")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Gamma scintigraphic study of the hydrodynamically balanced matrix tablets of Metformin HCl in rabbits
Authors Razavi M, Karimian H, Yeong CH, Ahmad Sarji S, Chung LY , Nyamathulla S, Noordin MI
Received 15 February 2015
Accepted for publication 30 March 2015
Published 19 June 2015 Volume 2015:9 Pages 3125—3139
DOI https://doi.org/10.2147/DDDT.S82935
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Mahboubeh Razavi,1 Hamed Karimian,1 Chai Hong Yeong,2 Sazilah Ahmad Sarji,2 Lip Yong Chung,1 Shaik Nyamathulla,1,3 Mohamed Ibrahim Noordin1,3
1Department of Pharmacy, Faculty of Medicine, 2Department of Biomedical Imaging and University of Malaya Research Imaging Centre, Faculty of Medicine, 3Department of Chemistry, Centre for Natural Products and Drug Discovery (CENAR), Faculty of Science, University of Malaya, Kuala Lumpur, Malaysia
Abstract: The purpose of this study is to evaluate the in vitro and in vivo performance of gastro-retentive matrix tablets having Metformin HCl as model drug and combination of natural polymers. A total of 16 formulations were prepared by a wet granulation method using xanthan, tamarind seed powder, tamarind kernel powder and salep as the gel-forming agents and sodium bicarbonate as a gas-forming agent. All the formulations were evaluated for compendial and non-compendial tests and in vitro study was carried out on a USP-II dissolution apparatus at a paddle speed of 50 rpm. MOX2 formulation, composed of salep and xanthan in the ratio of 4:1 with 96.9% release, was considered as the optimum formulation with more than 90% release in 12 hours and short floating lag time. In vivo study was carried out using gamma scintigraphy in New Zealand White rabbits, optimized formulation was incorporated with 10 mg of 153Sm for labeling MOX2 formulation. The radioactive samarium oxide was used as the marker to trace transit of the tablets in the gastrointestinal tract. The in vivo data also supported retention of MOX2 formulation in the gastric region for 12 hours and were different from the control formulation without a gas and gel forming agent. It was concluded that the prepared floating gastro-retentive matrix tablets had a sustained-release effect in vitro and in vivo, gamma scintigraphy played an important role in locating the oral transit and the drug-release pattern.
Keywords: gastro-retentive drug delivery, natural polymer, gamma scintigraphy, sustain release formulation, salep, tamarind seed
Introduction
Oral drug delivery is the preferred route of drug therapy and has a high patient compliance. Bioavailability of the oral dose formulation can be affected by several factors, such as the type of dosage form, gastric emptying behavior, gastrointestinal (GI) transit time, and the site of drug absorption. In recent years, several techniques have been developed to overcome difficulties of oral dose administration.1 Drug delivery systems that deliver drugs to the site of action have been designed to enhance drug bioavailability and achieve optimum drug concentration in the blood plasma. The controlled oral drug delivery systems reduce the frequency of drug administration and avoid drug fluctuations in blood plasma levels, which are often associated with an unwanted side effect.2
Gastro-retentive drug delivery systems are designed to release drug at the upper part of the GI tract for a prolonged and predictable period of time in order to improve its bioavailability.3 Excipients are “inert” materials added to the drug formulations for further processing of the materials into its final dosage form and to achieve an optimum absorption and therapeutic effect.4 They can be used as diluents, binders, disintegrants, adhesives, glidants, and sweeteners in pharmaceutical preparations.5
Apart from the pharmacologically active drugs, inactive and inert materials play an important role in drug formulation development. Recently, researchers are more interested in natural excipients as many of the synthetic excipients are toxic, hence posing problems for drug approval by regulatory authorities.6 Herbal excipients are ideal because of their natural origin, ease of availability, fewer side effects, biocompatibility, non-toxicity, bioacceptability, better patient acceptance, and low cost.7 Every year new drug with varying physicochemical properties is being introduced into the market, however, there are limited discovery of new excipients. Existing excipients are either unsuitable or incompatible with physicochemically diverse chemical entities. Therefore, research for new compatible excipients is a necessity to develop an optimum formulation with desired outcomes.8
Polymers are high-molecular-weight compounds composed of repeating structural units (monomers) that are used as excipients in the pharmaceutical industry. They affect the manufacture processing, stability, solubility, and bioavailability of dosage forms, enhancing the safety, effectiveness, and patient compliance.9 Plants are a good source of polymers and two of them used in the present study were Tamarindus indica and Orchis morio. T. indica Linn., a member of the Leguminosae family, is an evergreen tree that grows all over Southeast Asia and India is the largest producer. It is a multipurpose tree, as almost every part of the tree is used.10 Each fruit of this plant contains up to six seeds, where 60%–65% of each seed make up the kernel and the remaining 35%–40% is the husk. The tamarind seed extract has good gelling properties and is used as gelling, stabilizing, and thickening agents in the food industry.11 The polysaccharides present in seed form a mucilaginous dispersion in the presence of cold water.12
Salep is a hydrocolloid obtained from dried roots and tubers of O. morio var. mascula from the Orchidaceae family.13 This plant is widespread in Iran and Turkey. Two different types of Orchis species are cultivated in Iran, one with branched (palmate) and the other with rounded (unbranched) tubers. These tubers consist of 48% mucilage which is used as a gelling agent, a thickening agent and a stabilizer agent in ice cream preparations.14 The palmate tubers of salep were used in this study.
Metformin HCl is a biguanide antihyperglycemic agent with the ability to reduce blood glucose, it is widely used to treat type II diabetes.15 Metformin HCl is freely soluble in water and it belongs to the Biopharmaceutics Classification System class III, with an absolute bioavailability of 50%–60%. The absorption site for Metformin HCl is the proximal part of the small intestine.16 This characteristic makes the formulation less ideal as it is slowly and incompletely absorbed from the GI tract. In order to increase the bioavailability of Metformin, several approaches of controlled release and gastro-retentive formulations have been developed.16–19
The objective of the present work was to develop a new gastro-retentive formulation using a combination of different ratios of three natural polymers using a wet granulation process and their evaluation using in vivo gamma scintigraphic studies in animal models. The primary focus was to prepare gastro-retentive matrix tablets of Metformin HCl with short floating lag time (FLT), good floating, and swelling ability and 100% drug release within 12 hours. The effects of combining natural gums on gel-forming, floating properties, and release characteristics of Metformin HCl were evaluated.
Materials and methods
Materials
Metformin HCl BP 98 (99.61% purity, White crystalline powder, Batch No 2011MP0244) was obtained from Euro Chemo-Pharma Sdn. Bhd (Selangor, Darul Ehsan, Malaysia). The palmate tubers of salep were procured from the Agricultural Research Centre, Boushehr, Iran, with a voucher number of 4310/266/1. Amritum Bio-Botanica Herbs Research Laboratory Pvt. Ltd (Indore, India) kindly supplied the tamarind seeds. Sodium bicarbonate (NaHCO3, M=84.01 g/mol), cellulose microcrystalline (C6H10O5)n, pure xanthan, hydrochloric acid 37% (HCl, M=6.46 g/mol), talc, potassium chloride (KCl, M=74.56 g/mol), and magnesium stearate were purchased from R&Mchemicals, Essex, UK.
Collection and preparation of powders
The pulp was removed from the T. indicia seeds and the size of the dried seeds was reduced using a cutter mill machine (Retsch-Allee, Haan, Germany). Then, the small seed’s particle was crushed to powder using a blender. The obtained powder was named tamarind seed powder (TSP).
The husk of the small particles of the tamarind seeds obtained from a cutter mill machine was removed by a knife, the brownish coat of seed was completely removed, and the kernel portion was grounded into powder form using a blender and the powder was called tamarind kernel powder (TKP).
Dried palmate tubers of salep were size reduced using the cutter mill (Retsch-Allee). Salep fine powder was obtained by running the blender for several times.
The prepared salep, TKP, and TSP powders were passed through the sieve number 100 and no further treatment was carried out to the powders and stored in well-closed polypropylene containers in a desiccator.
Preparation of matrix tablets of Metformin HCl using different polymers
Gastro-retentive matrix tablet containing 500 mg Metformin HCl was prepared by the wet granulation method, according to the design shown in Table 1. Three different combinations of polymers were prepared and each formulation contained the active ingredient (Metformin HCl), release-retarding polymers (xanthan with TKP/TSP/salep), gas-generating agent (NaHCO3), diluent (microcrystalline cellulose [MCC]), lubricant (magnesium stearate) and the glidant (talc). Before weighing, all the ingredients were sieved using a sieve number 100. The tablet coding was carried out based on which polymer had been used in the formulation: the formulation containing salep (Orchis Mascula) was coded as MOX, the MKX coded formulation contained TKP, and the MTX coded formulation contained TSP. The specific amount of each ingredient, excluding the lubricant and glidant, was mixed well using the geometrical dilution method. Xanthan solution (1%) was used as a binder solution to granulate the homogeneously mixed blends. The wet mass was passed through a sieve number 10 and wet granules were dried at 45°C for 30 minutes. The dried granules were then passed again through a sieve number 18 and were lubricated with talc and magnesium stearate. Finally, an specific amount of each granular mixture equivalent to the weight of single tablet was weighed and fed manually into the die of a 12 mm round, concave single punch tableting machine (MTCM-I [Manual Tablet Compaction Machine], GlobePharma, North Brunswick, NJ, USA) and compressed at a compression pressure of 2,200 psi.
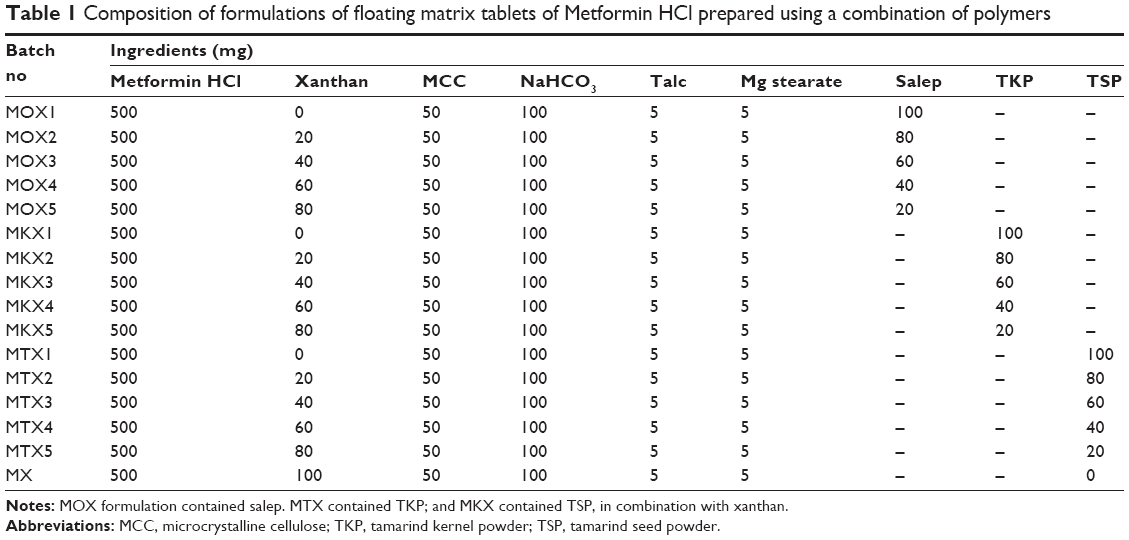 | Table 1 Composition of formulations of floating matrix tablets of Metformin HCl prepared using a combination of polymers |
Prepared matrix tablets evaluation
Tablets prepared were assessed for weight variation, hardness, friability, drug content uniformity, floating lag time (FLT) and total floating time (TFT).
Matrix tablets weight variation
Randomly selected tablets were weighed separately (n=20) by an analytical balance (Model MS-S/MS-L Models; Mettler Toledo, Greifensee, Switzerland) and then the tablets average weight were calculated. Each tablets weight was compared with the average weight and the weight variation was calculated. As per USP36, the weight range of the tablets should be within 5% variation from the average weight and the result is summarized in Table 2.
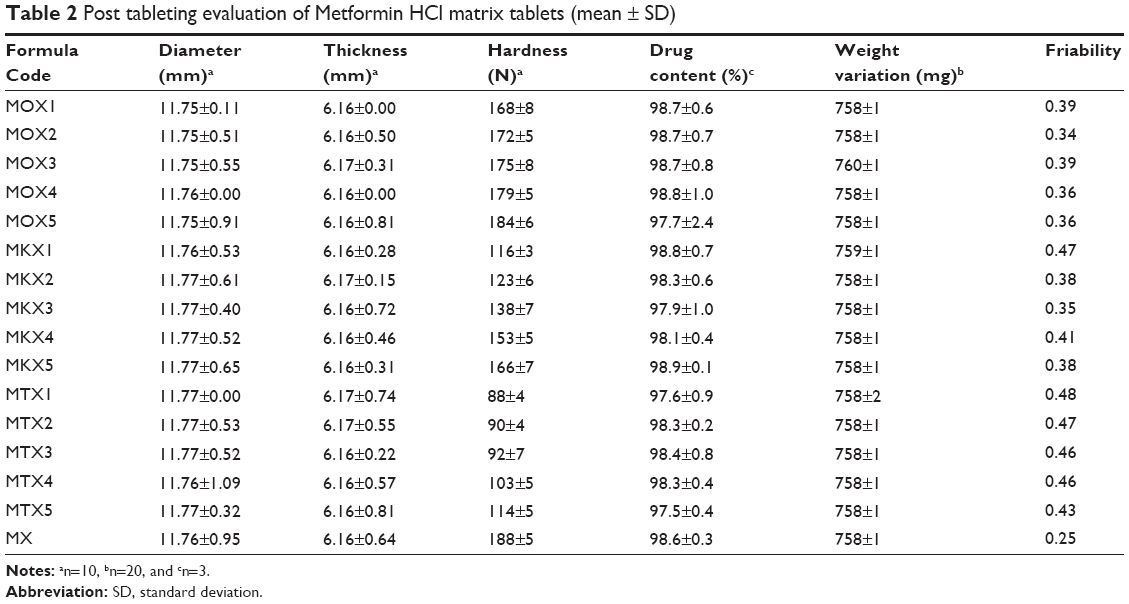 | Table 2 Post tableting evaluation of Metformin HCl matrix tablets (mean ± SD) |
Diameter and thickness of the matrix tablets
From each formulation, ten tablets were selected randomly. The diameter and thickness were measured by adjusting the instrument to measure thickness and diameter using a digital tablet hardness tester (Model 6D, Dr Schleuniger Pharmatron, Solothun, Switzerland). The results are recorded and compared (Table 2).
Hardness of the matrix tablets
The hardness of ten randomly selected tablets was determined by using a digital tablet hardness tester (Model 6D, Dr Schleuniger Pharmatron). The result was recorded and compared among the different formulations (Table 2).
Friability of the matrix tablets
Tablets equal to 6.5 g (n=10) were weighed and the weight was recorded as W1.20 The friability of the tablets was measured using a friability tester (Model Tar10, Erweka, Ensenstam, Germany) with 25 rotations per minute for 4 minutes. After 100 rotations, the tablets were de-dusted and reweighed. The final weight was recorded as W2 (Table 2). The acceptable range of the friability test is less than 1%. The following equation was used to calculate the friability:
Friability = (W1 - W2)/W1 ×100 | (1) |
Drug content of the matrix tablets
From each batch, three tablets were selected, and a mortar and pestle were used to crush them into powder form and 100 mg was weighed from crushed tablets and diluted to 100 mL of distilled water in a volumetric flask. The solution was then filtered and the absorbance was measured at 233 nm using a UV spectrophotometer and was recorded as shown in Table 2 (Lambda 35 UV Vis Spectrometer, PerkinElmer Inc., Waltham, MA, USA).
Total floating time and floating lag time of the tablets
The FLT can be defined as the time taken for a tablet to rise to the surface of the dissolution medium, and duration of the time taken by a tablet to remain constantly floating on the surface of the medium is called the TFT. These two properties are used to measure buoyancy. Randomly selected tablets from each formulation were placed in a beaker containing 500 mL of simulated gastric fluid at pH 1.2.21
In vitro drug release studies
In vitro release studies were carried out using the USP-II dissolution test apparatus (Copley Scientific Limited, Nottingham, UK). The dissolution basket was filled with 900 mL of 0.1 N HCl (pH 1.2) with a paddle speed of 50 rpm and the temperature was maintained at 37°C±0.5°C. At predetermined time intervals for 12 hours, 5 mL samples were withdrawn at 0, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10 and 12 hours, and at each time point was replaced with 5 mL of the fresh medium kept at the same temperature. The sample absorbance was measured at 233 nm using a UV spectrophotometer. Fresh media was used as a blank. The dissolution profiles were obtained by plotting the cumulative percentage of drug released on the y-axis and time (hours) in the x-axis.
The drug release kinetics
In order to determine the release mechanism and the order of drug release, the in vitro dissolution test data were fitted into mathematical models representing i) zero-order, ii) first-order, iii) Higuchi’s, and iv) Hixson–Crowell equations.22
Fourier transform infrared spectroscopy of MOX2 formulation
A small volume of Metformin HCl, polymers (Xanthan and salep), and powder from crushed tablets of the optimum formulation were mixed with potassium bromide and made into a pellet form separately. Pellets were then analyzed using a Fourier transform infrared (FTIR) spectrophotometer (model: Nicolet IS10, Thermo Scientific, WI, USA).
X-ray diffraction analysis of the optimum formulation
An X-ray diffraction (XRD) diffractometer (model: BTX324, Inxitu, CA, USA) was used to analyze the XRD spectra of the optimum formulation. Pellets were formed by pressing of the dried samples and their diffractograms were recorded using Cu-Kα radiation (40 kV, 60 mA). The scanning speed of 2°/min and a chart speed of 2°/2 cm per 2θ were run in a diffractogram for pure Metformin HCl, polymer, and powder from crushed tablet of the optimum formulation.
In vivo studies of optimum formulation in rabbits
Six adult male New Zealand White rabbits, aged 4 months and weighing approximately 2.5–3.0 kg were used in this study. Experiments for evaluation of the optimum gastro-retentive matrix tablet of Metformin HCl in New Zealand White rabbits were approved by the Institutional Animal Care and Use Committee (IACUC) of the Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia (2013-12-03/PHAR/R/MR).
The objective of this study was to demonstrate the floating ability of the prepared formulation in a 12-hour period and further to evaluate the drug release in vivo. Approximately, 10 mg of stable nuclide, 152Sm2O3, was added to each tablet composition during the tablet preparation as mentioned in the “Preparation of matrix tablets of Metformin HCl using different polymers” section. The neutron activity of the tablets were accounted at a nuclear reactor facility.
Each male rabbit was housed in an individual clean cage, maintained in a controlled temperature (22°C–26°C), relative humidity (60%±10%), and 12 hours alternate light and dark cycle environment. They were fed with a standard diet and ad libitum. Animals were divided into two groups, each containing three rabbits. One group received the optimized test formulation (MOX2) and the other group received control, tablets without polymers and NaHCO3 (Table 3). However, tablet dose was decreased to 250 mg, to inhibit hypoglycemia and to make administration easy to the rabbits. In all, 250 mg of in vivo test tablets shows a similar percentage of drug release and floating time as that of the 500 mg formulation at different time periods. The test formulations were prepared using the method described in the “Preparation of matrix tablets of Metformin HCl using different polymers” section. The rabbits had access to the water the night before and during the entire study.
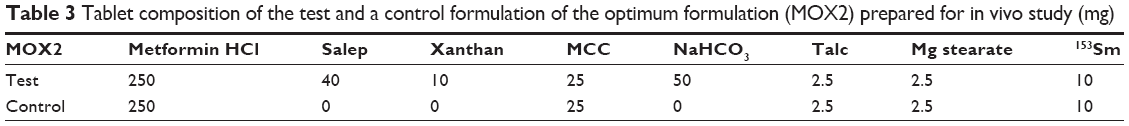 | Table 3 Tablet composition of the test and a control formulation of the optimum formulation (MOX2) prepared for in vivo study (mg) |
Neutron activation of MOX2 tablets for in vivo study
The neutron activation of the tablets was carried out at the nuclear reactor facility at Malaysian Nuclear Agency (Bangi, Selangor, Malaysia). This facility has a 250 kW open pool-type research reactor (Triga Mark II, General Atomics, CA, USA), which utilizes uranium zirconium hydride assembly with a low-enriched uranium (20 wt% 235U) fuel source. Each tablet was heat sealed into individual polyethylene vial and packed into a polyethylene ampoule (commonly known as a “rabbit”). The ampoule was then delivered to the reactor core by a pneumatic transport system. The tablet was irradiated in a neutron flux of 1×1013 cm−2 s−1 for 5 minutes to achieve a nominal radioactivity of 5 MBq at 48 hours after neutron activation. Gamma spectroscopy was carried out 24 hours after neutron activation using a coaxial, p-type, 40% relative efficiency germanium detector (Canberra, Meriden, CT, USA) and gamma spectrum analysis software (Genie™ 2000 V 3.2, Canberra) to detect any radioactive impurities. The tablets were then kept in a radioactive storage room until the study day to allow for the decay of the unwanted activated by-products.23
In vivo gamma scintigraphy study in rabbits
The optimum formulation (MOX2) was selected to evaluate the in vivo residence time of the swelling matrix tablets in the gastric region of rabbits. Rabbits from the test and control groups were orally administrated with neutron activated MOX2 formulations, the test and control tablets contained 10 mg radioactive samarium oxide (153Sm2O3).
The FLT and TFT study were carried out for the size-reduced MOX2 formulation contained 153Sm2O3. The TFT and FLT of the test formulations had no significant variation from the unreduced optimized formulation. The study was carried out in the same manner as described in the “Total floating time and FLT of the tablets” section.
The tablets were administered to rabbits orally through a pill dispenser followed by flushing with 30 mL of drinking water. Scintigraphic imaging was started immediately using a dual-detector single photon emission computed tomography-computed tomography (SPECT-CT) system (BrightView XCT, Philips, Eindhoven, the Netherlands) mounted with a low-energy high-resolution collimator. The computed tomography (CT) images were acquired using 120 kVp, 247 mAs, and 1 mm slice thickness at 512×512 matrix size, followed by SPECT imaging (128×128 matrix size, 64 angulations for 360°, 15 seconds per angulation). The SPECT-CT imaging was performed at 0, 1, 2, 4, 6, 8, 10, and 12 hours after administration of the tablets. During the imaging process, the rabbits were restrained in a towel to reduce movement (Figure 1).
 | Figure 1 A rabbit was undergoing SPECT-CT imaging for approximately 15 minutes. |
The 3D SPECT-CT images were reconstructed using dedicated software and viewed in three different planes, ie, sagittal, coronal, and axial planes. During data analysis, the region of interest was placed in the stomach region of the rabbit and the 153Sm activity was measured as the mean count value. As 153Sm was well mixed with the tablet ingredients during manufacturing, the rate of release of the 153Sm activity was considered as the rate of release of the active drug. By considering the maximum counts at 0 hour as 100% retention of the tablet, the percentage release of the drug can be calculated for each time point.
Results and discussion
Drug release can be influenced by physicochemical properties. The shape, size and thermodynamic compatibility of the ingredients will determine the success of a formulation. Therefore, before tablet preparation polymers TKP, TSP, and salep were evaluated for physicochemical properties, thermal stability, chemical interaction, and surface morphology using XRD, DSC, FTIR, and SEM.
Evaluation of the tablets
The floating gastro-retentive matrix tablets of Metformin HCl were prepared by a wet granulation technique using Metformin HCl, MCC, sodium bicarbonate, magnesium stearate, talc, and xanthan gum in combination with salep in MOX formulations, TKP in MKX formulations, and TSP in MTX formulations. Physical characteristics of floating tablets were determined for all of the 16 prepared formulation, and the results are shown in Table 2. The physical evaluation of formulations revealed a uniform thickness and weight. The thickness of formulations ranged from 6.16 to 6.17 mm and the diameter ranged from 11.75 to 11.77 mm. The weight of the formulations was within an acceptable range of 758–759 mg. The drug content uniformity test was carried out for the formulations and the result indicated that the tablets were uniform in drug content, the percentage of drug was ranging from 97.56% to 98.91%. The hardness of the formulations was measured and the result indicated that there was enough hardness to withstand packaging and transportation. It was also noticed that the formulation contains salep had a higher hardness than TKP and TSP formulations. This might be due to the shape of the particles as it is manifested in the case of needle and fibrous particles,24 and the better gelling and viscosity properties of salep compared with TKP and TSP. Friability values were found to be within the accepted range of 0.25%–0.48% across all formulations. These results indicated that the tablets had sufficient mechanical strength (Table 2).
Floating behavior of prepared matrix tablets
Sodium bicarbonate was used as a gas-generating agent in the formulation of gastro-retentive drug delivery systems. FLT and TFT for all the formulations were evaluated, and the results of FLT are illustrated in Figure 2. Although all the tablets were prepared using the same binder solution and the same procedure for granulation and compression had been followed, there was a difference between the FLT of each formulation. Sodium bicarbonate can affect the FLT by forming gas bubbles after reacting with the acidic medium, however, in this study, the same amount of NaHCO3 had been used as in all the formulations. The difference in FLT among the different formulations could be due to the polymers used, their molecular weight, swelling, viscosity, and the gelling property of each polymer.
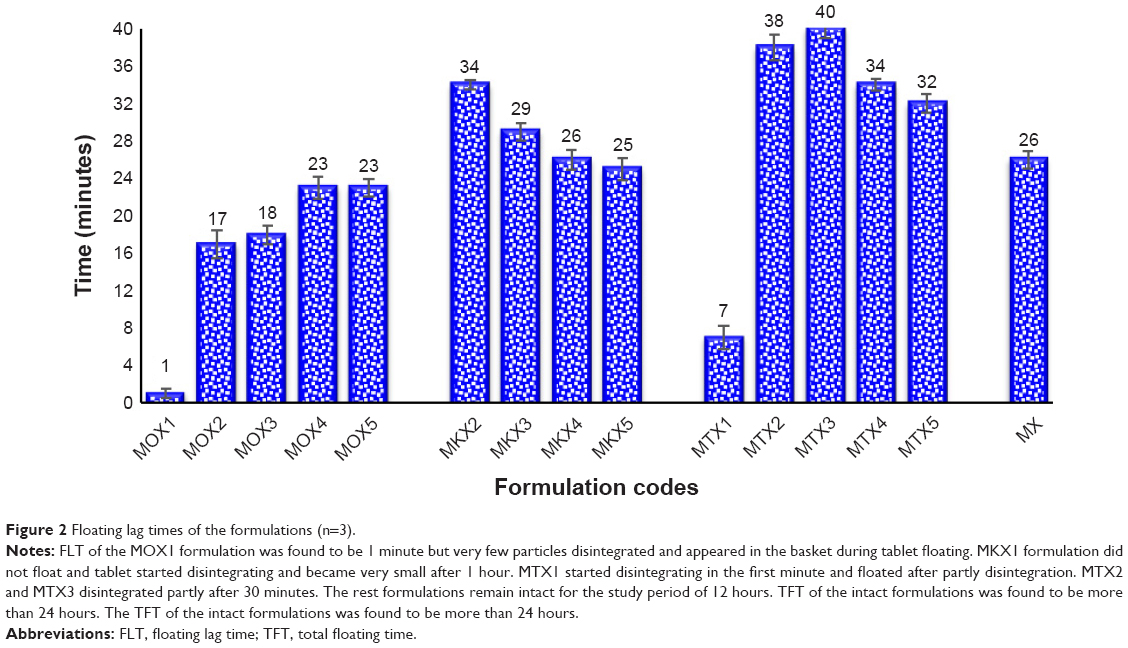 | Figure 2 Floating lag times of the formulations (n=3). |
The floating study showed that the MOX formulations have shorter FLT compared with the MTX and the MKX formulations. In the MOX formulations, FLT was increased by decreasing the salep and increasing the xanthan concentration. Because salep takes a shorter time compared with xanthan to swell and form a high viscous matrix layer in contact with aqueous media, the matrix layer could entrap the gas bubble and float. The formulations contained salep that had the shorter FLT compared with the MKX and MTX formulations, this can be explained because the strong matrix is formed in the MOX formulations, FLT depends on the matrix layer as the strong matrix can entrap the CO2, and this causes tablet floating, but in the formulation with weak gelling and swelling properties the matrix formed was not enough strong for CO2 entrapment.25,26
MKX1, MKX2, MTX1, and MTX2 formulations disintegrated either completely or partially before floating. The floating patterns were different for MTX and MKX formulations, and FLT was reduced by decreasing TKP and TSP concentrations and increase the xanthan concentration. In the previously published study, it was shown that xanthan has a better gelling and swelling ability compared with tamarind powders, and can form a better matrix layer.24
In vitro dissolution study of Metformin HCl matrix tablets
The in vitro dissolution for the MOX formulations showed that all the formulations were able to sustain the release of drug for 12 hours, and the release pattern was slower in the formulation contained higher concentrations of xanthan. This could be due to the stronger matrix layer formed by xanthan compared with salep, however, all of the formulations had more than 90% release within 12 hours, MOX1 had the highest release of 97%, followed by MOX2 (96.9%), MOX3 (93.9%), MOX4 (92.9%), MOX5 (91.7%), and MX (91.6%) (Figures 3 and 4). When compared with a commercially available formulation of Metformin HCl, we found that the MOX2 formulation had a similarity factor, f2, of 70%. Therefore, MOX2 was selected as the optimum formulation in the MOX batch. All the MOX formulations remained floating for more than 24 hours.
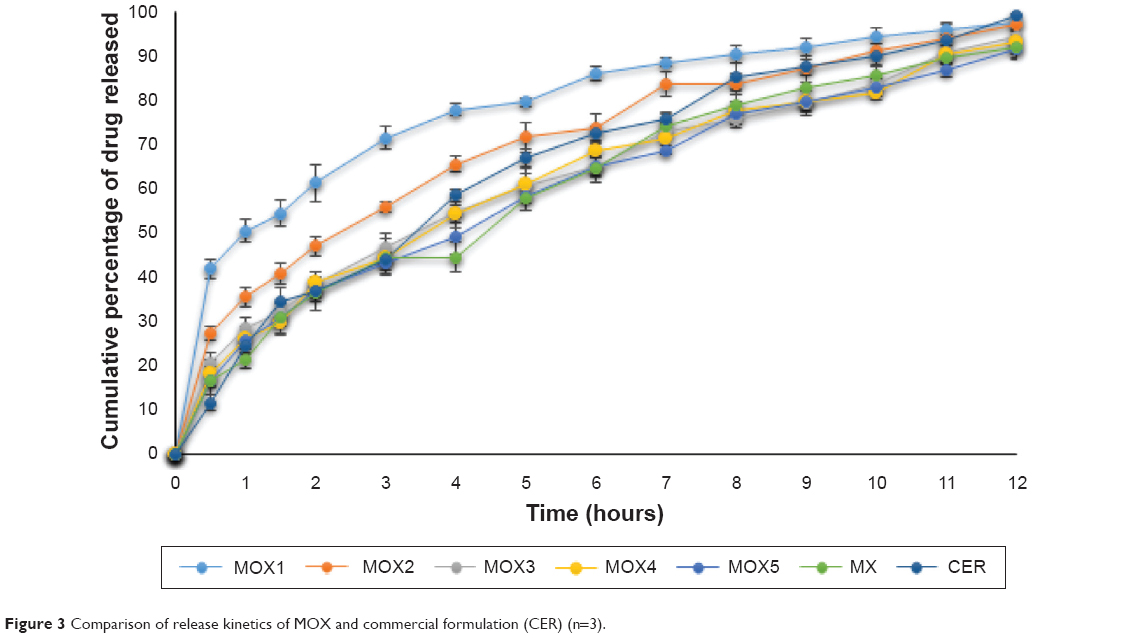 | Figure 3 Comparison of release kinetics of MOX and commercial formulation (CER) (n=3). |
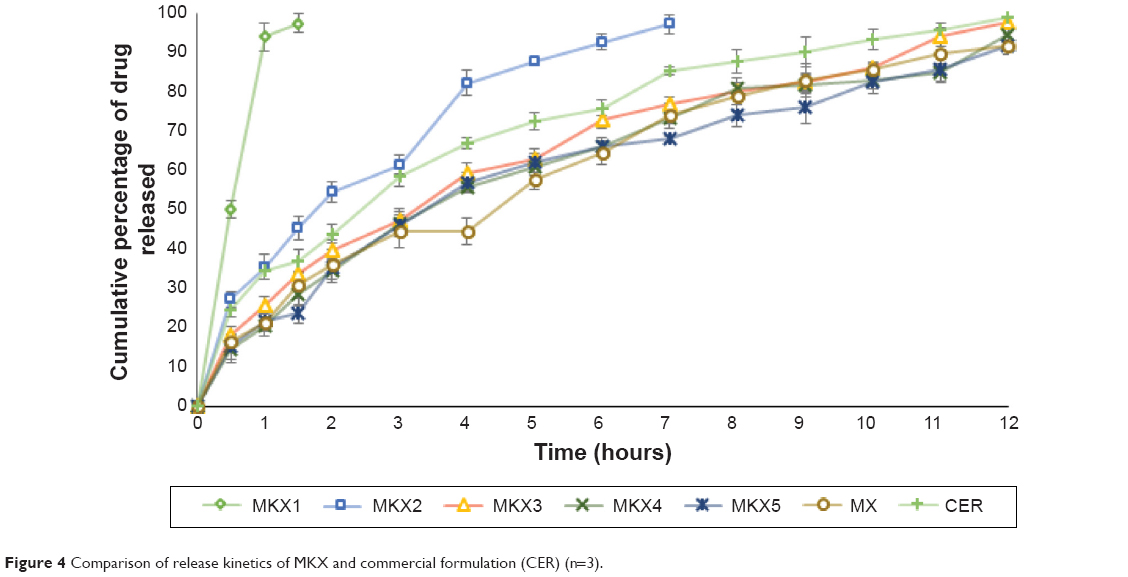 | Figure 4 Comparison of release kinetics of MKX and commercial formulation (CER) (n=3). |
In the MKX formulations, MKX1 disintegrated and drug was completely released in 3 hours. The result shows that TKP is unable to sustain the release when used alone in the formulation. MKX2 also partly disintegrated and 95% of the drug was released in 8 hours. The difference in the release patterns of MKX1 and MKX2 may be due to 20 mg of xanthan used in MKX2. Xanthan may have improved the release compared with TKP. The remaining four formulations could sustain the release up to 12 hours, wherein MKX3 up to 98%, followed by MKX4 (94.70%), MKX5 (91.7%), and MX (91.6%). MKX3 was found to be the optimum formulation in this group and it had the highest similarity factor (f2=60%) with the commercial extended release formulation. The intact formulation of MKX3 remained floating for more than 24 hours (Figure 4).
The drug release profiles of the MTX formulations showed the inability of TSP powder in sustaining release when it was used alone or in combination with low concentrations of xanthan. This shows the failure of TSP to produce a uniform and strong gel which caused the rapid drug release, this might be due to slow hydration of the TSP matrix to form a thick gel layer, but the result of salep and TKP shows when these polymers are exposed to aqueous medium, it undergoes rapid hydration and chain relaxation to form a viscose gelatinous layer to control the release. Only MTX4 and MTX5 were able to control the release for 12 hours. These results indicated that MTX4 which had the highest similarity factor (f2=61%) with the commercial formulations was the optimum formulation of this batch. The intact formulations remained floating for more than 24 hours (Figure 5).
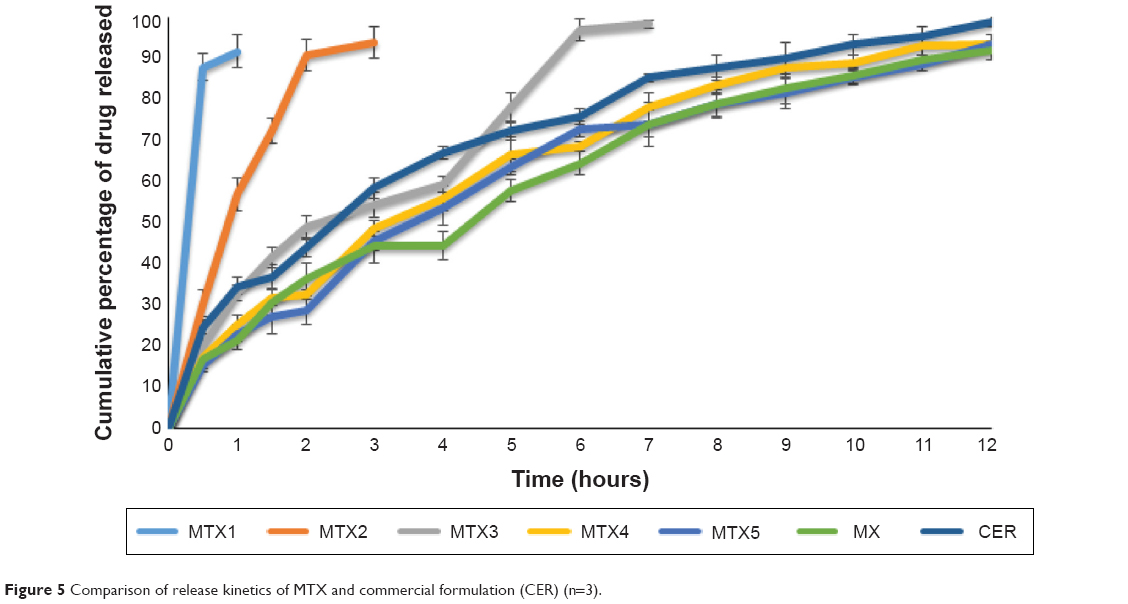 | Figure 5 Comparison of release kinetics of MTX and commercial formulation (CER) (n=3). |
Thus, based on the in vitro drug release, it can be concluded that salep’s ability in sustaining the drug release is comparable to that of xanthan, as the MOX1 formulation contained 100 mg salep, remained intact and the release was controlled in 12 hours (97%). MX formulation contained 100 mg of xanthan showed 91.6 released,24 and salep gelling, swelling and viscosity properties are better than that of the tamarind powders.22 On the other hand, TKP and TSP can be used in combination with a strong gel-forming compound like xanthan to improve the sustained release properties. Among all the formulations of salep, TKP, and TSP, MOX2 was found to be the optimum formulation as its profile is close to the commercial formulation. MOX2 had a shortest FLT, with floating durations of more than 24 hours, and the ability of sustaining drug release for 12 hours by forming a strong and jelly-like matrix layer (Figure 6). Therefore, MOX2 formulation was chosen for further validation through in vivo gamma scintigraphy study in rabbits.
 | Figure 6 MOX formulation after dissolution study. |
FTIR spectroscopy of the MOX2 formulation
The characterization of salep, xanthan, and optimum formulation were carried out by taking FTIR spectra, most of the functional groups (C–O) were measured at the fingerprint region (950–1,200 cm−1), where the salep’s peak was obtained at 3,339.5 cm−1 and xanthan’s peak was obtained at 3,374.4 cm−1. Other than fingerprint regions, pure salep showed a few peaks due to the manifestation of functional groups which were obtained at 2,925, 1,652.5, 1,376.9, 1,066.1, 1,023.7, and 868 cm−1, whereas for xanthan, the peaks were detected at 1,291.9, 1,718.2, 1,605.2, 1,408.9, 1,272.5, and 1,053.8 cm−1. For pure Metformin HCl, peaks were detected at 3,369.4, 3,295.7, 3,157, 1,623.5, 1,565, 1,475.6, 1,064.2, 938.1, and 737 cm−1. The prepared matrix tablet of MOX2 formulation exhibited peaks at 3,369.1, 3,295.6, 3,155.6, 1,623.1, 1,564.8, 1,475, 1,063.7, and 936.1 cm−1 (Figure 7). New peaks were not identified in the formulation spectrum, showing that no changes in the chemical structure had been occurring during the preparation process.
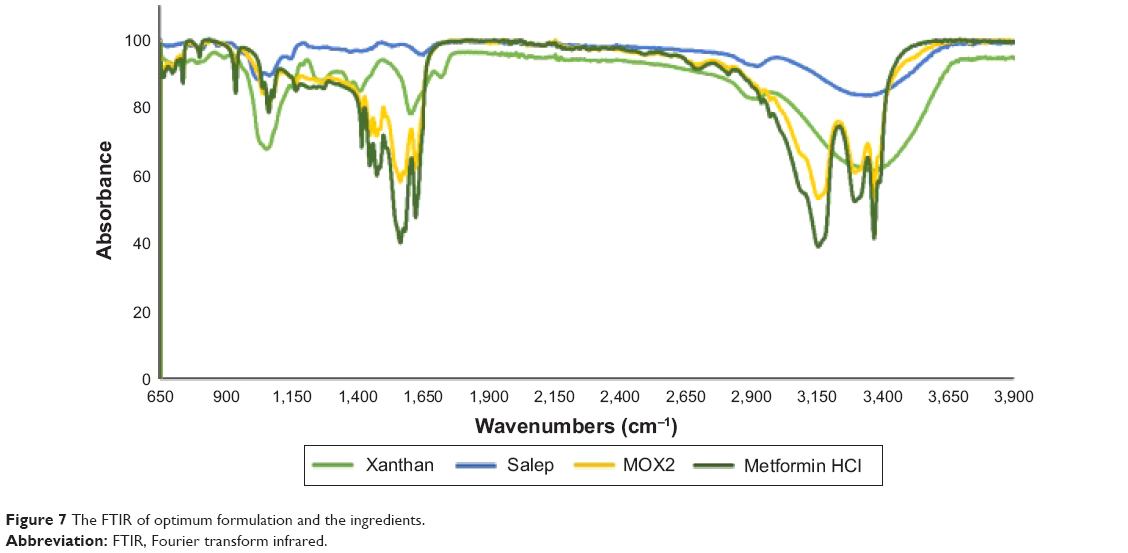 | Figure 7 The FTIR of optimum formulation and the ingredients. |
XRD analysis of the MOX2 formulation
XRD is used to achieve structural information about crystalline solids. The X-ray diffractograms of Metformin HCl, salep, xanthan, and the optimum formulation (MOX2) are presented in Figure 8. The X-ray diffractogram of pure Metformin HCl showed sharp peaks at 12.7°, 17.8°, 22.35°, 23.3°, 24.55°, 27.4°, 28.5°, 29.5°, 31.4°, 35.85°, and 37.35° (2θ) representing the crystallinity of the drug. Salep and xanthan indicated broad peaks at 20.4° and 20.2° (2θ), respectively, showing that the polymer is amorphous in nature. In X-ray diffractogram of MOX2 formulation, there was a decline in crystallinity of the Metformin HCl as reflected in the diffractogram by a reduction in the intensity of the peaks. No missing peaks were detected in the MOX2 formulation. The insignificant differences detected in the XRD can be due to physical interactions occurred during tablet preparation and compression.
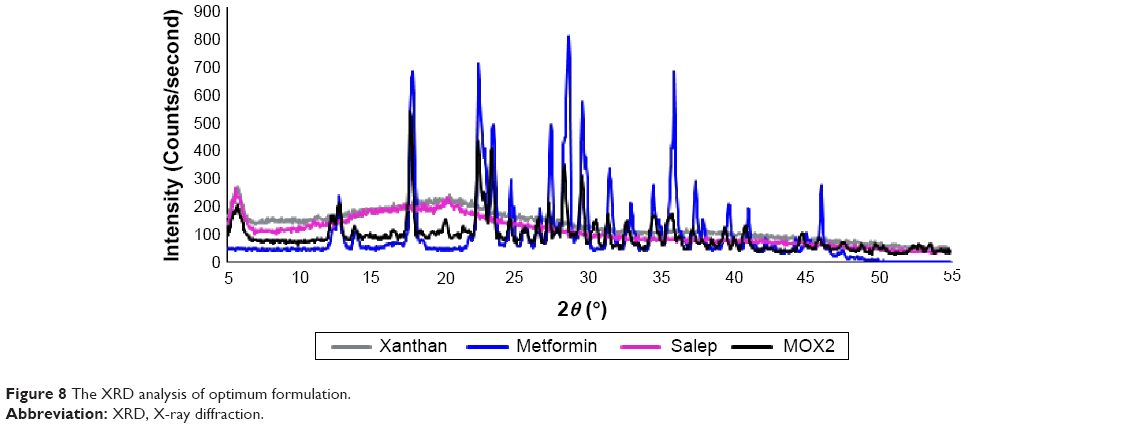 | Figure 8 The XRD analysis of optimum formulation. |
Drug release kinetics
When the maximum correlation coefficient values (R) were considered, the release data of formulations were observed to fit better with the first-order and Higuchi models. The highest regression value (r) obtained for MOX2 formulation was fitted into the first-order (R=0.97) and Higuchi (R=0.99) models. Drug release kinetics in all the formulations, except MTX3 had a higher linearity of first-order kinetic plots when compared with the zero-order kinetic plots. The first-order release kinetics describes the dependency on the drug concentration in the polymeric networks. It was concluded that these formulations showed drug concentration dependency up to a certain limit of loaded drug. The results showed that Higuchi was the dominant drug release mechanism for all the formulations, except MTX1 and MTX2, because the Higuchi plot showed a higher linearity compared with the Hixson–Crowell. These results indicated that drug release characteristics of polymer matrix tablets follow Higuchi square root time kinetics and the mechanism of drug release was diffusion and relaxation; it was likely because of the strong gel formed from a high proportion of polymer with narrow gap space, resulted in an incomplete release of drugs through diffusion (Table 4).
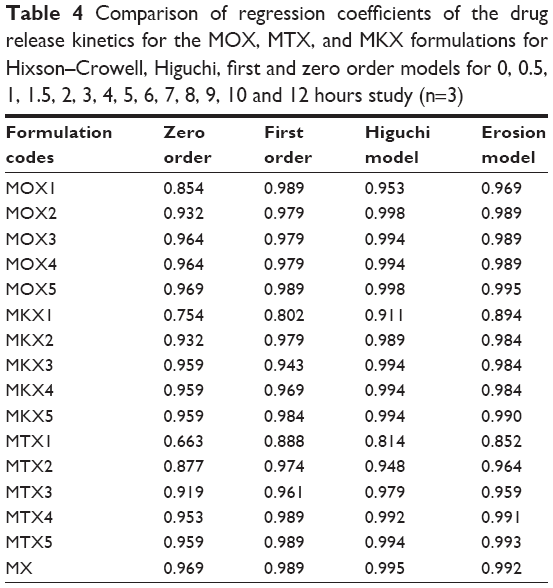 | Table 4 Comparison of regression coefficients of the drug release kinetics for the MOX, MTX, and MKX formulations for Hixson–Crowell, Higuchi, first and zero order models for 0, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10 and 12 hours study (n=3) |
In vivo gamma scintigraphic imaging of optimum formulation
In the present study, 153Sm2O3 was labeled to newly developed dose formulations to trace the kinetics of the pharmaceutical in the in vivo studies. 153Sm2O3 is a radionuclide that decays with a physical half-life of 46.3 hours by emitting beta and gamma radiations. In the last two decades, 153Sm2O3 has been used as a radiotracer in several pharmacoscintigraphic studies because of its availability, suitable gamma energies, easy production, and minimal radiation exposure.27 In addition, 153Sm2O3 is chemically stable and water insoluble, hence, it will not be absorbed into the GI tract and blood plasma during its transit in the body. 153Sm2O3 is excreted from the body through feces and eventually decays to a stable nuclide (153Eu); hence, special radioactive waste management is not required.28
The objective of in vivo gamma scintigraphic study was to provide a proof of concept that the floating capability of the MOX2 gastro-retentive tablet was useful for increasing the gastric residence time of the dosage form. 153Sm2O3 acts as the marker for locating the transit of the tablet in the GI tract. From the gamma spectroscopy data, it was shown that neutron activation of the tablet did not produce any unnecessary radioactive by products (Figure 9). Hybrid imaging of SPECT and CT was chosen in this study because the CT images provide excellent anatomical information to aid in the identification of the organ structures (Figure 10A). The examples of the SPECT-CT images of the rabbits at different times of acquisition are shown in Figure 10B. It was evident from the images that the MOX2 formulation was retained in the stomach for more than 12 hours and the drug was gradually released in the intestinal region over time. The results also indicated that the gastric peristalsis properties did not affect the tablets retention in the stomach and the active drug was released gradually within 12 hours.
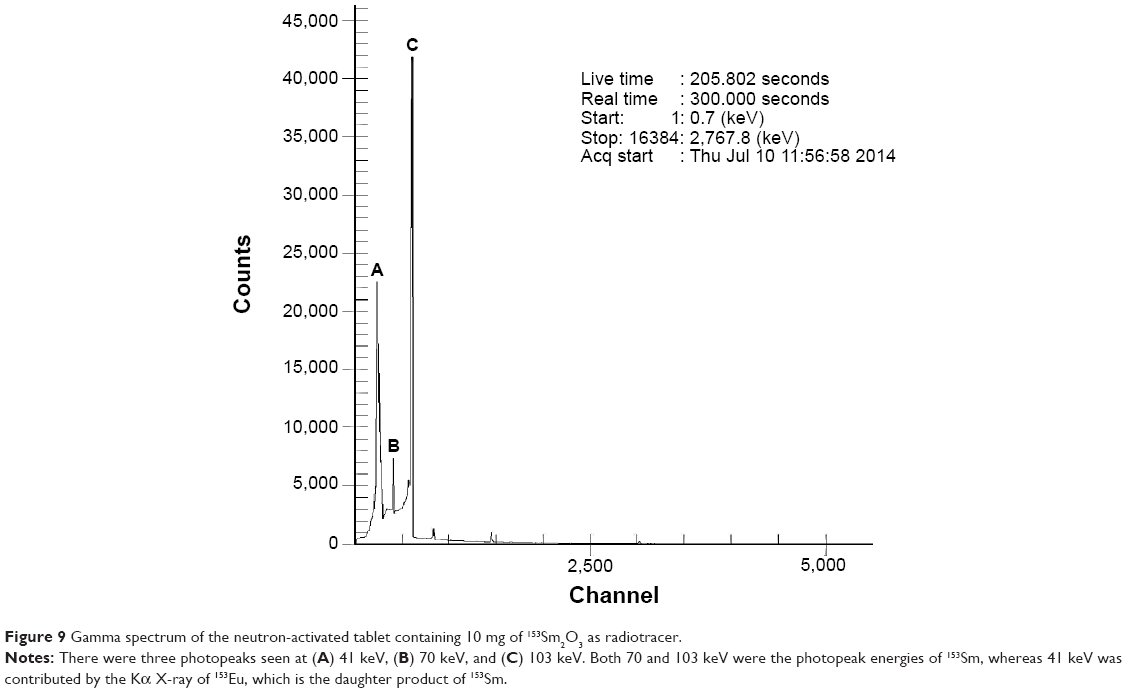 | Figure 9 Gamma spectrum of the neutron-activated tablet containing 10 mg of 153Sm2O3 as radiotracer. |
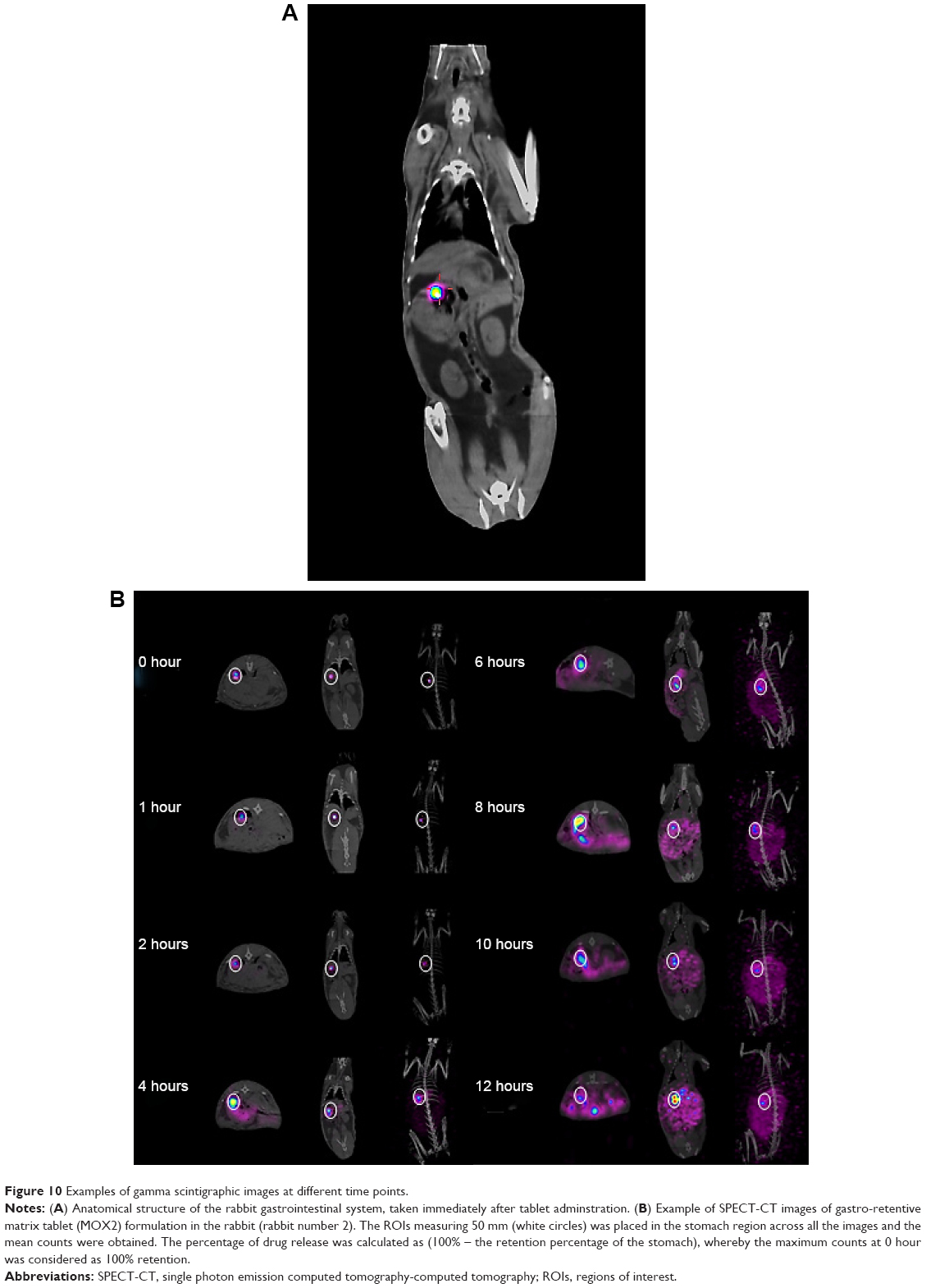 | Figure 10 Examples of gamma scintigraphic images at different time points. |
The FLT of the size-reduced MOX2 containing 153Sm2O3 was measured. The test was performed under the same conditions as in the other batches. The result indicated that size reduction and 153Sm2O3 did not affect the FLT of the tablets.
Gamma scintigraphy studies were carried out to determine the location of floating tablet in the gastric region after oral administration and the extent of its transit through the GI tract (Figure 10A). Scintigraphic data provide information regarding gastric retention time, the release behavior, and measurement of in vivo formulation performance. The in vivo results from gamma scintigraphy correlated to the obtained results from the in vitro dissolution. The images obtained from a gamma scintigraphy study showed that gastric emptying did not affect the retention properties of the formulations in the stomach due to its floating property (Figure 10B). The comparison of the test and control images shows the effects of the polymer (salep and xanthan) in sustaining the release of formulation. It was observed that the drug in MOX2 formulation was released in a sustained manner in comparison to the rapid and burst release in the control formulation (Figures 10B and 11). The 153Sm2O3 activity decreased in the stomach region over time, while the activity was distributing gradually over the intestines. The release profile showed the sustained release pattern and the ability of formulation to control the release. The percentages of drug release for MOX2 were found to be 0%, 26%, 38.6%, 59.5%, 68.7%, 78.2%, 89.2%, and 92.9%, while the control formulation release was found to be 55.2%, 80.3%, 85.0%, 88.5%, 91.5%, 94.8%, and 95.0% from 0 to 12 hours (Figure 12).
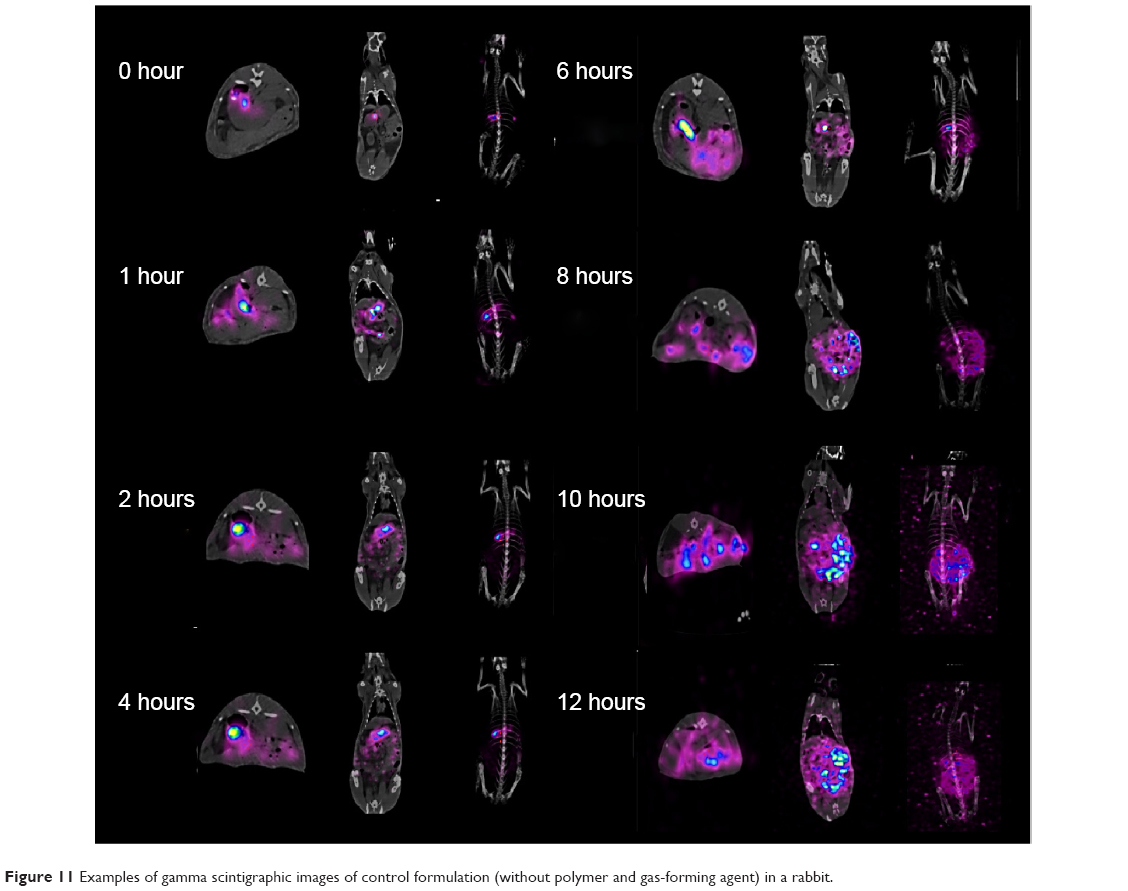 | Figure 11 Examples of gamma scintigraphic images of control formulation (without polymer and gas-forming agent) in a rabbit. |
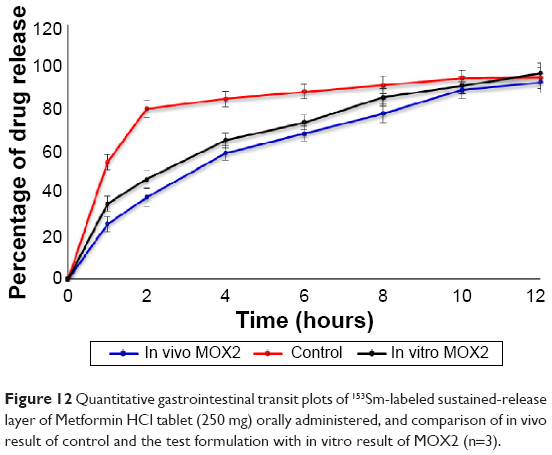 | Figure 12 Quantitative gastrointestinal transit plots of 153Sm-labeled sustained-release layer of Metformin HCl tablet (250 mg) orally administered, and comparison of in vivo result of control and the test formulation with in vitro result of MOX2 (n=3). |
The results showed that more than 80% of drug released in 2 hours in the control formulation, but it took 10 hours for the same amount of the drug to be released from the test formulation. The similarity factor between in vivo and in vitro formulations was 53%.
Conclusion
Floating gastro-retentive delivery system of Metformin HCl was developed using natural polymers having gelling ability. A study was carried out to achieve the retention of optimized formulation (MOX2) in the upper part of the stomach for at least 12 hours, a region in which Metformin HCl has maximum permeability and absorption. The results support that there is a significant correlation between in vitro drug release and in vivo drug release profiles of prepared optimized formulation as observed in the rabbit gamma scintigraphy study. The results support the capability of natural hydrocolloids in the development of the gastric-retentive delivery system of Metformin HCl.
Acknowledgment
The authors gratefully acknowledge the financial support from the University of Malaya for the research grant number PG052-2012B.
Disclosure
The authors report no conflicts of interest in this work.
References
Singh BN, Kim KH. Floating drug delivery systems: an approach to oral controlled drug delivery via gastric retention. J Control Release. 2000;63(3):235–259. | ||
Pawar VK, Kansal S, Garg G, Awasthi R, Singodia D, Kulkarni GT. Gastroretentive dosage forms: a review with special emphasis on floating drug delivery systems. Drug Deliv. 2011;18(2):97–110. | ||
Dehghan M, Kha F. Gastroretentive drug delivery systems: a patent perspective. Int J Health Res. 2009;2(1):23–44. | ||
Badawy SIF, Gray DB, Zhao F, Sun D, Schuster AE, Hussain MA. Formulation of solid dosage forms to overcome gastric pH interaction of the factor Xa inhibitor, BMS-561389. Pharmaceut Res. 2006;23(5):989–996. | ||
Kalasz H, Antal I. Drug excipients. Curr Med Chem. 2006;13(21):2535–2563. | ||
Tekade BW, Yogita A. Gums and mucilages: excipients for modified drug delivery system. J Adv Pharm Educ Res. 2013;3(4):359–367. | ||
Shirwaikar A, Shirwaikar A, Prabu SL, Kumar GA. Herbal excipients in novel drug delivery systems. Ind J Pharmaceut Sci. 2008;70(4):415. | ||
Marwaha M, Sandhu D, Marwaha RK. Coprocessing of excipients: a review on excipient development for improved tabletting performance. Int J Appl Pharmaceut. 2010;2(3):41–47. | ||
Beneke CE, Viljoen AM, Hamman JH. Polymeric plant-derived excipients in drug delivery. Molecules. 2009;14(7):2602–2620. | ||
Bhadoriya SS, Ganeshpurkar A, Narwaria J, Rai G, Jain AP. Tamarindus indica: extent of explored potential. Pharmacogn Rev. 2011;5(9):73. | ||
Bhatta R, Krishnamoorthy U, Mohammed F. Effect of tamarind (Tamarindus indica) seed husk tannins on in vitro rumen fermentation. Anim Feed Sci Technol. 2001;90(3):143–152. | ||
Nishinari K, Kim B, Fang Y, Nitta Y, Takemasa M. Rheological and related study of gelation of xyloglucan in the presence of small molecules and other polysaccharides. Cellulose. 2006;13(4):365–374. | ||
Ece Tamer C, Karaman B, Utku Copur O. A traditional turkish beverage: salep. Food Rev Int. 2006;22(1):43–50. | ||
Dogan M, Kayacier A. Rheological properties of reconstituted hot salep beverage. Int J Food Prop. 2004;7(3):683–691. | ||
Bastaki A. Diabetes mellitus and its treatment. Int J Diabetes Metab. 2005;13(3):111. | ||
Basak S, Rahman J, Ramalingam M. Design and in vitro testing of a floatable gastroretentive tablet of metformin hydrochloride. Die Pharmazie Int J Pharmaceut Sci. 2007;62(2):145–148. | ||
Stepensky D, Friedman M, Srour W, Raz I, Hoffman A. Preclinical evaluation of pharmacokinetic–pharmacodynamic rationale for oral CR metformin formulation. J Control Release. 2001;71(1):107–115. | ||
Boldhane SP, Kuchekar BS. Gastroretentive drug delivery of metformin hydrochloride: formulation and in vitro evaluation using 32 full factorial design. Curr Drug Deliv. 2009;6(5):477–485. | ||
Ali J, Arora S, Ahuja A, et al. Formulation and development of hydrodynamically balanced system for metformin: in vitro and in vivo evaluation. Eur J Pharmaceut Biopharmaceut. 2007;67(1):196–201. | ||
Rau A, Stadolik D, Corcino M, Acosta F. Tablets with improved friability. US Patent 20,140,315,719; 2014. | ||
Weh FH, Razavi M, Erh CH, et al. Formulation and in vitro evaluation of hydrodynamically balanced matrix tablets of famotidine using pectin as controlled release polymer. Latin Amer J Pharm. 2014;33(3):420–431. | ||
Razavi M, Nyamathulla S, Karimian H, Noordin MI. Novel swellable polymer of orchidaceae family for gastroretentive drug delivery of famotidine. Drug Des Dev Ther. 2014;8:1315. | ||
Yeong CH, Perkins AC. Sm-153 in gastrointestinal imaging extended use of Sm-153 in oral drug delivery and diagnostic imaging. LAP Lambert Academic Publishing; Saarbrücken, Germany. 2012. | ||
Razavi M, Nyamathulla S, Karimian H, Moghadamtousi SZ, Noordin MI. Hydrogel polysaccharides of tamarind and xanthan to formulate hydrodynamically balanced matrix tablets of famotidine. Molecules. 2014;19(9):13909–13931. | ||
Mostafavi A, Emami J, Varshosaz J, Davies NM, Rezazadeh M. Development of a prolonged-release gastroretentive tablet formulation of ciprofloxacin hydrochloride: pharmacokinetic characterization in healthy human volunteers. Int J Pharmaceut. 2011;409(1):128–136. | ||
Siraj S. Recent trends in gastroretentive drug delivery system. IJPRD. 2013;5(9):80–88. | ||
Yeong C-H, Abdullah BJJ, Ng K-H, et al. Production and first use of 153SmCl3-ion exchange resin capsule formulation for assessing gastrointestinal motility. Appl Radiat Isotopes. 2012;70(3):450–455. | ||
Yeong C-H, Abdullah BJJ, Ng K-H, et al. Neutron-activated 153Sm-ion-exchange resin as a tracer for gastrointestinal scintigraphy. Nucl Med Commun. 2011;32(12):1256–1260. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.