in targeting GSTπ, the newer GSTπ inhibitors TLK117/TLK199 and NBDHEX may prove to be more promising. Antitumor agents targeting GSTπ in context are listed in Table 2.
 | Table 2 Antitumor agents targeting GSTπ in context |
EA and its analogs
EA represents the first clinical application of GSTπ inhibitors. Previously, it was widely used for decades as a diuretic in clinical research. EA works to halt GSTπ activity through a number of mechanisms. First, it is able to bind directly to substrate-binding sites of isozymes to inhibit GSTπ. Second, it is able to induce the combination of α,β-unsaturated ketones and GSH through the nucleophilic addition reaction, depleting GSH and reducing the amount of GSH available to combine with chemotherapeutic agents, thus producing an overall GSTπ-inhibitory effect by sensitizing a cell to chemotherapeutic agents.57 However, the clinical applications of EA have been limited, due to its diuretic properties and lack of enzyme specificity, with long-term intake possibly risking water and salt imbalance.18
Zhao et al attempted to modify EA using thiazole derivatives of uric acid to strengthen its GSTπ-inhibitory effects. The team demonstrated that these derivatives had higher GSTπ-inhibitory activity in comparison to unmodified EA when administered to acute myeloid leukemia parental cells (HL60).58 In addition, the combination of EA and GSH has also been proven to possess superior inhibitory activity over EA alone and is able functionally to inhibit many GST isoenzymes. However, this compound also possesses limited clinical viability, given its tendency toward dissociation by γ-glutamyltransferase.59 Burg et al synthesized modified peptidomimetic glutathione analogs of these EA–GSH compounds, which were hypothesized to be stabler against peptidase-mediated dissolution. Unfortunately, these analogs instead reduced its GSTπ-inhibitory activity, despite demonstrating increased resilience toward γ-glutamyltransferase compared to the unmodified EA–GSH compounds.57 Taken together, EA and its analogs still represent novel avenues of research in the search for more efficacious antitumor drugs.
TLK117 and TLK199
Telintra (ezatiostat hydrochloride, TER199, TLK199) is a small-peptide, glutathione-analog molecule and was developed by Telik. Upon entering the body, TLK199 undergoes esterase hydrolysis, which releases TLK117, its activated form that has anti-GSTπ activity. TLK199 is able to enhance the potency of various antineoplastic agents against various tumor cell lines. The agent is also able to inhibit MRAP1 and prevent the combination of GSTπ and JNK, resulting in high JNK production that triggers tumor-cell apoptosis.60
Furthermore, clinical studies have found TLK199 to be able to promote the maturation of hematopoietic progenitor cells, induce cancer-cell death, and inhibit myeloproliferative diseases.61–63 In 2013, TLK199 successfully passed a US Food and Drug Administration audit and was approved to treat low- to intermediate-risk myelodysplastic syndrome. Long-term observation studies have highlighted the ability of TLK199 to enhance bone-marrow maturation and cellularity.64
NBDHEX and its analogs
NBDHEX (6-[7-nitro-2,1,3-benzoxadiazol-4-ylthio]hexanol) is a recently developed compound designed as a “mechanism-based inhibitor” that exerts potent effects on GSTπ. Since its first reports by the Tor Vergata University of Rome,65 numerous preclinical studies have shown that NBDHEX exerts high GSTπ-inhibitory activity across a wide range of tumor types. Pasello et al reported that this agent effectively reversed cisplatin resistance in osteosarcoma, alluding toward potentially improved clinical outcomes when using a combination of NBDHEX and cisplatin.66 NBDHEX GSTπ-inhibitory effects have also been observed in HL60 cells and their chemotherapy-resistant phenotype HL60/DNR.67 Other cell lines that have demonstrated NBDHEX sensitivity include Ewing sarcoma,68 the human mesothelioma cell lines MPP89, MMB1, MSTO211H, and Mero48a,69 melanoma cell lines Me501 and A375,42 and the non-small-cell lung cancer cell line H69AR.70 Further research indicates that aside from inherent antimelanoma activity, NBDHEX also has the ability to enhance the function of temozolomide, with the two able to work synergistically to suppress tumor growth.71
NBDHEX employs several methods in combating malignant cells. First, it is able to accumulate specifically in tumor cells, remaining unaffected by MRP while maintaining good cell-membrane permeability. Second, this agent can decompose the GSTπ–JNK complex and promote activation of the apoptosis pathway.42,65,67,70–72 Further research has also suggested that TRAF2 plays a key role in facilitating NBDHEX-mediated apoptosis. NBDHEX simultaneously activates JNK and TRAF2, interfering with the effect of GSTπ via two different pathways, leading to cell-cycle arrest and cell death.34 Intriguingly, researchers have provided further evidence to demonstrate that NBDHEX can also act as an autophagy inhibitor in tumor cells.40
Despite NBDHEX’s promising anticancer activity, consideration should be given to the relatively low GSTπ-target selectivity and poor water-solubility.73 In efforts to counter this limitation, a study group designed, synthesized, and screened 40 new NBDHEX analogs.74 The group added one or two oxygen atoms on the hydroxyl chain of the NBD bone, forming two NBDHEX analogs, MC3181 and MC3165, that were able to display higher selectivity and better external activity by forming a stable σ-complex with the active site of GSTπ. After extensive experiments, MC3181 was deemed the more promising compound of the two, due to it having a 50-fold rise in aqueous solubility and higher selectivity toward GSTπ. This novel compound yielded positive results when administered to several distinct human melanoma cell lines, particularly when used in BRAFV600E-mutation melanoma cells.75 Moreover, both intravenous and oral treatment of MC3181 in animals with different types of human melanoma xenografts resulted in astonishing curative effects and a satisfactory safety profile.75 In summary, we conclude that NBDHEX and its analogs may serve as potential treatment strategies in the management of patients with melanoma.
Prodrugs of GSTπ
Both conventional chemotherapeutic and targeted agents have well-established toxicity profiles, with a wide range of adverse effects, eg, bone-marrow suppression, gastrointestinal toxicity, immunosuppression, gastrointestinal toxicity, hepatotoxicity, and cardiotoxicity.76 GSTπ prodrugs appear to have a more favorable adverse-event profile, given that they are ingested as an inactive compound and undergo breakdown to release cytotoxic metabolites only in the presence of high concentrations of enzymes that occur in the proximity of tumor cells, thereby reducing collateral damage to healthy cells.2 Additionally, tumor cells generally have heightened expression of GSTπ, providing ideal conditions for GSTπ-activated prodrugs. There are two primary classes of GSTπ prodrugs. The first of these are GSH or GSH derivatives, such as canfosfamide (Telcyta, TER286, TLK286), which have cytotoxic drug segments. When catalyzed by GSTπ, the prodrug releases cytotoxic compounds. The other type, such as JS-K, consists of a similar structure, but without GST analogs. Upon being catalyzed by GSTπ, it forms an intermediate with GSH and releases its cytotoxic drug segments. Currently, prodrugs under research comprise TLK286, purine analogs,77 sulfonamides,78 and brostallicin.
TLK286
L-γ-Glutamyl-3-(bis[bis(2-chloroethyl)amino-phosphinyl]oxy)ethylsulfonyl-L-alanyl-2-phenyl-[2R]-glycine hydrochloride salt (TLK286) represents the most promising GSTπ-prodrug candidate. It can generate a GSH analog and a phosphorodiamidate, the latter an active, alkylating agent.53 Following activation, the former competitively inhibits molecules that stimulate drug resistance, while the nitrogen-mustard segment induces apoptotic activity by influencing the activities of MAPK, p38 kinase, JNK, MKK4, and caspase 3.53,79–81
In vitro studies have revealed that the GSTP1-null cell lines show a different degree of resistance in response to TLK286 compared with GSTP1+/+ cells. These differences were abrogated by cotransfecting these cells with GSTπ. Similar findings were reflected in in vivo analyses with nude mice. These data support the rationale that tumors with elevated GSTπ expression are more sensitive to the cytotoxic effects of TLK286.82
The promising mechanism of action of TLK286 and its positive preclinical data have sparked a series of clinical trials where the prodrugs have been applied alone or in combination with other standard chemotherapeutic agents. Completed Phase II and III clinical trials have indicated that the agent has a nonoverlapping toxicity profile and synergistic effects with carboplatin, paclitaxel, and anthracycline, has no cross-drug resistance and is well tolerated, with patients mostly reporting fatigue and nausea.83–88
A completed Phase I/IIA multicenter dose-ranging clinical trial that sought to assess the safety and efficacy of TLK286 found that the compound was highly efficacious. Patients who underwent TLK286 maintenance treatment experienced prolonged median survival of 16.8 months compared to the 8.8 months experienced by those who did not receive the agent.89 These clinical trials provide sound scientific evidence that supports the therapeutic efficacy of TLK286 in managing different types of malignancies.
Nitric oxide (NO) prodrugs
Another class of prodrugs are the NO prodrugs. These medications work by binding to intracellular GSTπ and undergoing GSTπ-mediated catalysis. This process releases NO molecules that go on to exert antitumor activity. NO is an ephemeral but pleiotropic molecule. It has been shown to have the capacity to affect several vital functions of the body.90 As such, this molecule has been investigated keenly for its role in carcinogenesis, tumor progression, invasion, angiogenesis, and other key biological processes.91 However, available experimental evidence suggests that NO is a double-edged sword when used to manage tumor diseases. NO itself is a source of cytotoxic molecules, and its deleterious effects are enhanced by its ability to concentrate locally around tumor cells and the tumor microenvironment.92 At modest concentrations, NO exerts a protumorigenic response that may benefit tumor growth and survival. Nevertheless, at fairly high concentrations, NO takes on antitumor-agent properties to accelerate tumor-cell death and to inhibit tumor-cell angiogenesis.93 Based on these observations, it is clear that NO has a role to play in combating tumor-cell resistance.
An example of an NO prodrug is O2-(2,4-dinitrophenyl)1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate (JS-K), devised by Keefer et al from the National Cancer Institute.94 The antineoplastic properties of JS-K rest on two modes of action: first, it can combine with GSTπ and then be activated to release NO at a high concentration to directly kill tumor cells; second, it binds to cellular GST/GSH, depleting its intracellular content to weaken the efflux of chemotherapy drugs in tumor cells.95 Furthermore, other literature suggests that JS-K inhibits angiogenesis,96 induces cell apoptosis (a process related to PARP, caspase 8 and 9 cleavage, and cell differentiation),95,97 destroys double-stranded DNA,95,97–99 and is also able to interfere with the cell cycle and its respective signaling pathways.92,100–102 These mechanisms are highly codependent, and function in an integrated manner to exert antitumor effects.
Remarkably, flow-cytometry findings have shown that JS-K can improve the formation of acidic vesicle organelles, underscoring its ability to induce autophagy.102 Furthermore, electron-microscopy observations have indicated that JS-K induces autophagic death in cells.102 Nevertheless, JS-K was able to spare surrounding healthy mammary epithelial cells. JS-K has been shown to be effective in several types of cancers, of which leukemia and myeloma appear to be the most susceptible.92,96,100,103 In addition, it is also efficacious in the treatment of solid tumors, such as breast cancer,102,104 lung cancer,97,103,105 glioma,103,106 prostate cancer,107 kidney cancer,108 bladder cancer,109 colon cancer,110 and hepatocellular carcinoma.111 Furthermore, JS-K is tolerated well by healthy tissue. These data indicate that further investigation into JS-K as an alternative chemotherapeutic agent is much needed.102–104,109
It is worth noting that JS-K works synergistically with chemotherapy drugs, such as cytarabine,98 bortezomib,95 cisplatin, and arsenic.112 JS-K acts as a dose-sparing agent when used with typical chemotherapeutic agents and is able to alleviate the severity of adverse effects as a consequence. While JS-K has been shown to be advantageous in treating cancer, its clinical use has been hindered with reports of poor solubility. Structural modification of JS-K is able to prolong its half-life, and combining JS-K with special nanoparticles can greatly improve its solubility and stability,103,113 further improving its prospects for clinical applications. JS-K and many other NO prodrugs represent an innovative biological approach in the development of anticancer therapeutics.
Conclusion
Multidrug resistance to chemotherapy drugs is one of the main obstacles in human cancer chemotherapy and has prompted intense research into discovering novel and innovative mechanisms that can overcome this barrier.6,7,20 There is an intimate connection between abnormal GST expression and multidrug resistance, unequivocally implicating GSTπ, a member of the GST family in tumor-drug resistance.4 GSTπ inhibitors and prodrugs are crucial agents that can help reverse multidrug resistance in tumors and increase the therapeutic index of anticancer drugs, which collectively decreases the physical and economic burden of cancer patients. Nevertheless, while the rapid growth of research on development of medication based on GSTπ inhibition has resulted in clinical studies on several compounds, the drugs that make it to commercial consumption are few. Realistically, fundamental issues that stand in the way of large-scale drug production include the vast number of biological GST family members, with each subtype having their own structural and functional differences, in addition to the existence of several genetic modifiers. The situation is further compounded by the presence of posttranslational modifying factors, such as kinase activities and S-glutathionylation. More in-depth research that clarifies the roles of these components of the GST-detoxification system are much needed, in order to produce compounds that have minimal side effects and high GSTπ selectivity. While modulation of the GSH-antioxidant system has provided promising preclinical results, some of these compounds demonstrate unacceptable toxicity profiles (eg, buthionine sulfoximine). Having said that, GSH-based medication has also been successfully employed to protect against cisplatin induced nephrotoxicity.114,115 The ongoing development of chemical genomics, computer-aided drug design, and more extensive molecular and cellular biology research will serve to be extremely useful in contributing toward the preclinical and clinical development of more efficient GSTπ-targeting drugs.
Acknowledgments
This study was supported by the Jiangsu Provincial Medical Youth Talent (grant number QNRC2016653); the National Nature Science Foundation of Jiangsu Provience (grant number BK20151016); the National Natural Science Foundation of China (grant number 81602441); and the Jiangsu Provincial Key Research Development Program (grant number BE2016794); the Postgraduate Research & Practice Innovation Program of Jiangsu Province (grant number KYCX17_1296).
Disclosure
The authors report no conflicts of interest in this work.
References
Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother. 2003;57(3–4):145–155. | ||
Tew KD, Townsend DM. Regulatory functions of glutathione S-transferase P1-1 unrelated to detoxification. Drug Metab Rev. 2011;43(2):179–193. | ||
Sawers L, Ferguson MJ, Ihrig BR, et al. Glutathione S-transferase P1 (GSTP1) directly influences platinum drug chemosensitivity in ovarian tumour cell lines. Br J Cancer. 2014;111(6):1150–1158. | ||
Sato K, Tsuchida S, Tamai K. Anti-cancer drug resistance and glutathione S-transferases. Gan To Kagaku Ryoho. 1989;16(3 Pt 2):592–598. | ||
Ij S, Cheng AL, Tsai TF, Lay JD. Retinoic acid-induced apoptosis and regression of a refractory Epstein-Barr virus-containing T cell lymphoma expressing multidrug-resistance phenotypes. Br J Haematol. 1993;85(4):826–828. | ||
Qin F, Qin X, Zhang X, Jia HW. Expression and significance of P-glycoprotein, glutathione S-transferase-pi and Topoisomerase II in gastric carcinomas. Ai Zheng. 2002;21(2):167–170. | ||
Zhu XH, Li JY, Xia XM, et al. Multidrug resistance mechanisms in cell line HL-60/VCR. Ai Zheng. 2002;21(12):1310–1313. | ||
Townsend DM, Findlay VL, Tew KD. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005;401:287–307. | ||
Schecter RL, Alaoui-Jamali MA, Batist G. Glutathione S-transferase in chemotherapy resistance and in carcinogenesis. Biochem Cell Biol. 1992;70(5):349–353. | ||
Wu B, Dong D. Human cytosolic glutathione transferases: structure, function, and drug discovery. Trends Pharmacol Sci. 2012;33(12):656–668. | ||
Morel F, Rauch C, Petit E, et al. Gene and Protein Characterization of the Human Glutathione S-Transferase Kappa and Evidence for a Peroxisomal Localization. J Biol Chem. 2004;279(16):16246–16253. | ||
Deng Q, He B, Pan Y, et al. Polymorphisms of GSTA1 contribute to elevated cancer risk: evidence from 15 studies. J BUON. 2015;20(1):287–295. | ||
Russo A, Saide A, Smaldone S, Faraonio R, Russo G. Role of uL3 in Multidrug Resistance in p53-Mutated Lung Cancer Cells. Int J Mol Sci. 2017;18(3):547. | ||
Depeille P, Cuq P, Mary S, et al. Glutathione S-transferase M1 and multidrug resistance protein 1 act in synergy to protect melanoma cells from vincristine effects. Mol Pharmacol. 2004;65(4):897–905. | ||
Chen WY, Mao WM, Zhao L, et al. Expression of P-gp, GST-pi and Topo II alpha in gastric and colorectal cancers and their clinical significance. Zhonghua Zhong Liu Za Zhi. 2005;27(12):738–740. | ||
Asojo OA, Homma K, Sedlacek M, et al. X-ray structures of Na-GST-1 and Na-GST-2 two glutathione S-transferase from the human hookworm Necator americanus. BMC Struct Biol. 2007;7(1):42. | ||
Kelleher A, Zhan B, Asojo OA. Structure of monomeric Na-GST-3, a glutathione S-transferase from the major human hookworm parasite Necator americanus. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013;69(8):839–843. | ||
Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22(47):7369–7375. | ||
Vasieva O. The many faces of glutathione transferase pi. Curr Mol Med. 2011;11(2):129–139. | ||
Li G, Dai J, Wang Y, et al. Overexpression and its clinical significance of multi-drug resistance associated genes in lung cancer tissues. Zhongguo Fei Ai Za Zhi. 2002;5(1):35–37. | ||
Hsu C-H, Chen C-L, Hong R-L, Chen K-L, Lin J-F, Cheng A-L. Prognostic Value of Multidrug Resistance 1, Glutathione-S-Transferase-π and p53 in Advanced Nasopharyngeal Carcinoma Treated with Systemic Chemotherapy. Oncology. 2002;62(4):305–312. | ||
Collier JD, Bennett MK, Hall A, Cattan AR, Lendrum R, Bassendine MF. Expression of glutathione S-transferases in normal and malignant pancreas: an immunohistochemical study. Gut. 1994;35(2):266–269. | ||
Su F, Hu X, Jia W, Gong C, Song E, Hamar P. Glutathion s transferase π indicates chemotherapy resistance in breast cancer. J Surg Res. 2003;113(1):102–108. | ||
Ezzikouri S, Benjelloun S, Pineau P. Human genetic variation and the risk of hepatocellular carcinoma development. Hepatol Int. 2013;7(3):820–831. | ||
Bennaceur-Griscelli A, Bosq J, Koscielny S, et al. High Level of Glutathione-S-Transferase Expression in Mantle Cell Lymphomas. Clin Cancer Res. 2004;10(9):3029–3034. | ||
Depeille P, Cuq P, Passagne I, Evrard A, Vian L. Combined effects of GSTP1 and MRP1 in melanoma drug resistance. Br J Cancer. 2005;93(2):216–223. | ||
Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. 2012;2012(5):736837. | ||
Zw Y, Zhang J, Townsend DM, Tew KD. Oxidative stress, redox regulation and diseases of cellular differentiation. Biochim Biophys Acta. 1850;2015(8):1607–1621. | ||
Chen W, Seefeldt T, Young A, et al. Microtubule S-glutathionylation as a potential approach for antimitotic agents. BMC Cancer. 2012;12(1):245. | ||
Tew KD, Manevich Y, Grek C, Xiong Y, Uys J, Townsend DM. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free Radic Biol Med. 2011;51(2):z299–313. | ||
Seefeldt T, Zhao Y, Chen W, et al. Characterization of a novel dithiocarbamate glutathione reductase inhibitor and its use as a tool to modulate intracellular glutathione. J Biol Chem. 2009;284(5):2729–2737. | ||
Lee H-C, Kim D-W, Jung K-Y, et al. Increased expression of antioxidant enzymes in radioresistant variant from U251 human glioblastoma cell line. Int J Mol Med. 2004;13(6):883–887. | ||
Gate L, Majumdar RS, Lunk A, Tew KD. Increased Myeloproliferation in Glutathione S-Transferase π-deficient Mice Is Associated with a Deregulation of JNK and Janus Kinase/STAT Pathways. J Biol Chem. 2004;279(10):8608–8616. | ||
Sau A, Filomeni G, Pezzola S, et al. Targeting GSTP1-1 induces JNK activation and leads to apoptosis in cisplatin-sensitive and -resistant human osteosarcoma cell lines. Mol Biosyst. 2012;8(4):994–1006. | ||
Zhang L, Blackwell K, Altaeva A, Shi Z, Habelhah H. TRAF2 phosphorylation promotes NF-κB–dependent gene expression and inhibits oxidative stress-induced cell death. Mol Biol Cell. 2011;22(1):128–140. | ||
Chen D, Liu J, Rui B, et al. GSTpi protects against angiotensin II-induced proliferation and migration of vascular smooth muscle cells by preventing signal transducer and activator of transcription 3 activation. Biochim Biophys Acta. 2014;1843(2):454–463. | ||
Sau A, Pellizzari Tregno F, Valentino F, Federici G, Caccuri AM. Glutathione transferases and development of new principles to overcome drug resistance. Arch Biochem Biophys. 2010;500(2):116–122. | ||
de Luca A, Mei G, Rosato N, et al. The fine-tuning of TRAF2–GSTP1-1 interaction: effect of ligand binding and in situ detection of the complex. Cell Death Dis. 2014;5(1):e1015. | ||
Coatti GC, Marcarini JC, Sartori D, Fidelis QC, Ferreira DT, Mantovani MS. Cytotoxicity, genotoxicity and mechanism of action (via gene expression analysis) of the indole alkaloid aspidospermine (antiparasitic) extracted from Aspidosperma polyneuron in HepG2 cells. Cytotechnology. 2016;68(4):1161–1170. | ||
Palumbo C, de Luca A, Rosato N, Forgione M, Rotili D, Caccuri AM. c-Jun N-terminal kinase activation by nitrobenzoxadiazoles leads to late-stage autophagy inhibition. J Transl Med. 2016;14(1):37. | ||
Fulci C, Rotili D, de Luca A, et al. A new nitrobenzoxadiazole-based GSTP1-1 inhibitor with a previously unheard of mechanism of action and high stability. J Enzyme Inhib Med Chem. 2017;32(1):240–247. | ||
Pellizzari Tregno F, Sau A, Pezzola S, et al. In vitro and in vivo efficacy of 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) on human melanoma. Eur J Cancer. 2009;45(14):2606–2617. | ||
Hw L, Ali-Osman F. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance. Curr Opin Pharmacol. 2007;7(4):367–374. | ||
Bartolini D, Galli F. The functional interactome of GSTP: A regulatory biomolecular network at the interface with the Nrf2 adaption response to oxidative stress. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1019:29–44. | ||
Kou X, Chen N, Feng Z, Luo LAN, Yin Z. GSTP1 negatively regulates Stat3 activation in epidermal growth factor signaling. Oncol Lett. 2013;5(3):1053–1057. | ||
Li F, Fernandez PP, Rajendran P, Hui KM, Sethi G. Diosgenin, a steroidal saponin, inhibits STAT3 signaling pathway leading to suppression of proliferation and chemosensitization of human hepatocellular carcinoma cells. Cancer Lett. 2010;292(2):197–207. | ||
Townsend DM, Shen H, Staros AL, Gate L, Tew KD. Efficacy of a glutathione S-transferase pi-activated prodrug in platinum-resistant ovarian cancer cells. Mol Cancer Ther. 2002;1(12):1089–1095. | ||
di Pietro G, Magno LAV, Rios-Santos F. Glutathione S-transferases: an overview in cancer research. Expert Opin Drug Metab Toxicol. 2010;6(2):153–170. | ||
Harris MJ, Coggan M, Langton L, Wilson SR, Board PG. Polymorphism of the Pi class glutathione S-transferase in normal populations and cancer patients. Pharmacogenetics. 1998;8(1):27–31. | ||
Cadoni G, Boccia S, Petrelli L, et al. A review of genetic epidemiology of head and neck cancer related to polymorphisms in metabolic genes, cell cycle control and alcohol metabolism. Acta Otorhinolaryngol Ital. 2012;32(1):1–11. | ||
Huang W, Wang W, Zhou M, Chen S, Zhang X. Association of glutathione S-transferase polymorphisms (GSTM1 and GSTT1) with primary open-angle glaucoma: An evidence-based meta-analysis. Gene. 2013;526(2):80–86. | ||
Habdous M, Siest G, Herbeth B, Vincent-Viry M, Visvikis S. Glutathione S-transferases genetic polymorphisms and human diseases: overview of epidemiological studies. Ann Biol Clin. 2004;62(1):15–24. | ||
Dourado DF, Fernandes PA, Ramos MJ, Mannervik B. Mechanism of glutathione transferase P1-1-catalyzed activation of the prodrug canfosfamide (TLK286, TELCYTA). Biochemistry. 2013;52(45):8069–8078. | ||
Nasr A, Sami R, Ibrahim N, Darwish D. Glutathione S transferase (GSTP 1, GSTM 1, and GSTT 1) gene polymorphisms in Egyptian patients with acute myeloid leukemia. Indian J Cancer. 2015;52(4):490–495. | ||
Sun N, Sun X, Chen B, et al. MRP2 and GSTP1 polymorphisms and chemotherapy response in advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2010;65(3):437–446. | ||
Khrunin AV, Moisseev A, Gorbunova V, Limborska S. Genetic polymorphisms and the efficacy and toxicity of cisplatin-based chemotherapy in ovarian cancer patients. Pharmacogenomics J. 2010;10(1):54–61. | ||
Burg D, Filippov DV, Hermanns R, van der Marel GA, van Boom JH, Mulder GJ. Peptidomimetic Glutathione Analogs as Novel γGT Stable GST Inhibitors. Bioorg Med Chem. 2002;10(1):195–205. | ||
Li T, Liu G, Li H, Yang X, Jing Y, Zhao G. The synthesis of ethacrynic acid thiazole derivatives as glutathione S-transferase pi inhibitors. Bioorg Med Chem. 2012;20(7):2316–2322. | ||
Enoiu M, Aberkane H, Salazar JF, et al. Evidence for the pro-oxidant effect of gamma-glutamyltranspeptidase-related enzyme. Free Radic Biol Med. 2000;29(9):825–833. | ||
Nakajima T, et al. Reversal of Multiple Drug Resistance in Cholangiocarcinoma by the Glutathione S-Transferase-pi-Specific Inhibitor O1-Hexadecyl-gamma-glutamyl-S-benzylcysteinyl-D-phenylglycine Ethylester. J Pharmacol Exp Ther. 2003;306(3):861–869. | ||
Raza A, Galli N, Callander N, et al. Phase 1-2a multicenter dose-escalation study of ezatiostat hydrochloride liposomes for injection (Telintra(R), TLK199), a novel glutathione analog prodrug in patients with myelodysplastic syndrome. J Hematol Oncol. 2009;2(1):20. | ||
Raza A, Galili N, Smith S, et al. Phase 1 multicenter dose-escalation study of ezatiostat hydrochloride (TLK199 tablets), a novel glutathione analog prodrug, in patients with myelodysplastic syndrome. Blood. 2009;113(26):6533–6540. | ||
Raza A, Galili N, Mulford D, et al. Phase 1 dose-ranging study of ezatiostat hydrochloride in combination with lenalidomide in patients with non-deletion (5q) low to intermediate-1 risk myelodysplastic syndrome (MDS). J Hematol Oncol. 2012;5(1):18. | ||
Mahadevan D, Sutton GR. Ezatiostat hydrochloride for the treatment of myelodysplastic syndromes. Expert Opin Investig Drugs. 2015;24(5):725–733. | ||
Ricci G, de Maria F, Antonini G, et al. 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases. Mechanism of action of potential anticancer drugs. J Biol Chem. 2005;280(28):26397–26405. | ||
Pasello M, Michelacci F, Scionti I, et al. Overcoming glutathione S-transferase P1-related cisplatin resistance in osteosarcoma. Cancer Res. 2008;68(16):6661–6668. | ||
Ascione A, Cianfriglia M, Dupuis ML, et al. The glutathione S-transferase inhibitor 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol overcomes the MDR1-P-glycoprotein and MRP1-mediated multidrug resistance in acute myeloid leukemia cells. Cancer Chemother Pharmacol. 2009;64(2):419–424. | ||
Scotlandi K, Remondini D, Castellani G, et al. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol. 2009;27(13):2209–2216. | ||
de Luca A, Pellizzari Tregno F, Sau A, et al. Glutathione S-transferase P1-1 as a target for mesothelioma treatment. Cancer Sci. 2013;104(2):223–230. | ||
Filomeni G, Turella P, Dupuis ML, et al. 6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol, a specific glutathione S-transferase inhibitor, overcomes the multidrug resistance (MDR)-associated protein 1-mediated MDR in small cell lung cancer. Mol Cancer Ther. 2008;7(2):371–379. | ||
Tentori L, Dorio AS, Mazzon E, et al. The glutathione transferase inhibitor 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) increases temozolomide efficacy against malignant melanoma. Eur J Cancer. 2011;47(8):1219–1230. | ||
de Luca A, Federici L, de Canio M, Stella L, Caccuri AM. New Insights into the Mechanism of JNK1 Inhibition by Glutathione Transferase P1-1. Biochemistry. 2012;51(37):7304–7312. | ||
Awasthi YC, Sharma R, Singhal SS. Human glutathione S-transferases. Int J Biochem. 1994;26(3):295–308. | ||
Rotili D, de Luca A, Tarantino D, et al. Synthesis and structure–activity relationship of new cytotoxic agents targeting human glutathione-S-transferases. Eur J Med Chem. 2015;89:156–171. | ||
de Luca A, Rotili D, Carpanese D, et al. A novel orally active water-soluble Inhibitor of human glutathione transferase exerts a potent and selective antitumor activity against human melanoma xenografts. Oncotarget. 2015;6(6):4126–4143. | ||
Bregni G, Galli G, Gevorgyan A, de Braud F, di Cosimo S. Trastuzumab cardiac toxicity: a problem we put our heart into. Tumori. 2016;102(1):1–5. | ||
Yang A-J, Li C-C, Lu C-Y, et al. Activation of the cAMP/CREB/Inducible cAMP Early Repressor Pathway Suppresses Andrographolide-Induced Gene Expression of the π Class of Glutathione S-Transferase in Rat Primary Hepatocytes. J Agric Food Chem. 2010;58(3):1993–2000. | ||
Lin C-Y, Fu R-H, Chou R-H, et al. Inhibition of JNK by pi class of glutathione S-transferase through PKA/CREB pathway is associated with carnosic acid protection against 6-hydroxydopamine-induced apoptosis. Food Chem Toxicol. 2017;103:194–202. | ||
Satyam A, Hocker MD, Kane-Maguire KA, Morgan AS, Villar HO, Lyttle MH. Design, Synthesis, and Evaluation of Latent Alkylating Agents Activated by Glutathione S-Transferase. J Med Chem. 1996;39(8):1736–1747. | ||
Tew KD. TLK-286: a novel glutathione S-transferase-activated prodrug. Expert Opin Investig Drugs. 2005;14(8):1047–1054. | ||
Ramsay EE, Dilda PJ. Glutathione S-conjugates as prodrugs to target drug-resistant tumors. Front Pharmacol. 2014;5(118):181. | ||
Morgan AS, Sanderson PE, Borch RF, et al. Tumor efficacy and bone marrow-sparing properties of TER286, a cytotoxin activated by glutathione S-transferase. Cancer Res. 1998;58(12):2568–2575. | ||
Rosen LS, Brown J, Laxa B, et al. Phase I study of TLK286 (glutathione S-transferase P1-1 activated glutathione analog) in advanced refractory solid malignancies. Clin Cancer Res. 2003;9(5):1628–1638. | ||
Rosen LS, Laxa B, Boulos L, et al. Phase 1 study of TLK286 (Telcyta) administered weekly in advanced malignancies. Clin Cancer Res. 2004;10(11):3689–3698. | ||
Kavanagh JJ, Gershenson DM, Choi H, et al. Multi-institutional phase 2 study of TLK286 (TELCYTAtm, a glutathione S-transferase P1-1 activated glutathione analog prodrug) in patients with platinum and paclitaxel refractory or resistant ovarian cancer. Int J Gynecol Cancer. 2005;15(4):593–600. | ||
Edelman MJ. Novel cytotoxic agents for non-small cell lung cancer. J Thorac Oncol. 2006;1(7):752–755. | ||
Raez LE, Lilenbaum R. New developments in chemotherapy for advanced non-small cell lung cancer. Curr Opin Oncol. 2006;18(2):156–161. | ||
Kavanagh JJ, Levenback CF, Ramirez PT, et al. Phase 2 study of canfosfamide in combination with pegylated liposomal doxorubicin in platinum and paclitaxel refractory or resistant epithelial ovarian cancer. J Hematol Oncol. 2010;3(1):9. | ||
Sequist LV, Fidias PM, Temel JS, et al. Phase 1-2a multicenter dose-ranging study of canfosfamide in combination with carboplatin and paclitaxel as first-line therapy for patients with advanced non-small cell lung cancer. J Thorac Oncol. 2009;4(11):1389–1396. | ||
Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. 2015;36(3):161–178. | ||
Singh S, Gupta AK. Nitric oxide: role in tumour biology and iNOS/NO-based anticancer therapies. Cancer Chemother Pharmacol. 2011;67(6):1211–1224. | ||
Shami PJ, Saavedra JE, Wang LY, et al. JS-K, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol Cancer Ther. 2003;2(4):409–417. | ||
Burke AJ, Sullivan FJ, Giles FJ, Glynn SA. The yin and yang of nitric oxide in cancer progression. Carcinogenesis. 2013;34(3):503–512. | ||
Keefer L. Broad-Spectrum Anti-Cancer Activity of O2-Arylated Diazeniumdiolates. For Immunopathol Dis Therap. 2010;1(3):205–218. | ||
Kiziltepe T, Hideshima T, Ishitsuka K, et al. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 2007;110(2):709–718. | ||
Kiziltepe T, Anderson KC, Kutok JL, et al. JS-K has potent anti-angiogenic activity in vitro and inhibits tumour angiogenesis in a multiple myeloma model in vivo. J Pharm Pharmacol. 2010;62(1):145–151. | ||
Maciag AE, Chakrapani H, Saavedra JE, et al. The nitric oxide prodrug JS-K is effective against non-small-cell lung cancer cells in vitro and in vivo: involvement of reactive oxygen species. J Pharmacol Exp Ther. 2011;336(2):313–320. | ||
Shami PJ, Maciag AE, Eddington JK, et al. JS-K, an arylating nitric oxide (NO) donor, has synergistic anti-leukemic activity with cytarabine (ARA-C). Leuk Res. 2009;33(11):1525–1529. | ||
Xue R, Wu J, Luo X, et al. Design, Synthesis, and Evaluation of Diazeniumdiolate-Based DNA Cross-Linking Agents Activatable by Glutathione S-Transferase. Org Lett. 2016;18(20):5196–5199. | ||
Kaczmarek MZ, Holland RJ, Lavanier SA, et al. Mechanism of action for the cytotoxic effects of the nitric oxide prodrug JS-K in murine erythroleukemia cells. Leuk Res. 2014;38(3):377–382. | ||
Ren Z, Kar S, Wang Z, Wang M, Saavedra JE, Carr BI. JS-K, a novel non-ionic diazeniumdiolate derivative, inhibits Hep 3B hepatoma cell growth and induces c-Jun phosphorylation via multiple MAP kinase pathways. J Cell Physiol. 2003;197(3):426–434. | ||
Mcmurtry V, Saavedra JE, Nieves-Alicea R, Simeone AM, Keefer LK, Tari AM. JS-K, a nitric oxide-releasing prodrug, induces breast cancer cell death while sparing normal mammary epithelial cells. Int J Oncol. 2011;38(4):963–971. | ||
Kaur I, Terrazas M, Kosak KM, Kern SE, Boucher KM, Shami PJ. Cellular distribution studies of the nitric oxide-generating antineoplastic prodrug O(2)-(2,4-dinitrophenyl)1-((4-ethoxycarbonyl)piperazin-1-yl)diazen-1-ium-1,2-diolate formulated in Pluronic P123 micelles. J Pharm Pharmacol. 2013;65(9):1329–1336. | ||
Simeone AM, McMurtry V, Nieves-Alicea R, et al. TIMP-2 mediates the anti-invasive effects of the nitric oxide-releasing prodrug JS-K in breast cancer cells. Breast Cancer Res. 2008;10(3):R44. | ||
Kitagaki J, Yang Y, Saavedra JE, Colburn NH, Keefer LK, Perantoni AO. Nitric oxide prodrug JS-K inhibits ubiquitin E1 and kills tumor cells retaining wild-type p53. Oncogene. 2009;28(4):619–624. | ||
Weyerbrock A, Osterberg N, Psarras N, et al. JS-K, a glutathione S-transferase-activated nitric oxide donor with antineoplastic activity in malignant gliomas. Neurosurgery. 2012;70(2):497–510; discussion 510. | ||
Laschak M, Spindler KD, Schrader AJ, et al. JS-K, a glutathione/glutathione S-transferase-activated nitric oxide releasing prodrug inhibits androgen receptor and WNT-signaling in prostate cancer cells. BMC Cancer. 2012;12:130. | ||
Chakrapani H, Kalathur RC, Maciag AE, et al. Synthesis, mechanistic studies, and anti-proliferative activity of glutathione/glutathione S-transferase-activated nitric oxide prodrugs. Bioorg Med Chem. 2008;16(22):9764–9771. | ||
Qiu M, Chen L, Tan G, et al. A reactive oxygen species activation mechanism contributes to JS-K-induced apoptosis in human bladder cancer cells. Sci Rep. 2015;5:15104. | ||
Edes K, Cassidy P, Shami PJ, Moos PJ. JS-K, a nitric oxide prodrug, has enhanced cytotoxicity in colon cancer cells with knockdown of thioredoxin reductase 1. PLoS One. 2010;5(1):e8786. | ||
Liu L, Wang D, Wang J, Wang S. The Nitric Oxide Prodrug JS-K Induces Ca(2+)-Mediated Apoptosis in Human Hepatocellular Carcinoma HepG2 Cells. J Biochem Mol Toxicol. 2016;30(4):192–199. | ||
Liu J, Li C, Qu W, et al. Nitric oxide prodrugs and metallochemotherapeutics: JS-K and CB-3-100 enhance arsenic and cisplatin cytolethality by increasing cellular accumulation. Mol Cancer Ther. 2004;3(6):709–714. | ||
Kaur I, Kosak KM, Terrazas M, et al. Effect of a Pluronic® P123 formulation on the nitric oxide-generating drug JS-K. Pharm Res. 2015;32(4):1395–1406. | ||
Kuhlmann MK, Burkhardt G, Köhler H. Insights into potential cellular mechanisms of cisplatin nephrotoxicity and their clinical application. Nephrol Dial Transplant. 1997;12(12):2478–2480. | ||
Hospers GA, Eisenhauer EA, de Vries EG. The sulfhydryl containing compounds WR-2721 and glutathione as radio- and chemoprotective agents. A review, indications for use and prospects. Br J Cancer. 1999;80(5–6):629–638. | ||
Iorio A, Spinelli M, Polimanti R, et al. GSTA1 gene variation associated with gestational hypertension and its involvement in pregnancy-related pathogenic conditions. Eur J Obstet Gynecol Reprod Biol. 2015;194:34–37. | ||
Bonifazi F, Storci G, Bandini G, et al. Glutathione transferase-A2 S112T polymorphism predicts survival, transplant-related mortality, busulfan and bilirubin blood levels after allogeneic stem cell transplantation. Haematologica. 2014;99(1):172–179. | ||
Zhang W, Moden O, Mannervik B. Differences among allelic variants of human glutathione transferase A2-2 in the activation of azathioprine. Chem Biol Interact. 2010;186(2):110–117. | ||
Rodrigues DA, Martins JV, E Silva KS, et al. GSTM1 polymorphism in patients with clinical manifestations of atherosclerosis. Genetics and Molecular Research: GMR. 2017;16(1). | ||
Abdur Rehman MY, Kamal A, Taqi MM, Malik RN. Tracing biomarker of PAH-exposure and susceptibility factor (GSTM-polymorphism) among cancer patients in Pakistan. Chemosphere. 2017;178:384–390. | ||
Tanwar R, Iyengar AR, Nagesh KS, Patil S, Subhash BV. GSTM1 null polymorphism prevalence in tobacco users, oral leukoplakia and oral squamous cell carcinoma patients in South Indian population: A polymerase chain reaction study. Indian J Dent Res. 2016;27(4):353–358. | ||
Xu Y, Wang J, Dong W. GSTM3 A/B polymorphism and risk for head and neck cancer: a meta-analysis. PLoS One. 2014;9(1):e83851. | ||
Feng X, Dong CQ, Shi JJ, Zhou HF, He W, Zheng BS. Lack of association of glutathione S-transferase M3 gene polymorphism with the susceptibility of lung cancer. Asian Pac J Cancer Prev. 2012;13(9):4465–4468. | ||
Bhat A, Masood A, Wani KA, et al. Promoter methylation and gene polymorphism are two independent events in regulation of GSTP1 gene expression. Tumour Biol. 2017;39(4):1010428317697563. | ||
Huang XK, Huang YH, Huang JH, Liang JY. Glutathione S-transferase P1 Ile105Val Polymorphism and Male Infertility Risk: An Updated Meta-analysis. Chin Med J (Engl). 2017;130(8):979–985. | ||
Soares PO, Maluf Cury P, Mendoza Lopez RV, et al. GTSP1 expression in non-smoker and non-drinker patients with squamous cell carcinoma of the head and neck. PLoS One. 2017;12(8):e0182600. | ||
Basharat Z, Yasmin A. Energy landscape of a GSTP1 polymorph linked with cytological function decay in response to chemical stressors. Gene. 2017;609:19–27. | ||
Choi B, Kim MG, Han N, et al. Population pharmacokinetics and pharmacodynamics of busulfan with GSTA1 polymorphisms in patients undergoing allogeneic hematopoietic stem cell transplantation. Pharmacogenomics. 2015;16(14):1585–1594. | ||
Song Y, Shan Z, Luo C, et al. Glutathione S-Transferase T1 (GSTT1) Null Polymorphism, Smoking, and Their Interaction in Coronary Heart Disease: A Comprehensive Meta-Analysis. Heart Lung Circ. 2017;26(4):362–370. | ||
Jang SG, Kim IJ, Kang HC, et al. GSTT2 promoter polymorphisms and colorectal cancer risk. BMC Cancer. 2007;7:16. | ||
Chelvanayagam G, Wilce MC, Parker MW, Tan KL, Board PG. Homology model for the human GSTT2 Theta class glutathione transferase. Proteins. 1997;27(1):118–130. | ||
Xu YT, Wang J, Yin R, et al. Genetic polymorphisms in Glutathione S-transferase Omega (GSTO) and cancer risk: a meta-analysis of 20 studies. Sci Rep. 2014;4:6578. | ||
Polimanti R, Graziano ME, Lazzarin N, Vaquero E, Manfellotto D, Fuciarelli M. GSTO1 uncommon genetic variants are associated with recurrent miscarriage risk. Fertil Steril. 2014;101(3):735–739. | ||
Wang Z, Qu K, Huang Z, et al. Glutathione S-transferase O2 gene rs157077 polymorphism predicts response to transarterial chemoembolization in hepatocellular carcinoma. Tumour Biol. 2015;36(8):6463–6469. | ||
Stamenkovic M, Radic T, Stefanovic I, et al. Glutathione S-transferase omega-2 polymorphism Asn142Asp modifies the risk of age-related cataract in smokers and subjects exposed to ultraviolet irradiation. Clin Exp Ophthalmol. 2014;42(3):277–283. | ||
Langaee TY, Zhong G, Li W, et al. The influence of human GSTZ1 gene haplotype variations on GSTZ1 expression. Pharmacogenet Genomics. 2015;25(5):239–245. | ||
Shroads AL, Coats BS, McDonough CW, Langaee T, Stacpoole PW. Haplotype variations in glutathione transferase zeta 1 influence the kinetics and dynamics of chronic dichloroacetate in children. J Clin Pharmacol. 2015;55(1):50–55. | ||
Karakas-Celik S, Aras N, Ates C. Glutathione S-transferase Z1 (GSTZ1) gene polymorphism in gastric cancer: a preliminary study in a Turkish population. Lab Med. 2014;45(1):37–42. | ||
Burg D, Filippov DV, Hermanns R, van der Marel GA, van Boom JH, Mulder GJ. Peptidomimetic glutathione analogs as novel gammaGT stable GST inhibitors. Bioorg Med Chem. 2002;10(1):195–205. | ||
Li T, Liu G, Li H, Yang X, Jing Y, Zhao G. The synthesis of ethacrynic acid thiazole derivatives as glutathione S-transferase pi inhibitors. Bioorg Med Chem. 2012;20(7):2316–2322. | ||
Mahadevan D, Sutton GR. Ezatiostat hydrochloride for the treatment of myelodysplastic syndromes. Expert Opin Investig Drugs. 2015;24(5):725–733. | ||
Fulci C, Rotili D, De Luca A, et al. A new nitrobenzoxadiazole-based GSTP1-1 inhibitor with a previously unheard of mechanism of action and high stability. J Enzyme Inhib Med Chem. 2017;32(1):240–247. | ||
Rotili D, De Luca A, Tarantino D, et al. Synthesis and structure – activity relationship of new cytotoxic agents targeting human glutathione-S-transferases. Eur J Med Chem. 2015;89:156–171. | ||
De Luca A, Mei G, Rosato N, et al. The fine-tuning of TRAF2-GSTP1-1 interaction: effect of ligand binding and in situ detection of the complex. Cell Death Dis. 2014;5:e1015. | ||
Graziani G, Artuso S, De Luca A, et al. A new water soluble MAPK activator exerts antitumor activity in melanoma cells resistant to the BRAF inhibitor vemurafenib. Biochem Pharmacol. 2015;95(1):16–27. | ||
Ramsay EE, Dilda PJ. Glutathione S-conjugates as prodrugs to target drug-resistant tumors. Front Pharmacol. 2014;5:181. | ||
Tew KD. TLK-286: a novel glutathione S-transferase-activated prodrug. Expert Opin Investig Drugs. 2005;14(8):1047–1054. | ||
Vergote I, Finkler N, del Campo J, et al. Phase 3 randomised study of canfosfamide (Telcyta, TLK286) versus pegylated liposomal doxorubicin or topotecan as third-line therapy in patients with platinum-refractory or -resistant ovarian cancer. Eur J Cancer. 2009;45(13):2324–2332. | ||
Qiu M, Ke L, Zhang S, Zeng X, Fang Z, Liu J. JS-K, a GST-activated nitric oxide donor prodrug, enhances chemo-sensitivity in renal carcinoma cells and prevents cardiac myocytes toxicity induced by Doxorubicin. Cancer Chemother Pharmacol. 2017;80(2):275–286. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.