")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Ginkgo biloba extract mitigates liver fibrosis and apoptosis by regulating p38 MAPK, NF-κB/IκBα, and Bcl-2/Bax signaling
Authors Wang Y, Wang R, Wang Y, Peng R, Wu Y, Yuan Y
Received 4 August 2015
Accepted for publication 30 September 2015
Published 3 December 2015 Volume 2015:9 Pages 6303—6317
DOI https://doi.org/10.2147/DDDT.S93732
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Yuanyuan Wang, Rong Wang, Yujie Wang, Ruqin Peng, Yan Wu, Yongfang Yuan
Department of Pharmacy, Shanghai 9th People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
Background: Liver fibrosis is the consequence of diverse liver injuries and can eventually develop into liver cirrhosis. Ginkgo biloba extract (GBE) is an extract from dried ginkgo leaves that has many pharmacological effects because of its various ingredients and has been shown to be hepatoprotective.
Purpose and methods: Aimed to investigate the underlying protective mechanisms of GBE on carbon tetrachloride (CCl4)-induced liver fibrosis in rats. Male Sprague Dawley rats were randomly divided into four groups: control group (C), model group (M), low-dose group (L), and high-dose group (H). Liver fibrosis was induced by CCl4 groups M, L, and H: group C was administered saline. In addition, GBE at different doses was used to treat groups L and H.
Results: The results of hematoxylin and eosin staining, Masson’s trichrome staining, a liver function index, and a liver fibrosis index showed that GBE application noticeably mitigated fibrosis and improved the function of the liver. The western blotting and immunohistochemistry analyses indicated that GBE reduced liver fibrosis not only by inhibiting p38 MAPK and NF-κBp65 via inhibition of IκBα degradation but also by inhibiting hepatocyte apoptosis via downregulation of Bax, upregulation of Bcl-2, and subsequent inhibition of caspase-3 activation. Inflammation-associated factors and hepatic stellate cell (HSC)-activation markers further demonstrated that GBE could effectively inhibit HSC activation and inflammation as a result of its regulation of p38 MAPK and nuclear factor-kappa B/IκBα signaling.
Conclusion: Our findings indicated a novel role for GBE in the treatment of liver fibrosis. The potential mechanisms may be associated with the following signaling pathways: 1) the p38 MAPK and nuclear factor-kappa B/IκBα signaling pathways (inhibiting inflammation and HSCs activation) and 2) the Bcl-2/Bax signaling pathway (inhibiting the apoptosis of hepatocytes).
Keywords: rats, HSCs activation, inflammation, hepatoprotective, mechanism, pathways
Introduction
Liver fibrosis is a common condition preceded by chronic inflammation.1 Many studies have indicated that unlike cirrhosis, fibrosis is reversible. Fibrosis is characterized by excessive deposition of collagen and extracellular matrix (ECM) proteins. Current research have provided growing evidence, which show that the key source of the ECM is the hepatic stellate cells (HSCs).2,3 HSCs, the focus of the fibrogenic response, go through a transdifferentiation from quiescent vitamin A-storing cells to proliferative myofibroblasts that secrete superfluous ECM, finally leading to liver fibrosis.4 Upon the transformation of quiescent to active HSCs, nuclear factor-kappa B (NF-κB) is induced, which leads to the production of various inflammatory factors including Cox-2, IL-1, IL-6, TNF-α, and TGF-β.5,6 These factors can in turn activate HSCs in a p38 MAPK-dependent process.7,8
p38 MAPK, a MAPK family member, plays an essential role in regulating many cellular processes including inflammation, fibrosis, and apoptosis.9 It can be induced by inflammatory factors and stress.10 The activation of p38 MAPK is often achieved by autophosphorylation at Thr180/Tyr182 residues, nuclear translocation, and dephosphorylation.11 Furthermore, p38 MAPK activation has been proposed to regulate the activation of NF-κB through a mechanism that may involve the regulation of histone H3.12
The NF-κB family of transcription factors, RelA (p65), RelB, c-Rel, p50, and p52, mainly regulate inflammatory and apoptosis responses through the NF-κB/IκBα signaling pathway.13 Numerous studies have shown that the canonical NF-κB/IκBα signal transduction pathway is accompanied by the translocation of specific NF-κB dimers from the cytoplasm to the nucleus. The related mechanisms include IκB kinase (IKK) activation, IκB phosphorylation and ubiquitination, nuclear translocation of related NF-κB dimers, and then transcription of the NF-κB target genes.14,15 NF-κB activation can contribute to hepatocyte injury and subsequent inflammation. Subsequently, massive hepatocyte death and inflammation may activate HSCs leading to fibrosis.16 However, when NF-κB activation is inhibited, HSCs will undergo enhanced apoptosis.17
In addition to p38 MAPK- and NF-κB/IκBα-mediated signaling, the Bcl-2/Bax signaling pathway plays an important role in apoptosis and hence impacts the development of liver fibrosis. The pro-survival Bcl-2 and pro-apoptotic Bax are the two most-studied members of the Bcl-2 family and function as the primary regulators of the mitochondrial pathway of apoptosis. The classic Bcl-2/Bax signaling pathway includes the activation of the signal transduction proteins tBid and Bim, the anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax responses, mitochondrial outer membrane permeabilization, the release of cytochrome C and caspase-3 induced apoptosis.18
Currently, herbal products account for a substantial portion of the current interest in alternative treatments. Ginkgo biloba extract (GBE), a traditional Chinese natural herb, has attracted immense attention for its treatment of diverse diseases both in humans and animal models, including coronary heart disease, Alzheimer disease and multi-infarct dementia, cerebral insufficiency, and renal ischemia-reperfusion injury.19–21 These interests are mainly due to the various ingredients of GBE, which include two terpene lactones, flavonoids, and organic acids as the main active components.22 Recent experimental studies showed that GBE had obvious inhibitory effects on liver fibrosis with no significant side effects.23–25 However, most studies of the mechanism of the GBE-mediated alleviation of liver fibrosis have been focused on the suppression of oxidant stress and the TGF-β signaling pathway.26,27 The anti-apoptotic effect and p38 MAPK signaling as well as NF-κB/IκBα signaling effects of GBE are still unknown.
On the basis of the previous studies, in this study, we further investigated the effects of various doses of GBE on carbon tetrachloride (CCl4)-induced liver fibrosis in rats. Specifically, we studied the inhibitory effect of GBE on inflammation and the activation of HSCs, which involved the p38 MAPK and NF-κB/IκBα signaling, accompanied by anti-apoptotic effect of GBE acting through the Bcl-2/Bax signaling.
Materials and methods
Major reagents
GBE (the national drug standard substance) was purchased from the Chinese Food And Drug Inspection Institute, Beijing, People’s Republic of China. CCl4 was purchased from Shanghai Jinghua Scientific & Technological Research Institute (Shanghai, People’s Republic of China). Pentobarbital sodium was purchased from Shanghai BeiZhuo Biochemical & Technological Co., Ltd (Shanghai, People’s Republic of China). Thermo Fisher Scientific, Waltham, MA, USA, provided the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Product No 78833, Lot#PG201976A). Monoclonal anti-p38 MAPK, anti-p-p38 MAPK, anti-NF-κBp65, and anti-IκBα rabbit antibodies were purchased from Santa Cruz Biotechnology Inc., Dallas, TX, USA. Monoclonal anti-Bcl-2, anti-Bax, anti-caspase-3, β-tubulin, and histone H3 rabbit antibodies were purchased from Cell Signaling Technology, Danvers, MA, USA. The horseradish peroxidase (HRP)-labeled GAPDH antibody was purchased from Bioworld Technology Inc, St Louis Park, MN, USA. The secondary antibody, HRP-labeled goat anti-rabbit IgG, was purchased from Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA. The radioimmunoprecipitation assay lysis buffer, phenylmethanesulfonyl fluoride, and sodium dodecyl sulfate -polyacrylamide gel electrophoresis protein-loading buffer (5×) were provided by the Beyotime Biotechnology Corporation, Shanghai, People’s Republic of China. Thermo Fisher Scientific provided theTRIzol® reagent. The PrimeScript™ RT Master Mix reagent (Cat #RR036A), Premix Ex Taq™ Hot Start Version reagent (Cat #RR030A) and SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) reagent (Cat #RR820A) were prepared by TaKaRa Co., Ltd, Kyoto, Japan. The rabbit IgG immunohistochemistry (streptavidin-biotin complex) and diaminobenzidine colorimetric kits were obtained from BOSTER Biological Technology Ltd, Wuhan, People’s Republic of China.
Rats and treatments
Male Sprague Dawley rats (6 weeks; 180–220 g) were from the B&K Universal Group Ltd. (Shanghai, People’s Republic of China). All animal experiments were approved by the Ethical Committee (approval ID: 2013-0016) of North Shanghai 9th People’s Hospital, Shanghai Jiao Tong University School of Medicine (Shanghai, People’s Republic of China). A total of 48 Sprague Dawley rats were housed in polypropylene cages in a standard laboratory animal room under a 12:12 hour light–dark cycle at a constant temperature (25°C) and humidity of 55%±10%. The rats were fed standard lab chow and provided with water ad libitum. Prior to the start of experimental treatment, they were acclimated to these conditions for 2 weeks.
After 2 weeks, the rats were randomly divided into four groups: control group (C), model group (M), low-dose group (L), and high-dose group (H) and were identified by marking with chrysolepic acid. The rats of group C were administered saline. The rats in the other groups were subjected to intragastric administration of CCl4 (1:1 in olive oil) at a dose of 1 mL/kg body weight, twice a week for 8 weeks. During the CCl4 exposures, the rats in groups L and H were also given GBE by intraperitoneal injection once per day at doses of 15 mg/kg and 30 mg/kg body weight, respectively, for 8 weeks. At the end of the 8th week, the surviving rats were weighed (empty stomach) and anesthetized with pentobarbital sodium. The blood samples were taken from the abdominal aorta and centrifuged (1,000 rpm for 15 minutes at room temperature) to obtain the sera, which were stored at −80°C until use. After that, the animals were exsanguinated, and the liver was quickly weighed and divided into two portions: one portion was fixed in 10% neutral formalin for histological study, and the other was immediately frozen in liquid nitrogen, and then transferred to a −80°C freezer.
Calculation of the liver index
The liver index was calculated by the following formula:
Liver index = [Liver weight (g)/Body weight (g)] ×100.28 |
Liver function examination
The liver function examination included three liver function markers: total bilirubin (TBIL), aspartate aminotransferase (AST), and alanine aminotransferase (ALT). The serum levels of the two enzymes were measured using enzyme-coupled oxidation of the reduced form of nicotinamide adenine dinucleotide (NADH) to the oxidized form of nicotinamide adenine dinucleotide (NAD+) using a fully automatic biochemical analyzer (Beckman LX-2, Beckman Coulter Inc, Fullerton, CA, USA). According to the reflectance spectrophotometry principle, the rate of oxidation represented the activity of the liver function parameters.
Liver fiber serological examination
To obtain the serum liver fibrosis index, the amino terminal propeptide of type III procollagen (PIIINP), type-IV collagen (IV-C), hyaluronic acid (HA), and laminin (LN) were detected using magnetic particle chemiluminescence assay with kits from the liver fibrosis detection series (Autobio Diagnostics Co., Ltd, Zhengzhou, People’s Republic of China). All the procedures were performed according to the standard steps of the instructions. The chemiluminescence detector was used to create the standard curves. The concentrations of PIIINP, IV-C, HA, and LN were obtained from the luminescence values for the tested samples and the standard curve.
Histopathological and immunohistochemical examination
The liver pieces that had been fixed in 10% neutral formalin were dehydrated through a graduated ethanol series, embedded in paraffin blocks, cut into 5 μm-thick sections with a microtome and mounted on glass slides. The liver sections were dewaxed in xylene, then rehydrated through a decreasing ethanol series for 2–3 minutes at each concentration and then washed with distilled water. Afterward, some slices were stained with hematoxylin and eosin (H&E) reagents or Masson’s trichrome staining reagents following the standard steps in the instructions. The remaining slices were used for immunohistochemical staining according to a previously published protocol29 using anti-p38 MAPK, anti-NF-κBp65, anti-Bcl-2, and anti-Bax as the primary antibodies. The secondary antibody was goat anti-rabbit. Microscopic fields in all liver sections were randomly selected for examination using a light microscope (Ti-S Inverted fluorescence microscope, Type 108, Nikon Corporation, Tokyo, Japan). For the histopathological analysis, the liver fibrosis severity was scored based on the criteria: 0, normal; 1, fibrosis present (collagen fibers present that extended from portal triad or central vein to peripheral region); 2, mild fibrosis (mild collagen fibers present with extension but without compartment formation); 3, moderate fibrosis (moderate collagen fibers present with some pseudo lobe formation); 4, severe fibrosis (many collagen fibers present with thickening of partial compartments and frequent pseudo lobe formation).30 For the immunohistochemical analysis, the expression of the same target proteins was evaluated according to the location and degree of tissues with positive staining: 0, positive staining (0%–5%); 1, weakly positive staining (6%–35%); 2, moderately positive staining (36%–55%); and 3, intensely positive staining (56%–100%).The level of each of the relevant proteins was evaluated and reported as the average score of the staining for each protein.31
Western blotting analysis
Before protein extraction, liver tissue blocks taken from the −80°C freezer were pulverized using a high throughput tissue-grinding apparatus (1 minute, 1,000 rpm, TL-2020, Ding Hao Yuan Technology Co., Ltd., Beijing, People’s Republic of China). Nuclear and cytosolic proteins from the tissues from the various groups were prepared with Nuclear and Cytoplasmic Extraction Reagents according to the provided instructions. The total cellular protein was extracted as a radioimmunoprecipitation assay lysate containing the protease inhibitor phenylmethanesulfonyl fluoride. To determine the loading quantity of samples, the protein concentrations were measured using the Bio-Rad protein assay (Bio-Rad Laboratories Inc., Hercules, CA, USA). After that, Sodium dodecyl sulfate -polyacrylamide gel electrophoresis sample loading buffer was added to all the protein samples, and the samples were thermally denatured at 95°C for 5 minutes using a thermostatic heater (WiseTherm® HB, DAIHAN Scientific Co., Ltd., Wonju-si, Korea). Then, western blotting assays were performed as previously described with some modifications.32 Equal protein amounts were resolved by electrophoresis on 12%, 10%, or 8% sodium dodecyl sulfate polyacrylamide gels and then transferred to polyvinylidene difluoride membranes. After blocking in 5% non-fat dry milk in Tris-Buffered Saline with Tween 20 for 2 hours at room temperature, the blots from the 12% sodium dodecyl sulfate polyacrylamide gels were then incubated overnight at 4°C with primary antibodies (monoclonal rabbit antibodies) to Bcl-2, Bax, or caspase-3 at 1:1,000 dilutions. The blots from the 10% sodium dodecyl sulfate polyacrylamide gels were used for analysis with the primary monoclonal rabbit antibodies to anti-p38 MAPK, anti-p-p38 MAPK, anti-NF-κBp65, or anti-IκBα. The next day, the blots were washed with Tris-Buffered Saline with Tween 20 and then incubated with HRP-labeled goat anti-rabbit IgG (1:5,000 dilution) for 60 minutes at room temperature. The bands were detected using an enhanced chemiluminescence system (Fusion FX7 Spectra; Vilber Lourmat, Eberhardzell, Germany).
Semi-quantitative RT-PCR and real-time PCR analysis
Total RNA from the liver tissue samples was isolated using the TRIzol reagent, and the concentration and purity were estimated using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, Gene Company Limited, Shanghai, People’s Republic China). The PrimeScript™ RT Master Mix reagent kit was used to synthesize cDNA using a Veriti 96-Well Thermal Cycler (Thermo Fisher Scientific) according to the manufacturer’s protocol. Afterward, real-time PCR was performed using SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) reagent in a LightCycler 480 instrument (Hoffman-La Roche Ltd., Basel, Switzerland). To further confirm the real-time PCR results, semi-quantitative PCR was conducted using Premix Ex Taq™ Hot Start Version reagent by the same Veriti 96-Well Thermal Cycler. The primers were synthesized by Sangon Biotech Co. Ltd. (Shanghai, People’s Republic of China) and are listed in Table 1. All steps were based on the manufacturer’s operating instructions except for minor improvements in the reaction conditions. The semi-quantitative PCR products were analyzed by 2.0% gel electrophoresis and visualized using a gel documentation system (Fusion FX7 Spectra; Vilber Lourmat). To obtain the concentrations of the PCR products, the conventional real-time PCR mathematical method calculation was performed using the cycle threshold values and the values were plotted as the fold change compared with control groups (2−ΔΔCT method).The levels of the target genes were normalized to that of GAPDH.
 | Table 1 Gene primers sequences for mRNA amplification |
Statistical analysis
All data were analyzed using SPSS v17.0 (SPSS Inc., Chicago, IL, USA) and analyzed by the one-way analysis of variance, the Student–Newman–Keuls test and the Mann–Whitney test. The data are presented as the means ± standard deviation (SD). P-values of 0.05 or less were regarded as statistically significant.
Results
GBE reduced liver injury upon CCl4 administration
We firstly evaluated the effects of GBE on body weight, liver weight, and liver indices of rats. As shown in Table 2, the body weight significantly decreased in group M compared with group C (P<0.05). GBE treatment (group H and L) significantly increased body weight compared with group M (P<0.05). Comparing group H with group L, significant differences were observed (P<0.05). The liver weight and liver indices of rats significantly increased in group M compared with group C (P<0.05) and significant differences were observed comparing group H with group L (P<0.05). Liver indices significantly decreased in group H and L compared with group M (P<0.05). But, no significant differences in the liver weight between the groups C and H or between groups M and L were found (P>0.05).
 | Table 2 Effects of GBE on body weight, liver weight, and liver index of rats |
To assess the effect of GBE on liver function, three liver function markers (TBIL, ALT, and AST) were detected. The results are shown in Table 3. The serum concentrations of TBIL, ALT, and AST significantly increased in group M compared with group C (P<0.05). However, group H significantly decreased the levels of the three markers in the serum compared with those of group M (P<0.05). No significant difference between groups M and L in the concentrations of TBIL were found (P>0.05).
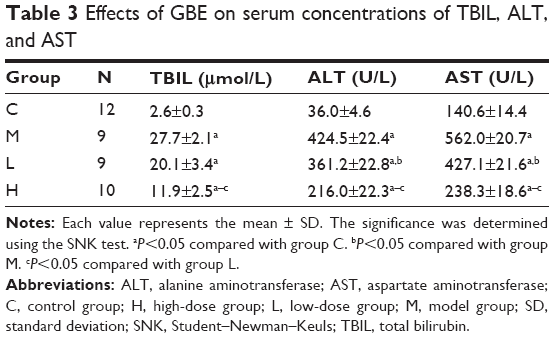 | Table 3 Effects of GBE on serum concentrations of TBIL, ALT, and AST |
To investigate the effect of GBE on liver fibrosis, the serum concentrations of PIIINP, IV-C, HA, and LN were detected, and the results are shown in Table 4. Compared with group C, the serum concentrations of PIIINP, IV-C, HA, and LN were significantly increased in group M (P<0.05). However, the treatment with GBE (group H and L) could significantly decrease the levels of the four markers (P<0.05). Comparing group H with group L, significant differences were observed (P<0.05). Collectively, these results supported that GBE not only reduced liver injury and liver fibrosis but also improved liver function upon CCl4 administration.
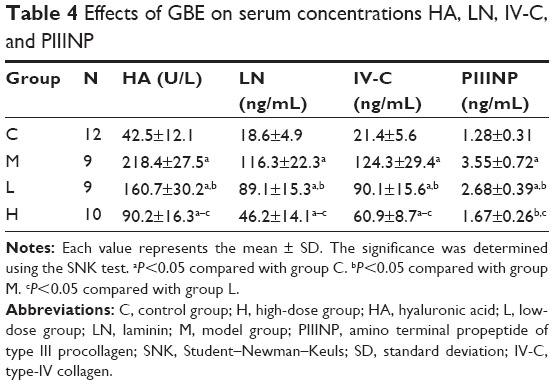 | Table 4 Effects of GBE on serum concentrations HA, LN, IV-C, and PIIINP |
GBE alleviated histopathological changes by inhibiting liver fibrosis and apoptosis
To further determine the protective effect of GBE against liver fibrosis in vivo, histological changes in the liver tissue of rats were detected by H&E and Masson’s trichrome staining. Representative photographs of the liver morphology are shown in Figures 1 and 2, and the degree of fibrosis for each group is shown in Table 5. In the livers of group C, the liver cells were radically arranged around the central vein accompanied by lobular architecture with normal integrity. No degeneration or necrosis of liver cells nor any other pathological features could be seen (Figures 1A and 2A). Compared with group C, in group M, the liver cells around the central vein were spotty or fully necrotic (P<0.05). The livers exhibited marked connective tissue hyperplasia, the ECM content was increased, and collagen accumulation surrounded the lobules, which resulted in large fibrous septa and pseudo-lobule formation (Figures 1B and 2B). However, in the GBE-supplemented groups L and H, these alterations in the liver sections were remarkably reduced, and the degree of liver fibrosis was decreased (P<0.05). Especially in group H, the hepatocyte degeneration and necrosis were not obvious (Figures 1D and 2D). Connective tissue hyperplasia, collagen deposition, and inflammatory cell infiltration were markedly decreased (P<0.05). Furthermore, the pseudo-lobule formation was not observed. However, the effect was less obvious in group L than in group H (Figures 1C and 2C). Analysis of the figures and data (Figures 1 and 2, Table 5) confirmed that GBE reduced liver fibrogenesis rather than completely preventing it, as indicated by the significant difference between group H and group C (P<0.05).
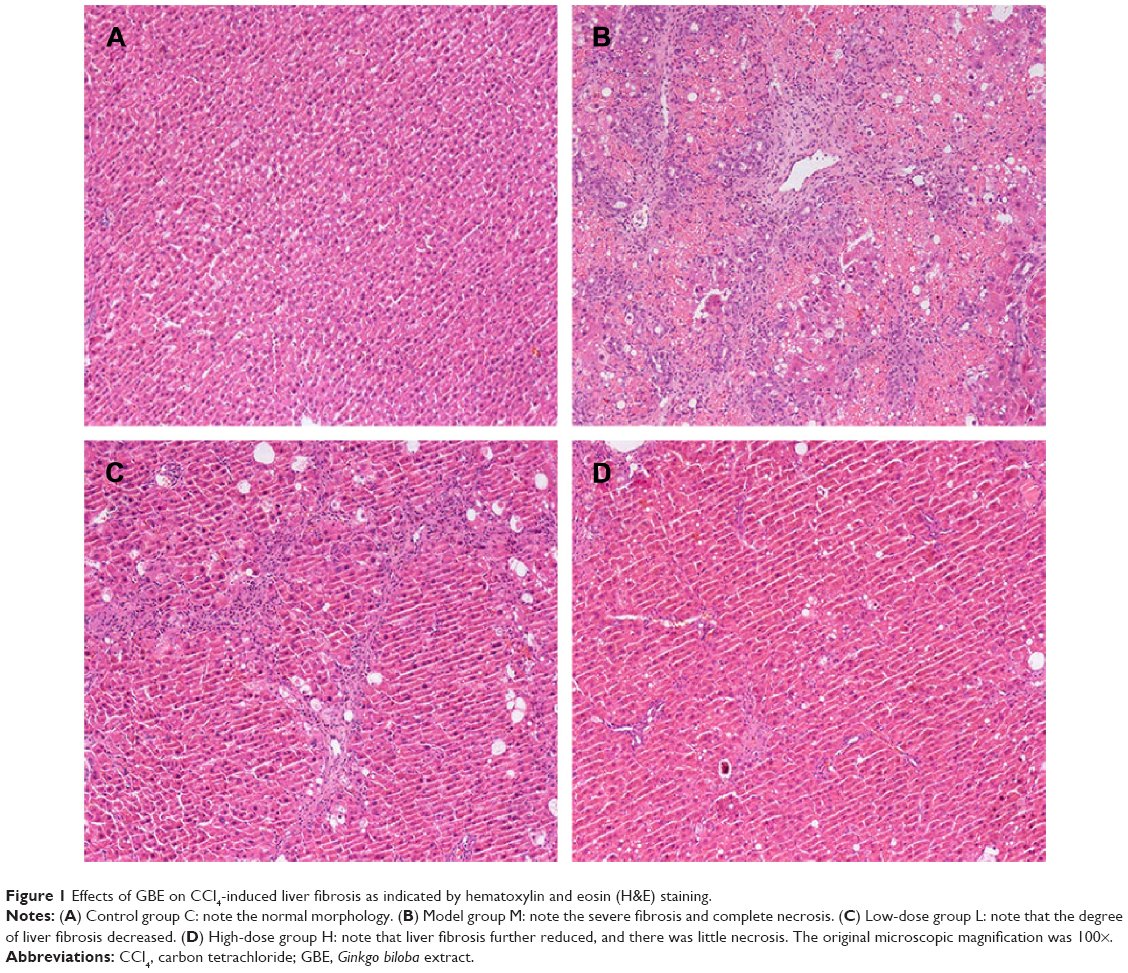 | Figure 1 Effects of GBE on CCl4-induced liver fibrosis as indicated by hematoxylin and eosin (H&E) staining. |
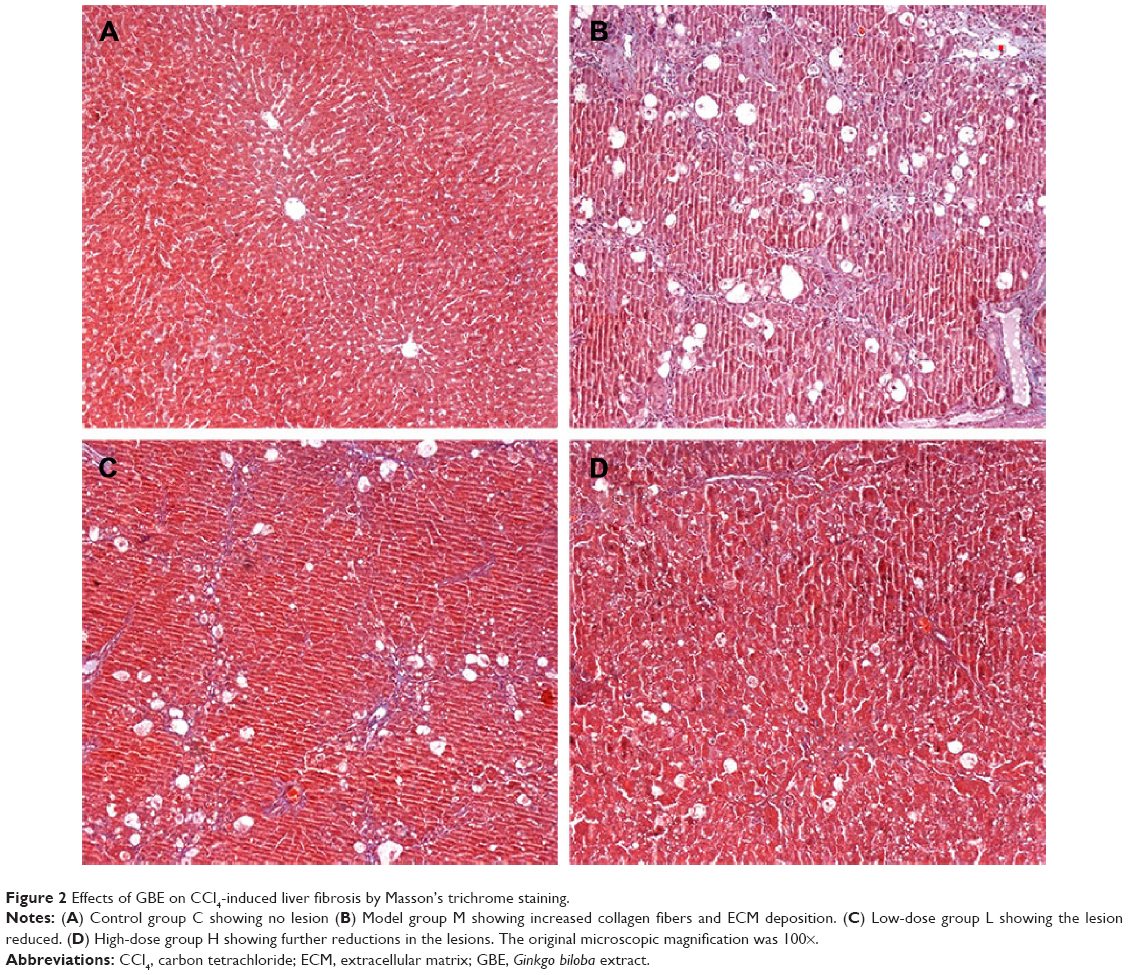 | Figure 2 Effects of GBE on CCl4-induced liver fibrosis by Masson’s trichrome staining. |
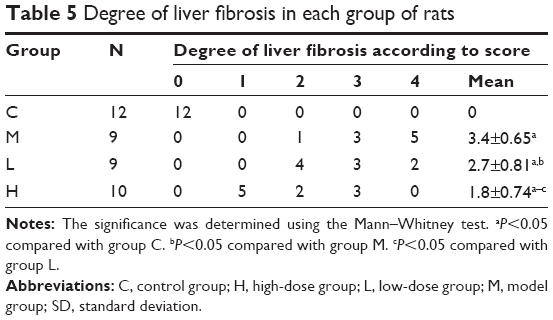 | Table 5 Degree of liver fibrosis in each group of rats |
GBE suppressed the expression levels of proteins involved in p38 MAPK, NF-κB/IκBα, and Bcl-2/Bax signaling
In this paper, we mainly focused on the p38 MAPK, NF-κB/IκBα, and Bcl-2/Bax signaling to study the potential mechanism by which GBE mitigated liver fibrosis and apoptosis. To determine whether the anti-fibrotic effect of GBE is associated with the p38 MAPK, NF-κB/IκBα, and Bcl-2/Bax signaling pathways, immunohistochemistry and western blotting analysis were performed. Images of the immunohistochemically stained sections and the staining scores of all groups are shown in Figures 3 and 4. Positive signals were indicated by a brown or yellow granular mass. Except for NF-κB, which appeared in both cytoplasm and nucleus, staining of the other proteins was mainly observed in the cytoplasm. For p38 MAPK, NF-κB, and Bax, the positive-staining scores were dose-dependently decreased in the GBE-treatment groups as shown in Figures 3B, D and 4D (P<0.05). In addition, the degree of staining in group M was the most intense, and group C was the lightest by comparison as demonstrated in the immunohistochemical staining images in Figures 3A, C and 4C (P<0.05). However, the trend for the positive-staining degree for Bcl-2 was completely the opposite: the scores were dose-dependently increased and group C was the highest of all, as shown in Figure 4A and B (P<0.05). These results indicated that GBE could effectively increase Bcl-2 and decrease Bax to inhibit apoptosis. Furthermore, the p38 MAPK and NF-κB downregulation suggested that GBE might influence p38 MAPK and NF-κB signaling.
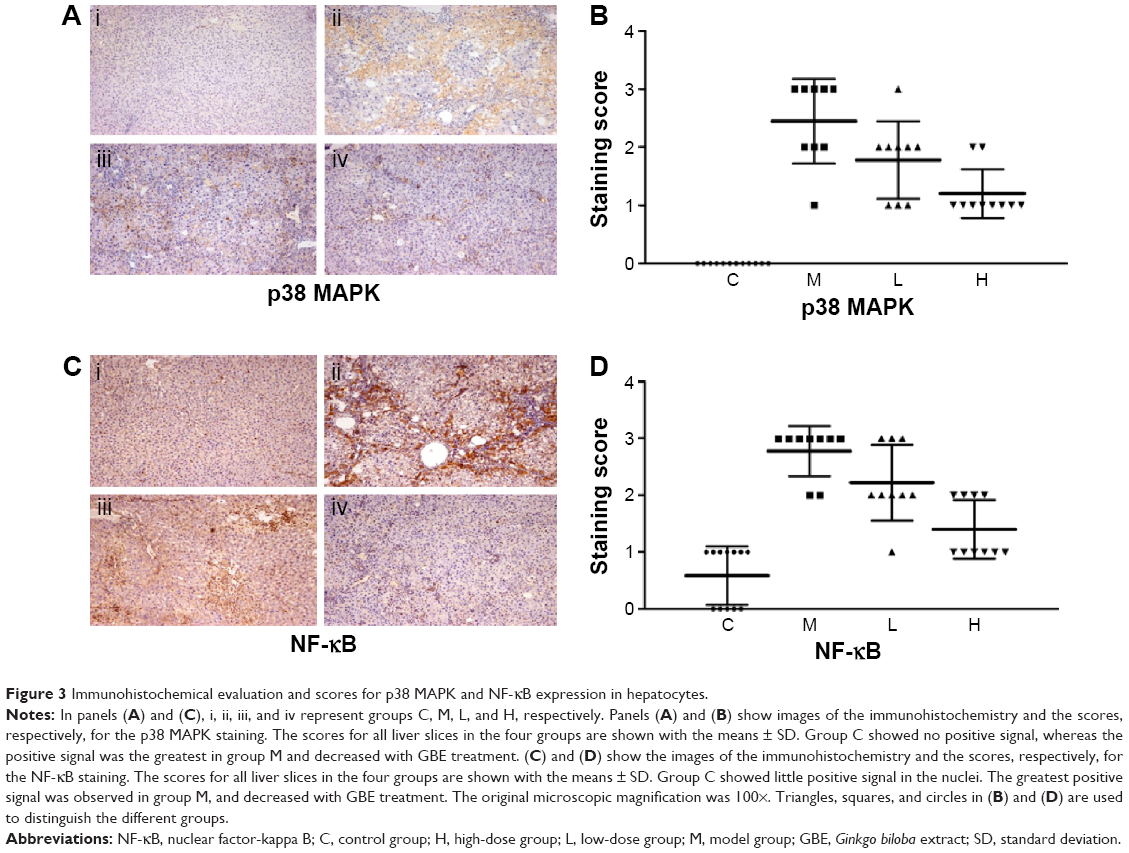 | Figure 3 Immunohistochemical evaluation and scores for p38 MAPK and NF-κB expression in hepatocytes. |
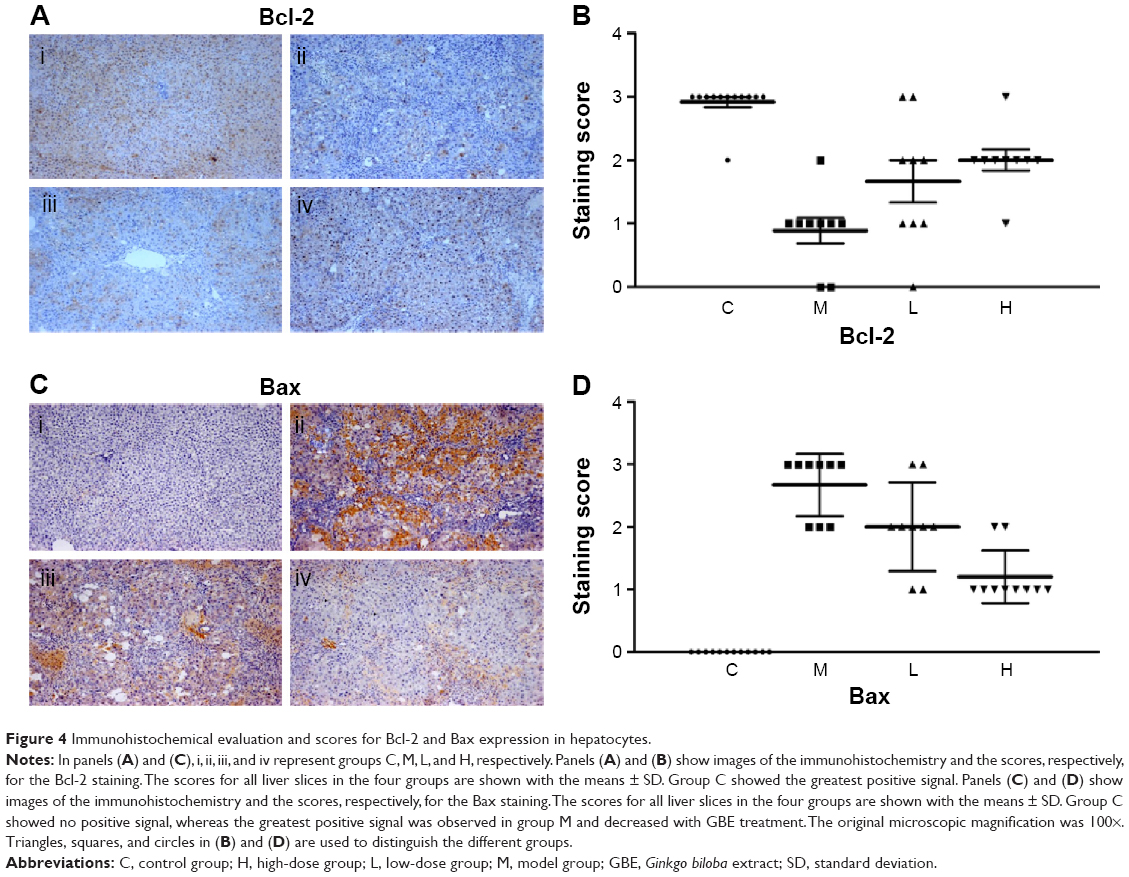 | Figure 4 Immunohistochemical evaluation and scores for Bcl-2 and Bax expression in hepatocytes. |
To verify these results further, the western blotting analyses were performed. The results of the western blotting analyses are shown in Figures 5, 6, 7, and 8. All western blotting data were consistent with the immunohistochemical results. As shown in Figure 5, the expression levels of p38 MAPK and p-p38 MAPK were dose-dependently downregulated by the GBE treatment, as was the level of NF-κBp65 in the nuclei (Figure 7). Compared with group C, the levels of these proteins were significantly increased in group M (P<0.05). As shown in Figure 6, the expression levels of the cytoplasmic proteins NF-κBp65 and IκBα were significantly decreased in group M compared with other groups (P<0.05). Despite failing to reach the levels found in group C, the levels of these proteins were dose-dependently increased in the GBE-treated groups (L and H) (P<0.05). In addition, the isolated cytoplasmic fraction displayed a high level of cytoplasmic internal reference β-tubulin but a low level of nuclear internal reference histone H3, which was opposite to the result of the isolated nuclear fraction as shown in Figure 6C. These indicated that the high purity of the separation of the cytoplasmic and the nucleus proteins have been got. As Figure 8 shows, the trends in the expression levels of Bcl-2 and Bax in the total cell protein extracts were opposite from each other. The GBE supplementation upregulated Bcl-2, downregulated Bax, and inhibited caspase-3 in a dose-dependent manner (P<0.05). Compared with group C, the expression level of Bcl-2 was significantly decreased in group M (P<0.05). However, the levels of Bax and caspase-3 were significantly increased in group M compared with group C (P<0.05).
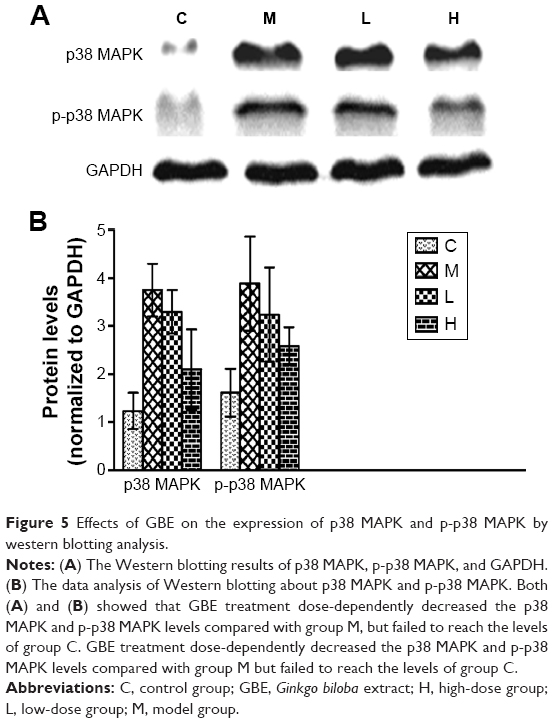 | Figure 5 Effects of GBE on the expression of p38 MAPK and p-p38 MAPK by western blotting analysis. |
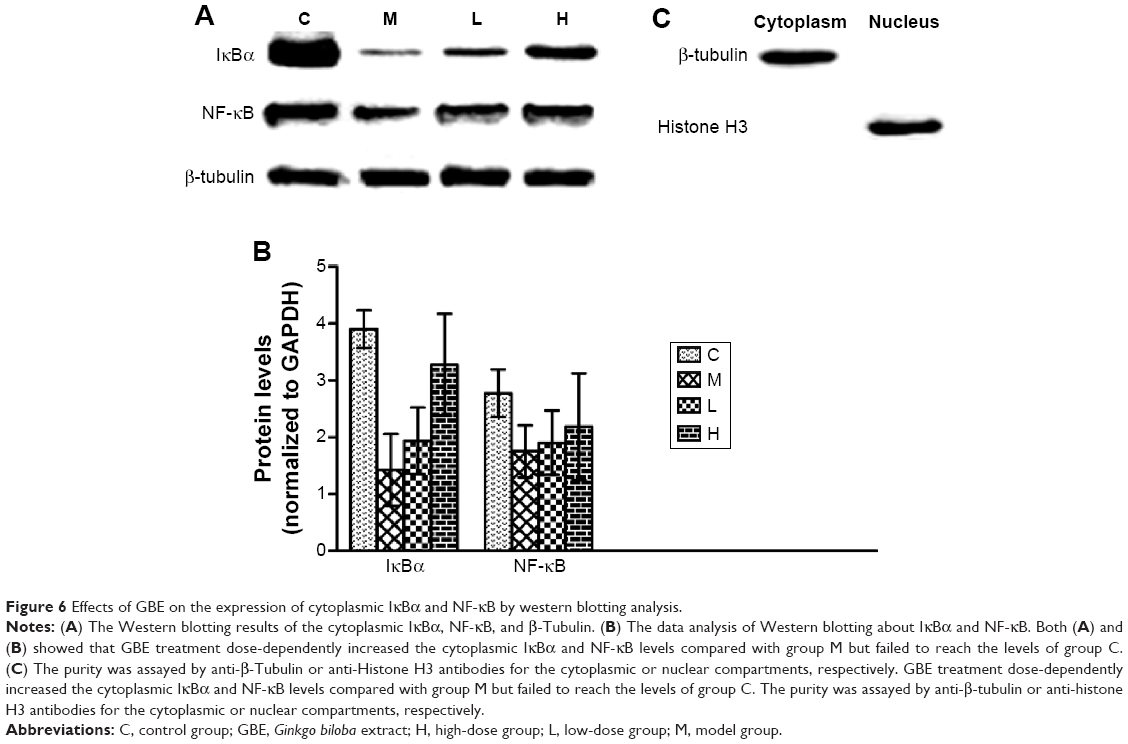 | Figure 6 Effects of GBE on the expression of cytoplasmic IκBα and NF-κB by western blotting analysis. |
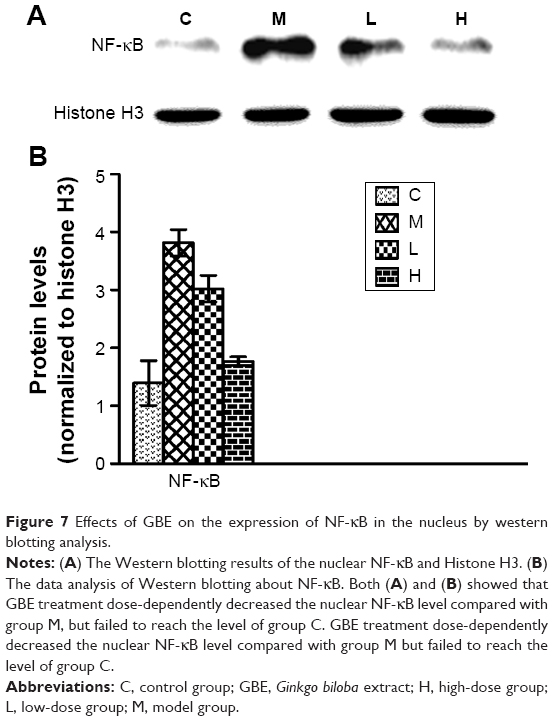 | Figure 7 Effects of GBE on the expression of NF-κB in the nucleus by western blotting analysis. |
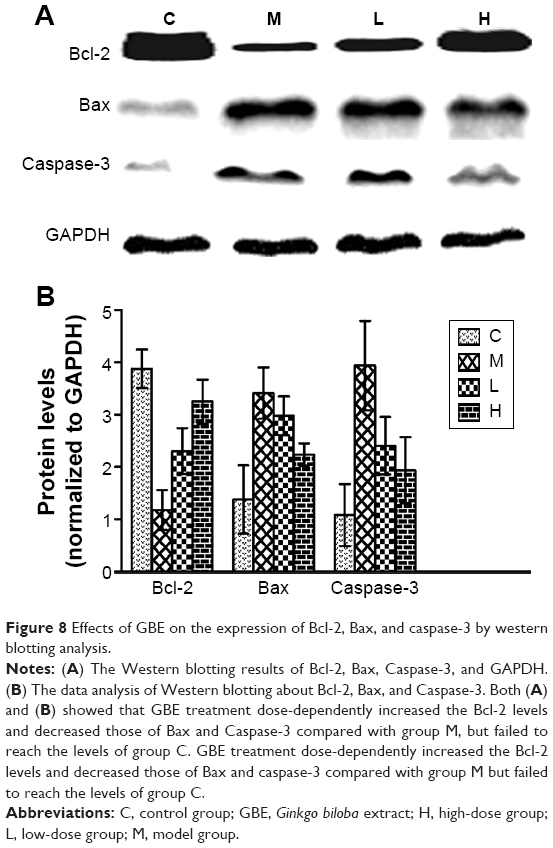 | Figure 8 Effects of GBE on the expression of Bcl-2, Bax, and caspase-3 by western blotting analysis. |
Taken the results of immunohistochemistry and western blotting analysis into consideration collectively, we concluded that GBE mitigated liver fibrosis and apoptosis by inhibiting the p38 MAPK, NF-κB/IκBα, and Bcl-2/Bax signaling pathways.
GBE reduced inflammation and HSC activation
Inflammation and HSC activation play important roles in the pathogenesis of hepatic fibrosis. To determine the impact of GBE on inflammation and HSC activation, inflammatory factors (Cox-2, IL-1, IL-6, TNF-α, and TGF-β) and HSC activation markers (Colla1, Colla2, MMP-1, TIMP-1, α-SMA, and desmin) were measured by semi-quantitative RT-PCR and real-time PCR. The results are shown in Figures 9 and 10. Except for gene MMP-1, the mRNA levels of these specific genes had the same trends both in Figures 9 and 10. They were markedly decreased in a dose-dependent manner in the GBE-treatment groups compared with group M (P<0.05). However, the levels in the high-dose group H did not reach the levels of group C (P<0.05). As for the mRNA levels of gene MMP-1, the result was just the opposite (Figure 10). MMP-1 was markedly increased in a dose-dependent manner in the GBE-treatment groups compared with group M (P<0.05). However, the mRNA level of MMP-1 in the high-dose group H did not reach the level in group C (P<0.05). In Figures 9B and 10B, the CT values were estimated from the real-time PCR experiments. According to the relative quantitative formula 2−ΔΔCT, the relative expression quantities of the target genes were normalized to that of GAPDH. As shown in Figures 9 and 10, the results of the real-time PCR analyses were consistent with those of the semi-quantitative RT-PCR. Both methods indicated that GBE could reduce the NF-κB-regulated inflammatory factors and HSC activation.
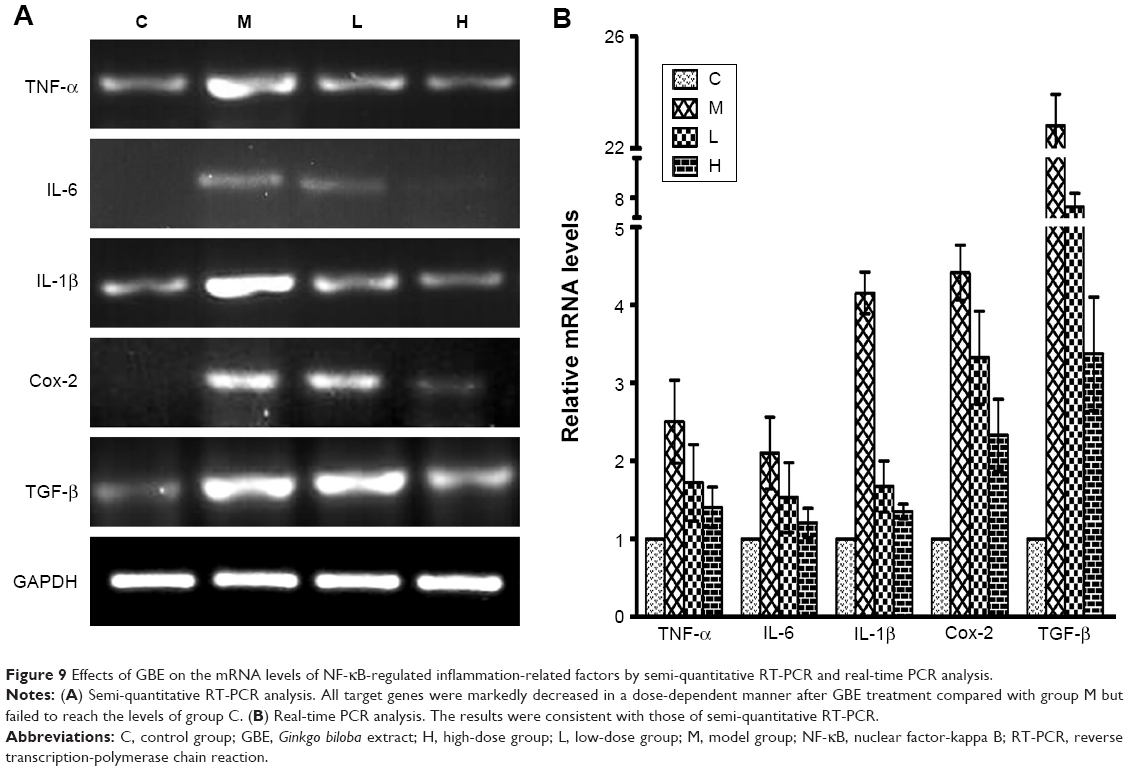 | Figure 9 Effects of GBE on the mRNA levels of NF-κB-regulated inflammation-related factors by semi-quantitative RT-PCR and real-time PCR analysis. |
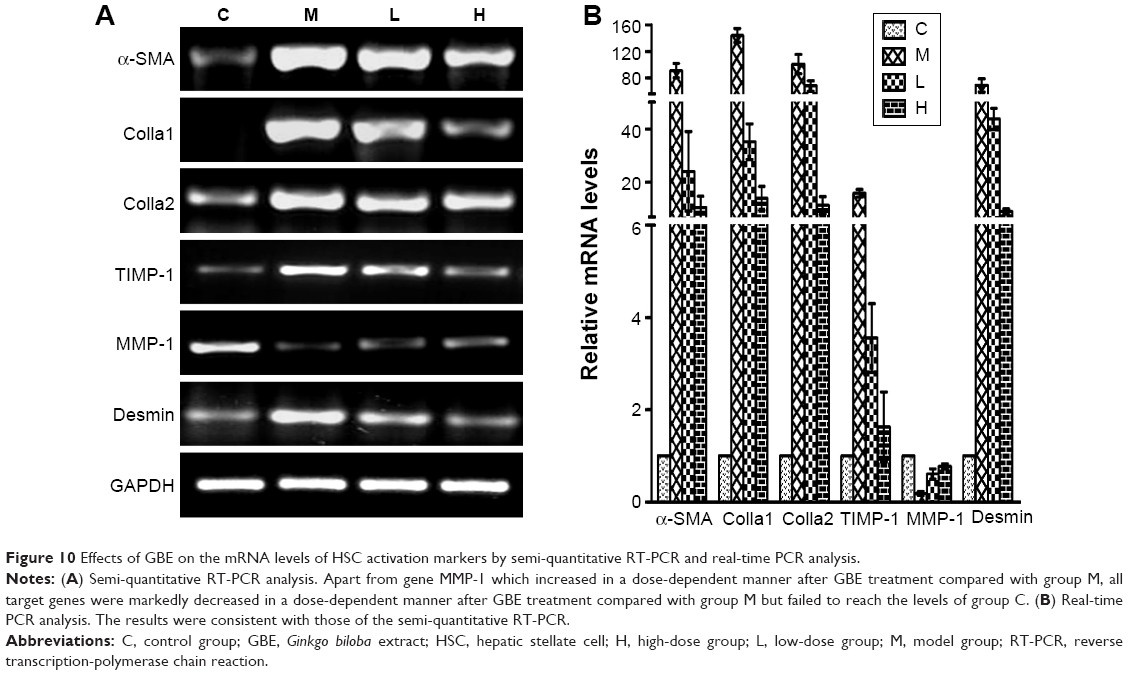 | Figure 10 Effects of GBE on the mRNA levels of HSC activation markers by semi-quantitative RT-PCR and real-time PCR analysis. |
Discussion
Liver fibrosis is a common clinical syndrome. In the past two decades, studies of liver fibrosis have increased the understanding of the molecular pathogenesis of the condition, which has been helpful for the discovery of potentially effective drugs to treat liver fibrosis. Previous studies have indicated that GBE could actively suppress hepatic fibrosis, and Zhang et al33 concluded that GBE could mitigate liver fibrosis through multiple mechanisms. However, the mechanisms involved in this process have not yet been completely elucidated.
GBE, a well-known and inexpensive herb, is one of the most often used herbal dietary supplements in the world. The present study was designed to determine its potential mechanism in the context of preventing liver fibrosis and apoptosis. To establish the liver fibrosis model in our study, intragastric administration of CCl4 was used to induce liver fibrosis. The model showed many of the same features as human liver fibrosis of various etiologies.34 Consistent with previous studies,35,36 intraperitoneal injection of GBE for 8 weeks produced inhibitory effects on the levels of the important liver function indicators TBIL, AST, and ALT. At the same time, GBE decreased the serum levels of PIIINP, IV-C, HA, and LN. These markers, especially serum HA and LN, are used to diagnose the degree of liver fibrosis.37,38 Based on the serological data in combination with the results of the H&E and Masson’s trichrome staining, we concluded that GBE could significantly ameliorate liver fibrosis.
There is now overwhelming evidence indicating that the activation of HSCs is vital to liver injury and liver fibrosis.39 Thus, controlling the HSC activation must be very important in preventing or reversing liver fibrosis. Studies have shown that activation of the NF-κBs, especially the NF-κBp65 subunit, is closely associated with HSC activation in liver fibrosis.40,41 In addition, the NF-κB/IκBα signal, constitutively expressed in all cell types and activated by various stimuli, inhibited the apoptosis of activated HSCs.42,43 These results indicate that a drug that could inhibit NF-κB/IκBα signaling might be a potential anti-fibrosis drug. In this paper, we first assumed that GBE suppressed liver fibrosis through the NF-κB/IκBα signaling pathway. It is very clear that in the unstimulated state, NF-κB is associated with IκBα in cytoplasm. Upon stimulation by stressors, IKK is activated leading to the phosphorylation of IκB, which is then degraded through the ubiquitin-proteasome pathway. Afterward, NF-κB is transported into the nuclei resulting in the activation of gene transcription.14,15 Our study, therefore, employed western blotting to detect the cytoplasmic proteins IκBα and NF-κBp65 in both the nuclei and cytoplasm. In addition, immunohistochemical staining was performed to further clarify the changes in the subcellular distribution of NF-κB. The data obtained from these experiments showed that consistent with our hypothesis, GBE could indeed inhibit NF-κB/IκBα signaling.
Inflammation and apoptosis are always associated with liver fibrosis in diseases caused by hepatic injuries.44,45 Hepatic injury results in a massive accumulation of recruited inflammatory cells and is often accompanied with sustained inflammation. These inflammatory cells can induce an immune response, interact with HSCs and/or myofibroblasts, activate HSCs, and inhibit ECM degradation by releasing inflammatory factors.46 Moreover, previous studies have shown that inflammatory factors, including Cox-2, IL-1, IL-6, TNF-α, and TGF-β are regulated by NF-κB. These factors clearly induce inflammation and liver fibrosis,47–49 which indicates their importance in liver fibrosis formation. Therefore, we measured the mRNA levels for these factors. The results of these experiments provided further evidence for the inhibitory effects of GBE on inflammation and liver fibrosis, especially those associated with the activation of NF-κB. The hepatocyte apoptosis process is always associated with an inflammatory response, which is an important mechanism of liver fibrosis. When hepatocytes undergo apoptosis and fail to regenerate, the apoptotic hepatocytes are replaced by abundant ECM.50 Considering that Bcl-2/Bax signaling is a key to the mitochondrial apoptosis pathway and an important regulator of the intrinsic apoptosis, we then studied the anti-apoptotic effect of GBE by investigating the protein levels of Bcl-2, Bax, and caspase-3. Importantly, the results of these experiments were consistent with those for many other anti-fibrotic agents, which have been shown to inhibit apoptosis by upregulating Bcl-2 and downregulating Bax, and caspase-3 through Bcl-2/Bax signaling.51,52
To further investigate if there were any other signaling molecules involved in the anti-fibrosis process of GBE, we analyzed the signaling pathways involved in liver fibrosis and concluded that p38 MAPK signaling may be closely associated with liver fibrosis. For example, Schnabl et al53 found that p38 MAPK had a major role in the process of HSC differentiation. Some studies have also indicated that inflammatory factors such as IL-1, TNF-α, and TGF-β could effectively promote HSC activation by regulating p38 MAPK signaling.7,8 Therefore, the effect of GBE on p38 MAPK signaling was studied in this project. We were pleasantly surprised to find that GBE could effectively suppress p38 MAPK activation. However, p38 MAPK has been shown to be required for IKK-independent IκBα phosphorylation and degradation. Mainly the p65 dimer has been reported to be released by such signaling.54 The mechanism by which p38 MAPK activates NF-κB as described by Saccani et al12 is that stimulus induces p38 MAPK phosphorylation as well as histone H3 phosphoacetylation, then p38-dependent H3 phosphorylation marks promoters for increased NF-κB recruitment. In addition, Olson et al55 confirmed that p38 MAPK activity regulates the transcriptional activation of NF-κBp65 by phosphorylating MSK1. As p38 MAPK has numerous direct and indirect interactions with NF-κB, an obvious direction for our future studies will be to investigate the possible interactions between the p38 MAPK and NF-κB/IκBα signaling in GBE-treatment models. Moreover, HSC activation markers such as Colla1, Colla2, MMP-1, TIMP-1, α-SMA, and desmin were inhibited by GBE in our study. From these previous studies and our data, we conclude that GBE mitigates liver fibrosis by regulating the p38 MAPK and NF-κB/IκBα signaling that activates the HSCs.
In addition, p38 MAPK can also induce mitochondria-dependent apoptosis. p38 MAPK activation not only promoted the mitochondrial translocation of Bax and Bim and inhibited Bcl-2 function by enhancing the phosphorylation of these factors but also triggered the activation of caspase-3, 9.56,57 Hence, we concluded that p38 MAPK signaling and Bcl-2/Bax signaling might influence each other and cooperatively contribute to the liver fibrosis-suppressing effect of GBE.
In summary, the present paper has addressed the novel mechanisms by which GBE might act as a potential anti-fibrotic agent by affecting p38 MAPK, NF-κB/IκBα signaling, and Bcl-2/Bax signaling pathways. These observations were not included in the previous studies. The new findings demonstrated decreased expression of the p38 MAPK, NF-κB-IκBα axis, and Bcl-2-Bax axis elements. These results showed that GBE could markedly ameliorate CCl4-induced liver fibrosis by regulating p38 MAPK and NF-κB/IκBα signaling to inhibit HSC activation and disrupting Bcl-2/Bax signaling to inhibit hepatocyte apoptosis. In summary, p38 MAPK, NF-κB/IκBα signaling, and Bcl-2/Bax signaling cooperatively support hepatocyte survival and inhibit the development of liver fibrosis. However, the role of GBE as a therapeutic agent in liver fibrosis needs to be further investigated. We expect a more comprehensive mechanism will be found for the anti-fibrotic effects of GBE.
Acknowledgments
This project was supported by the Science and Technology Commission of Shanghai Baoshan District (Grant No 14-E-5), the Xinhua Research Fund (Grant No 14XJ22011). We thank the WebShop of ELSEVIER (http://webshop.elsevier.com/languageservices/) for its linguistic assistance during the preparation of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25(2):195–206. | ||
Li D, Friedman SL. Liver fibrogenesis and the role of hepatic stellate cells: new insights and prospects for therapy. J Gastroenterol Hepatol. 1999;14(7):618–633. | ||
Friedman SL. Hepatic fibrosis-Overview. Toxicology. 2008;254(3):120–129. | ||
Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–1669. | ||
Lee KS, Buck M, Houglum K, Chojkier M. Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest. 1995;96(5):2461–2468. | ||
Schwabe RF, Schnabl B, Kweon YO, Brenner DA. CD40 activates NF-kappa B and c-Jun N-terminal kinase and enhances chemokine secretion on activated human hepatic stellate cells. J Immunol. 2001;166(11):6812–6819. | ||
Varela-Rey M, Montiel-Duarte C, Oses-Prieto JA, et al. p38 MAPK mediates the regulation of alpha1(I) procollagen mRNA levels by TNF-alpha and TGF-beta in a cell line of rat hepatic stellate cells(1). FEBS Lett. 2002;528(1–3):133–138. | ||
Zhang YP, Yao XX, Zhao X. Interleukin-1 beta up-regulates tissue inhibitor of matrix metalloproteinase-1 mRNA and phosphorylation of c-jun N-terminal kinase and p38 in hepatic stellate cells. World J Gastroenterol. 2006;12(9):1392–1396. | ||
Debacq-Chainiaux F, Boilan E, Dedessus Le Moutier J, Weemaels G, Toussaint O. p38(MAPK) in the senescence of human and murine fibroblasts. Adv Exp Med Biol. 2010;694:126–137. | ||
Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81(2):807–869. | ||
Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15(1):11–18. | ||
Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3(1):69–75. | ||
Napetschnig J, Wu H. Molecular basis of NF-kappaB signaling. Annu Rev Biophys. 2013;42:443–468. | ||
Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–362. | ||
Makino A, Fujino K, Parrish NF, Honda T, Tomonaga K. Borna disease virus possesses an NF-kB inhibitory sequence in the nucleoprotein gene. Sci Rep. 2015;5:8696. | ||
Shen H, Sheng L, Chen Z, et al. Mouse hepatocyte overexpression of NF-kappaB-inducing kinase (NIK) triggers fatal macrophage-dependent liver injury and fibrosis. Hepatology. 2014;60(6):2065–2076. | ||
Anan A, Baskin-Bey ES, Bronk SF, Werneburg NW, Shah VH, Gores GJ. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology. 2006;43(2):335–344. | ||
Shamas-Din A, Bindner S, Chi X, Leber B, Andrews DW, Fradin C. Distinct lipid effects on tBid and Bim activation of membrane permeabilization by pro-apoptotic Bax. Biochem J. 2015;467(3): 495–505. | ||
Kanowski S, Herrmann WM, Stephan K, Wierich W, Horr R. Proof of efficacy of the Ginkgo biloba special extract EGb 761 in outpatients suffering from mild to moderate primary degenerative dementia of the Alzheimer type or multi-infarct dementia. Phytomedicine. 1997;4(1):3–13. | ||
Kimbel KH. Ginkgo biloba. Lancet. 1992;340(8833):1474. | ||
Akdere H, Tastekin E, Mericliler M, Burgazli KM. The protective effects of Ginkgo biloba EGb761 extract against renal ischemia-reperfusion injury in rats. Eur Rev Med Pharmacol Sci. 2014;18(19):2936–2941. | ||
Birks J, Grimley Evans J. Ginkgo biloba for cognitive impairment and dementia. Cochrane Database Syst Rev. 2009;(1):CD003120. | ||
Al-Attar AM. Attenuating effect of Ginkgo biloba leaves extract on liver fibrosis induced by thioacetamide in mice. J Biomed Biotechnol. 2012;2012:761450. | ||
Zhang C, Zhu Y, Wan J, Xu H, Shi H, Lu X. Effects of Ginkgo biloba extract on cell proliferation, cytokines and extracellular matrix of hepatic stellate cells. Liver Int. 2006;26(10):1283–1290. | ||
Luo YJ, Yu JP, Shi ZH, Wang L. Ginkgo biloba extract reverses CCl4-induced liver fibrosis in rats. World J Gastroenterol. 2004;10(7): 1037–1042. | ||
He SX, Luo JY, Wang YP, et al. Effects of extract from Ginkgo biloba on carbon tetrachloride-induced liver injury in rats. World J Gastroenterol. 2006;12(24):3924–3928. | ||
Zhou ZY, Tang SQ, Zhou YM, Luo HS, Liu X. Antioxidant and hepatoprotective effects of extract of Ginkgo biloba in rats of non-alcoholic steatohepatitis. Saudi Med J. 2010;31(10):1114–1118. | ||
Wang YP, Cheng ML, Zhang BF, Mu M, Wu J. Effects of blueberry on hepatic fibrosis and transcription factor Nrf2 in rats. World J Gastroenterol. 2010;16(21):2657–2663. | ||
Liu H, Ren GP, Wang TY, Chen YX, et al. Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial-mesenchymal transition. Carcinogenesis. 2015;36(4):459–468. | ||
Li C, Luo J, Li L, Cheng M, et al. The collagenolytic effects of the traditional Chinese medicine preparation, Han-Dan-Gan-Le, contribute to reversal of chemical-induced liver fibrosis in rats. Life Sci. 2003;72(14):1563–1571. | ||
Li HL, Han L, Chen HR, et al. PinX1 serves as a potential prognostic indicator for clear cell renal cell carcinoma and inhibits its invasion and metastasis by suppressing MMP-2 via NF-kappaB-dependent transcription. Oncotarget. Epub 2015 May 27. | ||
Li Y, Jiang Y, Wan Y, et al. Medroxyprogestogen enhances apoptosis of SKOV-3 cells via inhibition of the PI3K/Akt signaling pathway. J Biomed Res. 2013;27(1):43–50. | ||
Zhang CF, Zhang CQ, Zhu YH, Wang J, Xu HW, Ren WH. Ginkgo biloba extract EGb 761 alleviates hepatic fibrosis and sinusoidal microcirculation disturbance in patients with chronic hepatitis B. Gastroenterol Res. 2008;1(1):20–28. | ||
Fujii T, Fuchs BC, Yamada S, et al. Mouse model of carbon tetrachloride induced liver fibrosis: histopathological changes and expression of CD133 and epidermal growth factor. BMC Gastroenterol. 2010; 10:79. | ||
Ding J, Yu J, Wang C, et al. Ginkgo biloba extract alleviates liver fibrosis induced by CCl in rats. Liver Int. 2005;25(6):1224–1232. | ||
Chávez-Morales RM, Jaramillo-Juárez F, Posadas del Río FA, Reyes-Romero MA, Rodríguez-Vázquez ML, Martínez-Saldaña MC. Protective effect of Ginkgo biloba extract on liver damage by a single dose of CCl(4) in male rats. Hum Exp Toxicol. 2011;30(3): 209–216. | ||
Li F, Zhu CL, Zhang H, et al. Role of hyaluronic acid and laminin as serum markers for predicting significant fibrosis in patients with chronic hepatitis B. Braz J Infect Dis. 2012;16(1):9–14. | ||
Parsian H, Rahimipour A, Nouri M, Somi MH, Qujeq D. Assessment of liver fibrosis development in chronic hepatitis B patients by serum hyaluronic acid and laminin levels. Acta Clin Croat. 2010;49(3):257–265. | ||
Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–172. | ||
Wang F, Liu S, DU T, Chen H, Li Z, Yan J. NF-kappaB inhibition alleviates carbon tetrachloride-induced liver fibrosis via suppression of activated hepatic stellate cells. Exp Ther Med. 2014;8(1):95–99. | ||
Kong D, Zhang F, Wei D, et al. Paeonol inhibits hepatic fibrogenesis via disrupting nuclear factor-kappaB pathway in activated stellate cells: in vivo and in vitro studies. J Gastroenterol Hepatol. 2013;28(7):1223–1233. | ||
Robinson SM, Mann DA. Role of nuclear factor kappaB in liver health and disease. Clin Sci (Lond). 2010;118(12):691–705. | ||
Oakley F, Meso M, Iredale JP, et al. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology. 2005;128(1): 108–120. | ||
Zhu L, Kong M, Han YP, et al. Spontaneous liver fibrosis induced by long term dietary vitamin D deficiency in adult mice is related to chronic inflammation and enhanced apoptosis. Can J Physiol Pharmacol. 2015;93(5):385–394. | ||
Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. 2014;147(3):577–594. | ||
Henderson NC, Iredale JP. Liver fibrosis: cellular mechanisms of progression and resolution. Clin Sci (Lond). 2007;112(5):265–280. | ||
Choi I, Kang HS, Yang Y, Pyun KH. IL-6 induces hepatic inflammation and collagen synthesis in vivo. Clin Exp Immunol. 1994;95(3):530–535. | ||
Yin M, Wheeler MD, Kono H, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117(4):942–952. | ||
Shirasaki T, Honda M, Shimakami T, et al. Impaired interferon signaling in chronic hepatitis C patients with advanced fibrosis via the transforming growth factor beta signaling pathway. Hepatology. 2014;60(5):1519–1530. | ||
Duval F, Moreno-Cuevas JE, Gonzalez-Garza MT, Rodriguez-Montalvo C, Cruz-Vega DE. Liver fibrosis and protection mechanisms action of medicinal plants targeting apoptosis of hepatocytes and hepatic stellate cells. Adv Pharmacol Sci. 2014;2014:373295. | ||
Pan Y, Fu H, Kong Q, et al. Prevention of pulmonary fibrosis with salvianolic acid a by inducing fibroblast cell cycle arrest and promoting apoptosis. J Ethnopharmacol. 2014;155(3):1589–1596. | ||
Wang Y, Gao J, Zhang D, Zhang J, Ma J, Jiang H. New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J Hepatol. 2010;53(1):132–144. | ||
Schnabl B, Bradham CA, Bennett BL, Manning AM, Stefanovic B, Brenner DA. TAK1/JNK and p38 have opposite effects on rat hepatic stellate cells. Hepatology. 2001;34(5):953–963. | ||
Tergaonkar V. NFkappaB pathway: a good signaling paradigm and therapeutic target. Int J Biochem Cell Biol. 2006;38(10):1647–1653. | ||
Olson CM, Hedrick MN, Izadi H, Bates TC, Olivera ER, Anguita J. p38 mitogen-activated protein kinase controls NF-kappaB transcriptional activation and tumor necrosis factor alpha production through RelA phosphorylation mediated by mitogen- and stress-activated protein kinase 1 in response to Borreliaburgdorferi antigens. Infect Immun. 2007;75(1):270–277. | ||
Liu J, Wu N, Ma LN, et al. p38 MAPK signaling mediates mitochondrial apoptosis in cancer cells induced by oleanolic acid. Asian Pac J Cancer Prev. 2014;15(11):4519–4525. | ||
Huang HL, Hsieh MJ, Chien MH, Chen HY, Yang SF, Hsiao PC. Glabridin mediate caspases activation and induces apoptosis through JNK1/2 and p38 MAPK pathway in human promyelocytic leukemia Cells. PLoS One. 2014;9(6). |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.