")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Evaluation of the efficacy and safety of three dosing regimens of agalsidase alfa enzyme replacement therapy in adults with Fabry disease
Authors Goláň L, Goker-Alpan O, Holida M , Kantola I, Klopotowski M, Kuusisto J, Linhart A, Musial J, Nicholls K , Gonzalez-Rodríguez D, Sharma R, Vujkovac B, Chang P, Wijatyk A
Received 14 January 2015
Accepted for publication 27 March 2015
Published 8 July 2015 Volume 2015:9 Pages 3435—3444
DOI https://doi.org/10.2147/DDDT.S80928
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Lubor Goláň,1 Ozlem Goker-Alpan,2 Myrl Holida,3 Ikka Kantola,4 Mariusz Klopotowski,5 Johanna Kuusisto,6 Aleš Linhart,1 Jacek Musial,7 Kathleen Nicholls,8 Derlis Gonzalez-Rodriguez,9 Reena Sharma,10 Bojan Vujkovac,11 Peter Chang,12 Anna Wijatyk12
1First Faculty of Medicine, Department of Cardiovascular Medicine, Charles University, Prague, Czech Republic; 2Lysosomal Research and Treatment Unit, Fairfax, VA, USA; 3Stead Family Department of Pediatrics, Division of Medical Genetics, University of Iowa Hospitals and Clinics, Iowa City, IA, USA; 4Division of Medicine, Turku University Hospital, Turku, Finland; 5Institute of Cardiology, Warsaw, Poland; 6Department of Medicine, Center for Medicine and Clinical Research, University of Eastern Finland and Kuopio University Hospital, Kuopio, Finland; 7Department of Internal Medicine, Jagiellonian University Medical College, Krakow, Poland; 8Department of Nephrology, Royal Melbourne Hospital and the University of Melbourne, VIC, Australia; 9Instituto Privado de Hematologia E Investigacion Clinica (IPHIC), Asuncion, Paraguay; 10Salford Royal NHS Foundation Trust, Salford, UK; 11General Hospital Slovenj Gradec, Slovenj Gradec, Slovenia; 12Shire, Lexington, MA, USA
Purpose: Efficacy and safety of agalsidase alfa at 0.2 mg/kg weekly were compared with 0.2 mg/kg every other week (EOW). Exploratory analyses were performed for 0.4 mg/kg weekly.
Patients and methods: This was a 53-week, Phase III/IV, multicenter, open-label study (NCT01124643) in treatment-naïve adults (≥18 years) with Fabry disease. Inclusion criteria were left ventricular hypertrophy at baseline, defined as left ventricular mass indexed to height >50 g/m2.7 for males and >47 g/m2.7 for females. Primary endpoint was reduction of left ventricular mass indexed to height as assessed by echocardiography. Secondary endpoints included cardiac (peak oxygen consumption, 6-minute walk test, Minnesota Living with Heart Failure Questionnaire, New York Heart Association classification), renal (Modification of Diet in Renal Disease, estimated glomerular filtration rate), and biomarker (plasma globotriaosylceramide) assessments. Safety endpoints were adverse events and anti–agalsidase alfa antibodies.
Results: Twenty patients were randomized to 0.2 mg/kg EOW (mean age, 50.3 years; 70% male), 19 to 0.2 mg/kg weekly (51.8 years; 53% male), and 5 to 0.4 mg/kg weekly (49.4 years; 40% male). The mean change in left ventricular mass indexed to height by Week 53 in the 0.2-mg/kg EOW and weekly groups was 3.2 g/m2.7 and 0.5 g/m2.7, with no significant difference between groups. No clinically meaningful changes by Week 53 were found within or between the 0.2-mg/kg groups for peak oxygen consumption, 6-minute walk test, or Minnesota Living with Heart Failure Questionnaire. Two patients in each group improved by ≥1 New York Heart Association classification. No significant differences were found between 0.2 mg/kg EOW and weekly for mean change in estimated glomerular filtration rate (-1.21 mL/min/1.73 m2 vs -3.32 mL/min/1.73 m2) or plasma globotriaosylceramide (-1.05 nmol/mL vs -2.13 nmol/mL), respectively. Infusion-related adverse events were experienced by 25% and 21% in the 0.2-mg/kg EOW and weekly groups. Tachycardia, fatigue, and hypotension were experienced by two or more patients overall. Anti–agalsidase alfa antibodies were detected in 11.4% of patients and neutralizing antibodies in 6.8%. Infusion-related reactions did not appear to be correlated with antibody status.
Conclusion: No efficacy or safety differences were found when the approved EOW dosage of agalsidase alfa was increased to weekly administration. Exploratory analyses for 0.4 mg/kg weekly showed similar results.
Keywords: adverse events, exercise tolerance, left ventricular hypertrophy, lysosomal storage disorder, quality of life, renal function
Introduction
Fabry disease (FD) is a rare, X-linked glycosphingolipid storage disorder caused by mutations in the GLA gene (location chromosome Xq22.1).1 The resulting functional deficiency in the alpha-galactosidase A enzyme (EC 3.2.1.22) leads to accumulation of glycosphingolipids, especially globotriaosylceramide (Gb3), in lysosomes.2 The effects of Gb3 accumulation are progressive; it occurs throughout the body and affects many tissues and organs, with symptom severity in both male and female patients increasing with age. Symptoms of FD include debilitating neuropathic pain, proteinuria and progressive renal failure, and cerebrovascular and cardiovascular diseases, particularly hypertrophic cardiomyopathy, ultimately leading to premature mortality.3–12 FD-associated cardiomyopathy occurs in both classic and cardiac variant clinical types and is characterized by progressive left ventricular hypertrophy (LVH), which is reflected by a progressive increase in left ventricular mass indexed to height (LVMI, g/m2.7).13 In females, the age of onset tends to be later and the rate of progression is slower.10
Agalsidase alfa has been approved for the treatment of FD in numerous countries for over 10 years. A number of studies have evaluated the efficacy of agalsidase alfa in the treatment of both children and adult patients with FD.14–20 A previous short-term clinical study (NCT00097890) found similar safety, tolerability, and pharmacodynamic response of the approved agalsidase alfa dosing regimen of 0.2 mg/kg every other week (EOW) compared with regimens ranging from 0.1 mg/kg to 0.4 mg/kg weekly or EOW.21 In order to fulfill a European postmarketing commitment, the current study was designed to evaluate the efficacy and safety of two agalsidase alfa dosing regimens (0.2 mg/kg EOW and 0.2 mg/kg weekly) in a specific population of adults with FD and LVH. Additional exploratory analyses were also undertaken with a dose of 0.4 mg/kg weekly.
Materials and methods
Study design and patients
This was a 1-year, Phase III/IV, multicenter, randomized, three-arm, open-label study. Eligible patients included treatment-naïve adults (≥18 years) with a confirmed diagnosis of FD (biochemical testing and/or genotyping for males, genotyping for females) and LVH defined as >50 g/m2.7 for males and >47 g/m2.7 for females,13 determined by echocardiography. Patients were excluded if they had documented New York Heart Association (NYHA) functional Class IV heart failure symptoms, clinically significant systemic hypertension (untreated resting blood pressure >160/110 mmHg or poorly controlled hypertension defined by a blood pressure >150/100, while receiving antihypertensive medication), hemodynamically significant valvular stenosis or regurgitation, morbid obesity (body mass index >39 kg/m2), and known autosomal dominant sarcoplasmic contractile protein gene mutation.
Patients provided signed informed consent forms that were approved by the treating centers’ Institutional Review Board/Independent Ethics Committee and were randomized in an approximate 4:4:1 ratio to one of three agalsidase alfa (Replagal®; Shire, Lexington, MA, USA) dosing regimens administered as an IV infusion: 0.2 mg/kg EOW, main comparator 0.2 mg/kg weekly (both administered over 40 minutes), exploratory dose of 0.4 mg/kg weekly (administered over 80 minutes).
Efficacy and safety outcomes
The primary efficacy endpoint was absolute change in LVMI from baseline to Week 53. Echocardiography using M-mode measurements guided by 2D imaging was standardized across the study sites. Echocardiograms were read by a single reader in a central laboratory, who was blinded to treatment and clinical information, and LVM was calculated using a published formula22 before being indexed to height (g/m2.7).13 The secondary efficacy endpoints were changes from baseline to Week 53 in peak oxygen consumption (peak VO2), evaluated using the standard exponential exercise protocol;23 the 6-minute walk test (6MWT);24 the Minnesota Living with Heart Failure Questionnaire;25 NYHA heart failure classification;26 estimated glomerular filtration rate (eGFR), tested in a centralized laboratory and calculated using the four-variable formula for Modification of Diet in Renal Disease eGFR, which takes into account the serum creatinine value (mg/dL) and patient age, sex, and race;27 proteinuria/albuminuria, using the urine albumin/creatinine ratio (A/Cr); and fasting plasma Gb3.28 An additional subset analysis of eGFR stratified by baseline chronic kidney disease (CKD) stage was performed: Stage 1A, ≥130 mL/min/1.73 m2 (hyperfiltrators); Stage 1B, 90 mL/min/1.73 m2 to <130 mL/min/1.73 m2; Stage 2, 60 mL/min/1.73 m2 to <90 mL/min/1.73 m2; Stage 3, 30 mL/min/1.73 m2 to <60 mL/min/1.73 m2; Stage 4/5, 0 to <30 mL/min/1.73 m2.
Safety endpoints included treatment-emergent adverse events ([AEs] that occurred on or after the first agalsidase alfa infusion); use of concomitant medications; and changes in laboratory test results, vital signs, and electrocardiogram results. Blood samples for anti–agalsidase alfa antibody determination were screened using an enzyme-linked immunosorbent assay and all positive samples were confirmed by a titration assay. Positive samples were isotyped and tested for neutralizing enzyme activity using an in vitro assay. Absorbance ratios ≥2.0 relative to baseline were considered antibody positive if absolute absorbance was >0.04.
Primary and secondary efficacy endpoints and safety were compared in the two 0.2-mg/kg treatment groups, whereas the 0.4-mg/kg treatment group was included for exploratory purposes only.
Statistical analyses
Statistical analyses were performed using SAS® statistical software version 9.3 (SAS Institute Inc., Cary, NC, USA). A two-sided, two-sample t-test, assuming a 5% Type I error rate and a 5-g/m2.7 common standard deviation, was performed to determine the sample size that would provide 80% power to detect a 5-g/m2.7 treatment difference in the primary efficacy endpoint (mean change from baseline in LVM)29 between the two 0.2-mg/kg dose groups. A 10% dropout rate was also assumed; thus, a total of 38 patients were required for the two 0.2-mg/kg groups, and the addition of five patients for the exploratory 0.4-mg/kg group made the total required for enrollment 43 patients.
Baseline demographics and clinical characteristics were summarized using descriptive statistics. Where comparisons were made between the two 0.2-mg/kg treatment groups, the two-sample t-test was used to analyze continuous baseline parameters, and the exact Cochran-Mantel-Haenszel general association test and row means score test were used to analyze nominal-scaled and ordinal-scaled categorical baseline parameters, respectively.
The primary efficacy endpoint was analyzed using a repeated-measures analysis of variance model with baseline LVMI as the covariate, treatment, time, and treatment-by-time interaction as fixed effects, and patients nested within-treatment group as a random effect. The treatment effect and comparison P-values were estimated from the indicated statistical model for comparison of changes between the two 0.2-mg/kg treatment groups. No treatment comparisons between each of the two 0.2-mg/kg groups versus the 0.4-mg/kg weekly group were performed.
Least squares means (LSM) for changes from baseline for each of the two 0.2-mg/kg groups, the differences in LSMs between the two groups, and the associated two-sided 95.005% (due to multiplicity adjustment) confidence intervals (CI) were estimated. Other continuous efficacy endpoints were analyzed using a similar approach.
With a few exceptions, P-values ≤0.05 were considered to indicate statistical significance (ie, 95% CIs were estimated). The exceptions were LVMI, eGFR, and plasma Gb3, where P-values ≤0.04995 were used (ie, 95.005% CIs were estimated) as the significance level due to multiplicity adjustment from performing a planned interim analysis. As each of the efficacy endpoints was analyzed using a repeated-measures analysis of variance model with baseline endpoint as the covariate, no imputation methods were used in the analyses.
Results
Patient population
Among the 65 patients, 21 failed screening, mostly due to having a baseline LVMI below the level of the inclusion criterion. The remaining 44 patients were randomized to treatment (Figure 1). The demographic and baseline clinical characteristics are summarized in Table 1; briefly, the 0.2-mg/kg EOW group contained more males (n=14/20; 70%) than the 0.2-mg/kg weekly group (n=10/19; 52.6%) and also a higher percentage of patients >45 years of age (75.0% vs 68.4%). Mean age was similar across the three treatment groups. Cardiac status in terms of NYHA classification appeared to be more advanced for the 0.2-mg/kg EOW group compared with the 0.2-mg/kg weekly group, with more patients classified as Class II or III (Table 1). Three patients (15%) in the 0.2-mg/kg EOW group were discontinued (one had participation terminated by the investigator, one was discontinued due to noncompliance, and one suffered fatal cardiac arrest, deemed unrelated to the study drug). One patient (5.3%) in the 0.2-mg/kg weekly group withdrew consent, and all five patients (100%) randomized to the 0.4-mg/kg weekly group completed the study.
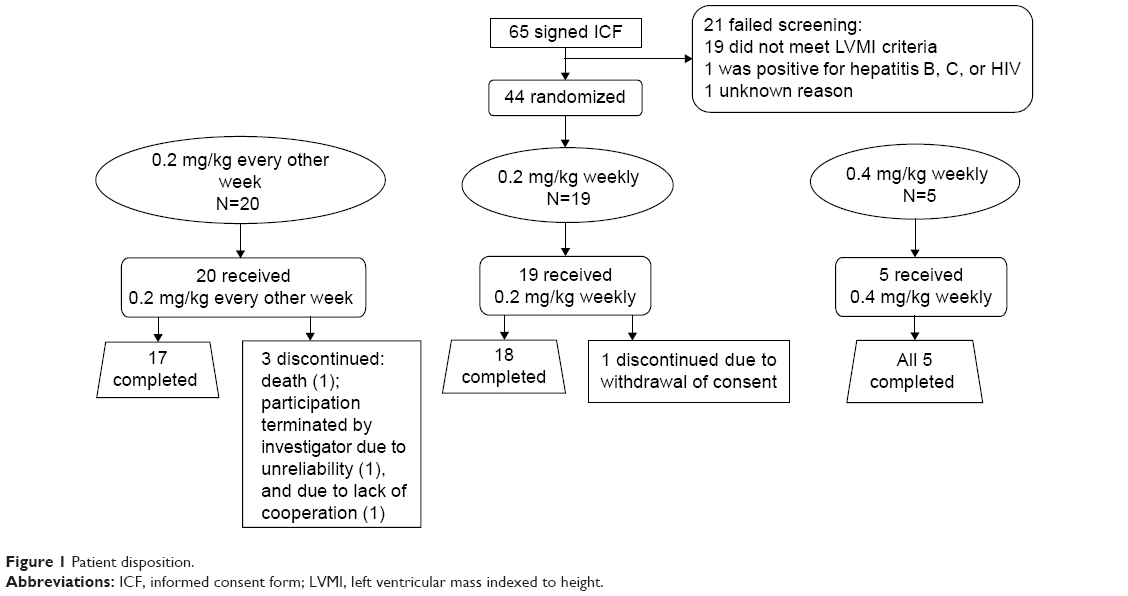 | Figure 1 Patient disposition. |
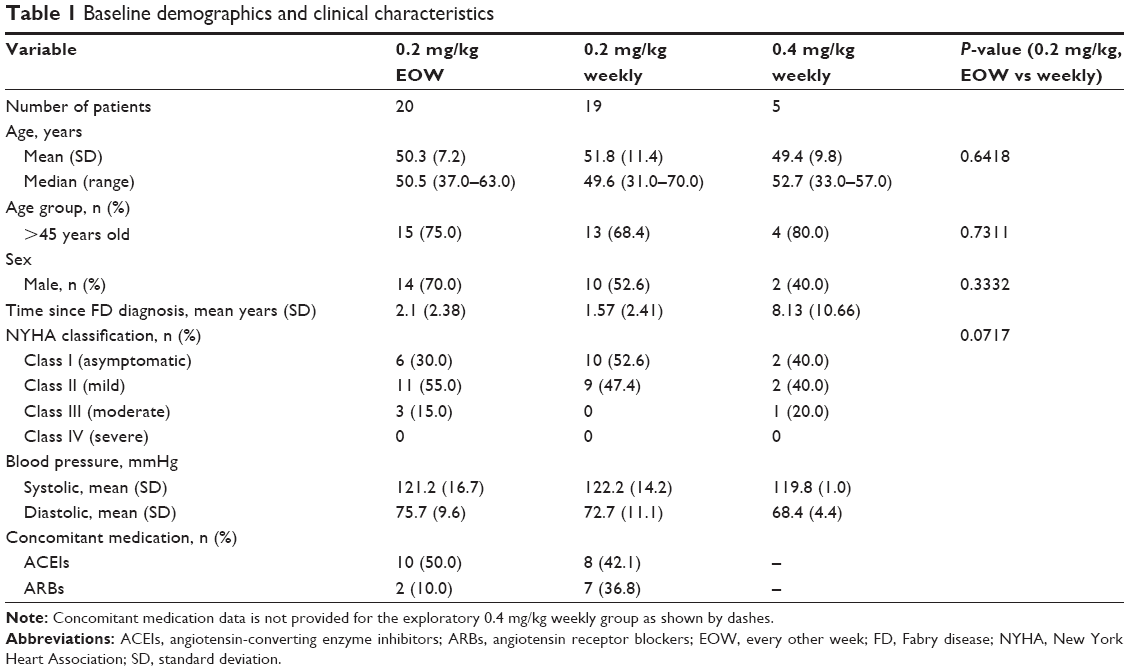 | Table 1 Baseline demographics and clinical characteristics |
Efficacy outcomes
The primary efficacy endpoint, LVMI, remained relatively stable over the study period in both 0.2-mg/kg dose groups (Table 2), as reflected by mean overall changes from baseline at Week 53 of 3.2 g/m2.7 (3.9%) for the 0.2-mg/kg EOW group and 0.5 g/m2.7 (3.4%) for the 0.2-mg/kg weekly group. The mean change in LVMI in the exploratory 0.4-mg/kg weekly group was −10.3 g/m2.7 (−9.3%). When assessed by sex, mean changes from baseline within each of the three dose groups for males and females were not statistically significant as the 95% CIs contained zero (Table 2). The difference in LVMI mean change from baseline between the two 0.2-mg/kg groups was not statistically significant (LSM difference −2.20g/m2.7; standard error 4.93; 95.005% CI: −12.26, 7.85; unadjusted P-value =0.6585).
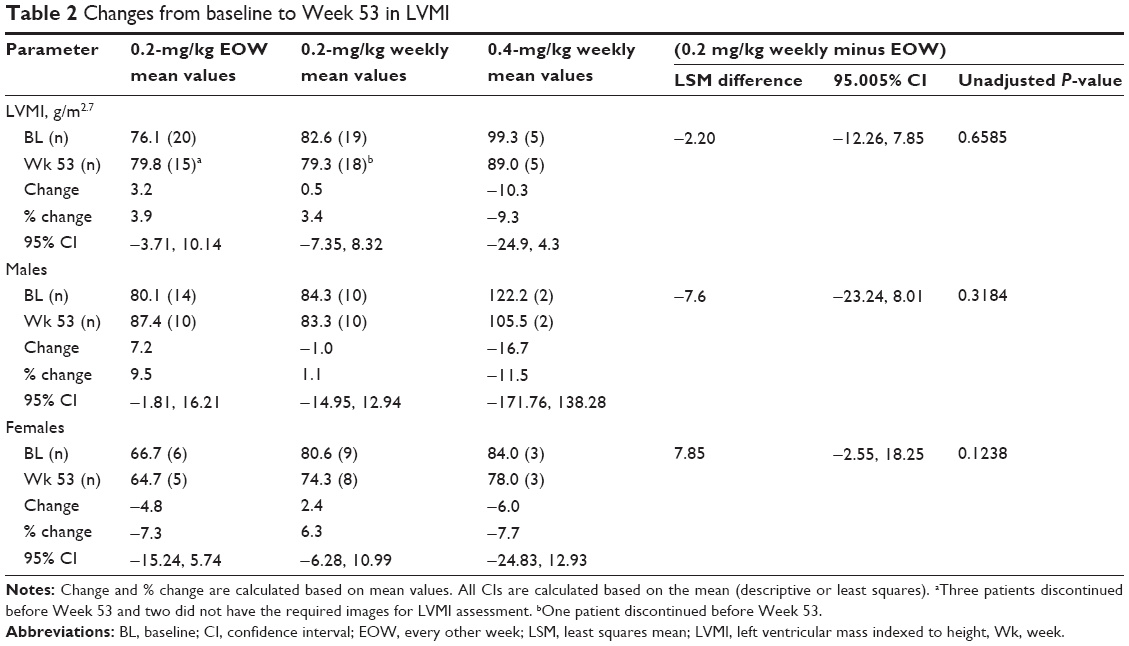 | Table 2 Changes from baseline to Week 53 in LVMI |
No statistically significant differences in changes from baseline to Week 53 between the 0.2-mg/kg EOW and weekly groups were found for exercise tolerance (6MWT and peak VO2), although an increase in mean distance walked in the 6MWT of 37.9 m (95% CI: 2.9, 73.0) was found for the 0.2-mg/kg weekly group (Table 3). A slight decrease in peak VO2 was noted in the group receiving 0.2 mg/kg EOW (change: −2.0 mL/kg/min, 95% CI: −3.91, −0.16; Table 3).
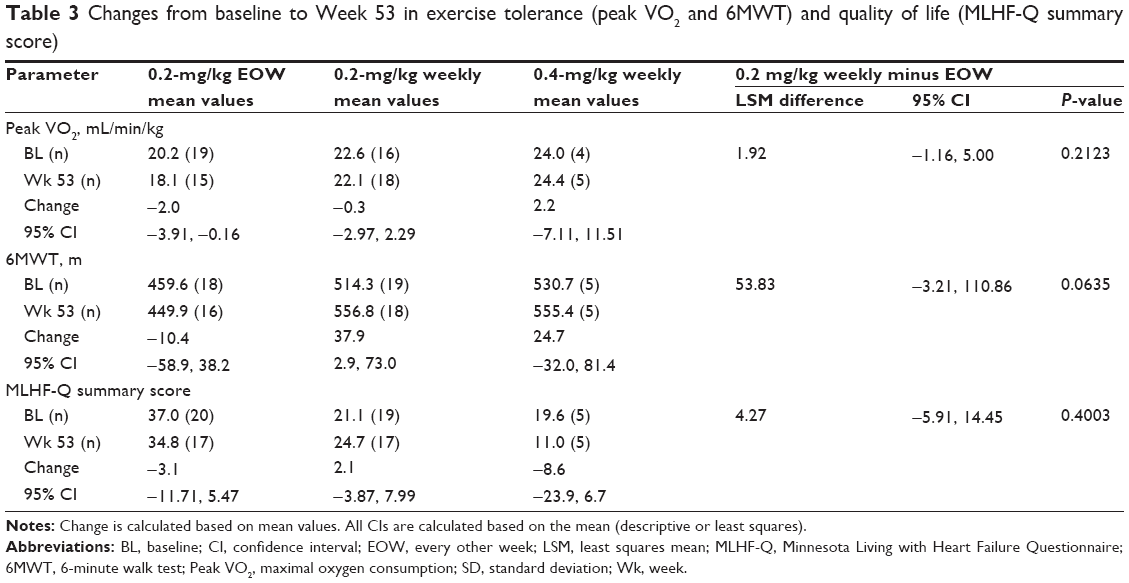 | Table 3 Changes from baseline to Week 53 in exercise tolerance (peak VO2 and 6MWT) and quality of life (MLHF-Q summary score) |
Regarding quality of life, the mean Minnesota Living with Heart Failure Questionnaire summary score at baseline for the 0.2-mg/kg EOW group was 37.0 compared with 21.1 for the 0.2-mg/kg weekly group, possibly suggesting a greater impact of disease burden at the start in this group (Table 3). By Week 53, however, the mean change within each group was not considered clinically meaningful, nor was it considered statistically significant when compared between the two groups. No significant or clinically meaningful changes from baseline to Week 53 in exercise tolerance or quality of life were noted for the 0.4-mg/kg weekly group (Table 3).
An improvement of one class or higher in the NYHA classification at Week 53 for patients who had NYHA Class II or III at baseline plus a Week 53 evaluation (baseline: nine mild and two moderate in the 0.2-mg/kg EOW group, and baseline: eight mild in the 0.2-mg/kg weekly group) was seen in two patients in the 0.2-mg/kg EOW group (18.2%) and two in the 0.2-mg/kg weekly group (25%). The difference (95% CI) in the proportion of patients who showed an improvement of one class or higher in NYHA classification between the 0.2-mg/kg EOW and weekly groups was −6.8% (−44.5%, 30.9%). In the 0.4-mg/kg weekly group, one patient in Class II (20%) and one patient in Class III (20%) at baseline improved to Class I by Week 53.
Regarding renal function, the baseline mean (standard deviation [SD]) eGFR was 82.0 mL/min/1.73 m2 (34.1) in the 0.2-mg/kg EOW group, 78.1 mL/min/1.73 m2 (33.8) in the 0.2-mg/kg weekly group, and 72.1 mL/min/1.73 m2 (31.2) in the 0.4-mg/kg weekly group (Table 4). The mean changes (95% CI) from baseline in eGFR at Week 53 were −1.21 mL/min/1.73 m2 (−7.46, 5.05) in the 0.2-mg/kg EOW group, −3.32 mL/min/1.73 m2 (−9.63, 2.99) in the 0.2-mg/kg weekly group, and −1.68 (−14.00, 10.64) mL/min/1.73 m2 in the 0.4-mg/kg weekly group. The estimated difference from the statistical model in mean change between the 0.2-mg/kg groups was not significant (−1.56 [95.005% CI: −9.23, 6.12]; unadjusted P-value =0.6830; Table 4). A subset analysis of eGFR by CKD stage showed that patients with Stage 1A CKD (eGFR ≥130 mL/min/1.73 m2) at baseline had a reduced mean eGFR at Week 53 (mean change [95% CI] −24.4 [−82.6, 34.2] in the 0.2-mg/kg EOW group and −24.4 [−71.4, 22.6] in the 0.2-mg/kg weekly group; Table 5). Patients with all other CKD stages at baseline remained stable throughout the study.
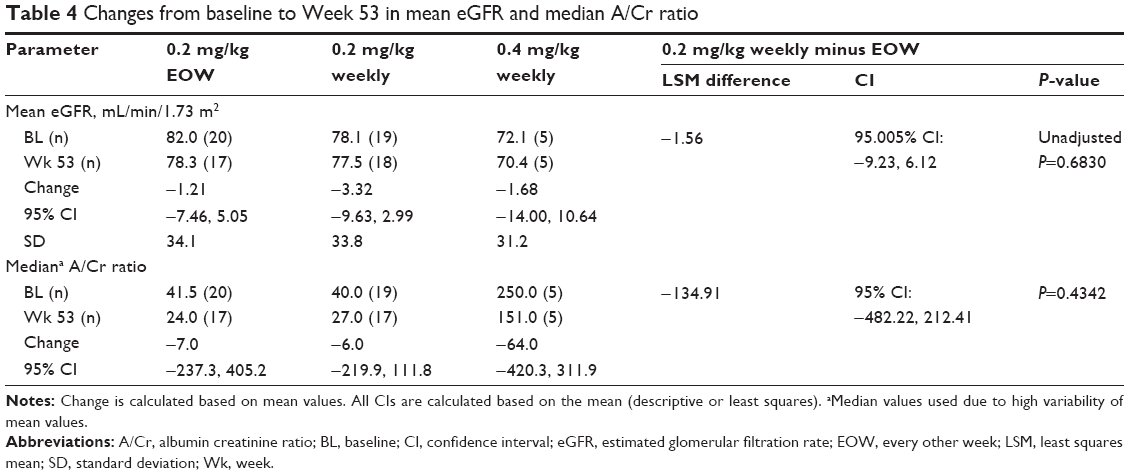 | Table 4 Changes from baseline to Week 53 in mean eGFR and median A/Cr ratio |
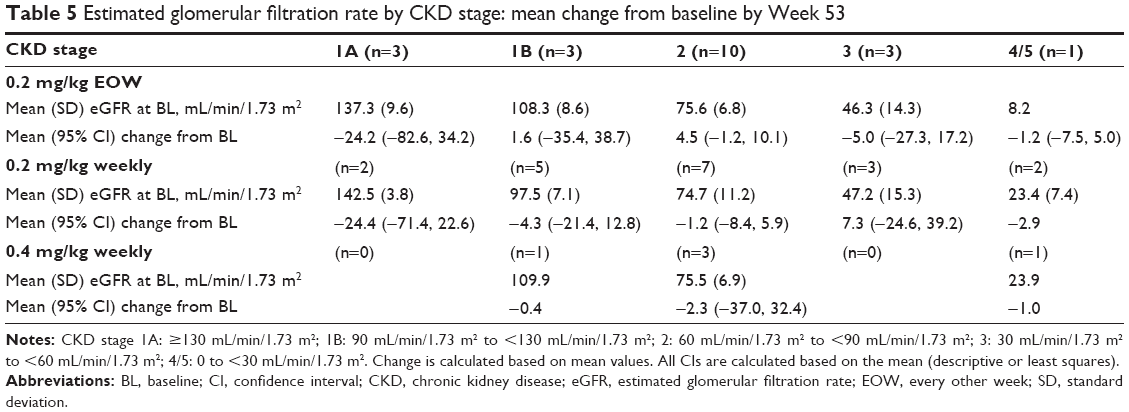 | Table 5 Estimated glomerular filtration rate by CKD stage: mean change from baseline by Week 53 |
Due to the high variability of A/Cr data, median values for the A/Cr ratio were evaluated (0.2 mg/kg EOW baseline 41.5 [range 4–2,394], median change by Week 53 −7.0; 0.2 mg/kg weekly baseline 40.0 [range 3–1,982], median change by Week 53 −6.0; 0.4 mg/kg weekly baseline 250 [range 81–486], median change by Week 53 −64.0; Table 4). The median A/Cr ratio in each 0.2-mg/kg group remained stable over the 53 weeks and, based on the statistical model, no statistically significant difference was found in the change from baseline to Week 53 between the two 0.2-mg/kg groups.
Mean baseline plasma Gb3 values were only slightly elevated above normal (upper limit of normal 3.9 nmol/mL) in all three treatment groups (0.2 mg/kg EOW, mean 6.1 nmol/mL [n=20]; 0.2 mg/kg weekly, mean 5.7 [n=19]; 0.4 mg/kg weekly, mean 5.3 [n=5]; Table 6). The mean changes (SD) from baseline at Week 53 in Gb3 were −1.0 nmol/mL (2.3) in the 0.2-mg/kg EOW group and −2.1 nmol/mL (4.4) in the 0.2-mg/kg weekly group. From the statistical analysis, the LSM difference between the two 0.2-mg/kg groups was −1.017 nmol/mL [95.005% CI: −2.127, 0.094]; unadjusted P-value =0.0715). An exploratory analysis of changes in Gb3 by sex also found no significant mean changes from baseline or differences between the 0.2-mg/kg regimens.
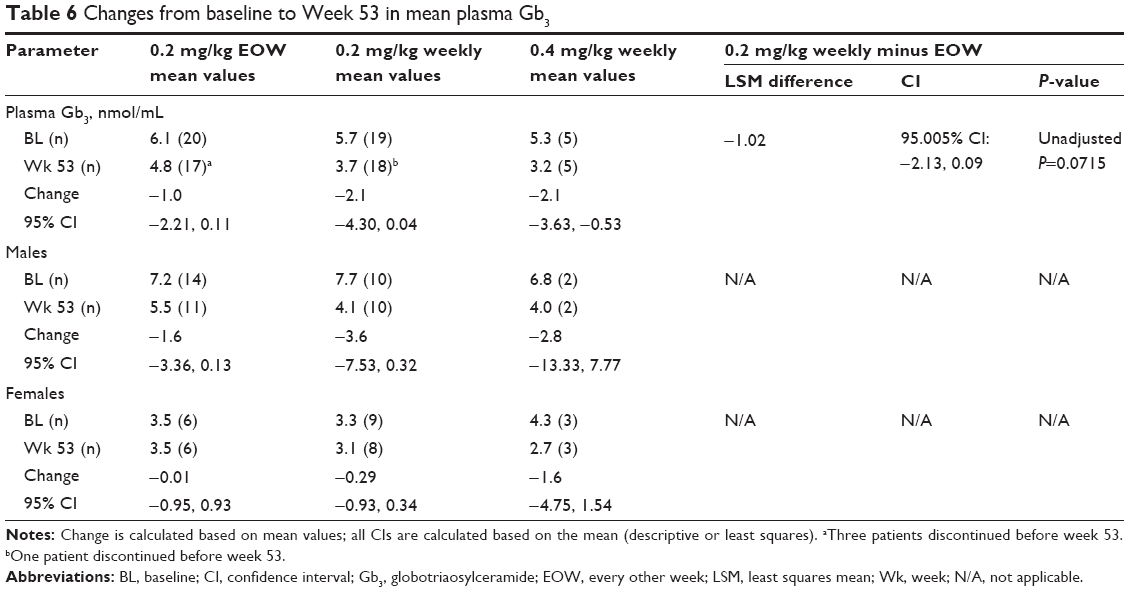 | Table 6 Changes from baseline to Week 53 in mean plasma Gb3 |
Safety outcomes
The frequencies of treatment-emergent AEs were similar in the three treatment groups (0.2 mg/kg EOW, 95% [n=19]; 0.2 mg/kg weekly, 89.5% [n=17]; 0.4 mg/kg, 100% [n=5]). Infusion-related reactions were mild or moderate and were reported in 25% (n=11) of patients overall: 5/20 (25.0%), 4/19 (21.1%), and 2/5 (40%) patients experienced at least one infusion-related AE in the 0.2-mg/kg EOW and weekly and the 0.4-mg/kg weekly groups, respectively. No patients reported severe or life-threatening, infusion-related AEs. Infusion-related AEs experienced by two or more patients overall were tachycardia (4.5% [n=2]), hypotension (4.5% [n=2]), and fatigue (9.1% [n=4]). Fewer patients in the 0.2-mg/kg weekly group experienced severe or life-threatening AEs (21.1% [n=4]) compared with the 0.2-mg/kg EOW group (40.0% [n=8]). No severe or life-threatening AEs were reported by those in the 0.4-mg/kg group. Furthermore, a lower percentage of patients in the 0.2-mg/kg weekly group experienced serious adverse events (SAEs) (26.3% [n=5]) compared with the EOW group (40.0% [n=8]) or the 0.4-mg/kg weekly group (40.0% [n=2]). An SAE of hypotension (also considered an infusion-related AE) was deemed possibly related to the study drug and was experienced by a patient in the 0.2-mg/kg EOW group; it resolved without sequelae. A life-threatening SAE of traumatic hematoma was experienced by another patient in the 0.2-mg/kg EOW group; this was deemed unrelated to the study drug and resolved without sequelae. A further patient in the 0.2-mg/kg EOW group suffered an SAE of cardiac arrest with a fatal outcome, which was also considered unrelated to the study drug.
Overall, anti–agalsidase alfa antibodies were detected (titers ≤130) in 11.4% (n=5) of patients: in three patients from the 0.2-mg/kg EOW group (1 immunoglobulin G [IgG], 1 IgM, and 1 IgG/IgM) and two from the 0.2-mg/kg weekly group (1 IgG, 1 IgM; none were detected in the 0.4-mg/kg weekly group). Neutralizing antibodies were detected in 6.8% (n=3) patients: in two patients from the 0.2-mg/kg EOW group (1 IgG, 1 IgG/IgM) and one from the 0.2-mg/kg weekly group (1 IgG). Seroconversion was noted in three patients in the 0.2-mg/kg EOW group (two persistently positive for IgG or IgG/IgM and neutralizing antibodies; one transiently positive for IgM but no neutralizing antibodies) and in two patients in the 0.2-mg/kg weekly group (one persistently positive for IgG and neutralizing antibodies, one persistently positive for IgM but no neutralizing antibodies). Infusion-related reactions did not appear to be correlated with antibody or neutralizing antibody status.
Concomitant medication usage and changes in laboratory parameters, vital signs, and electrocardiogram results were also monitored as part of the safety profile of each patient. No clinically meaningful trends over time were observed, and the results from these measures were consistent between treatment groups.
Discussion
This study reports the efficacy and safety outcomes of two potential alternative treatment regimens of agalsidase alfa, alongside the approved dose of 0.2 mg/kg EOW, in adult patients with FD and LVH. The approved dose has previously been shown to improve or stabilize the progression of clinical manifestations of FD,14,15,17,18 and the results herein indicate that the alternative regimens of 0.2 mg/kg and 0.4 mg/kg weekly also slowed the progression of some FD manifestations. The analysis of 0.4 mg/kg weekly was exploratory; statistical comparisons were only made between the two 0.2-mg/kg regimens.
Presence of LVH at baseline was an inclusion criterion of the study. Patients with advanced cardiomyopathy at baseline might not be expected to show great improvement during enzyme replacement therapy,30 although slowing of disease progression or disease stabilization would still be an important outcome. Consistent with this, LVMI remained stable over the 53-week study period and did not significantly differ between the 0.2-mg/kg EOW and weekly regimens. An improvement or stabilization in cardiac outcomes after treatment with the approved dose of 0.2 mg/kg agalsidase alfa EOW has previously been demonstrated.17,20 The apparent stabilization in cardiac status herein also seemed to be reflected by an improvement in NYHA classification for two patients in each of the 0.2-mg/kg treatment groups.
Regarding exercise tolerance, no universally accepted reference values exist as yet for the 6MWT, but one multicenter study of 444 healthy adults showed an overall mean (SD; range) distance walked of 571 (90; 380–782) m,31 and a single-center study on 102 healthy adults an overall mean (SD; range) distance of 614 (56; 459–738) m.32 The mean distances walked at baseline in the 0.2-mg/kg EOW and weekly groups of the present study were similar to these results in healthy adults, potentially indicating that there has not been significant impact from the disease on the mean distance walked and therefore clinically meaningful improvements would not be expected as a result of treatment. For peak VO2, a slight reduction was observed in the group receiving 0.2 mg/kg EOW, although the interpretation of peak VO2 data is confounded by missing baseline data and the fact that information was lacking on whether the patients had exercised to their maximum capacity. However, no significant differences in change from baseline were observed for peak VO2 between the two 0.2-mg/kg groups, and quality of life, as assessed by Minnesota Living with Heart Failure Questionnaire scores, also remained stable for both 0.2-mg/kg groups. Overall, the results of the current study do not indicate any additional cardiac benefits from increasing the dose to 0.2 mg/kg every week.
The patients were not selected or stratified based on baseline eGFR values or their renal status; these showed considerable variation, but changes within the 0.2-mg/kg EOW and weekly groups by study end were minimal and did not significantly differ between the two groups. When analyzed by CKD stage, reductions in mean eGFR in patients with hyperfiltration (Stage 1A, ≥130 mL/min/1.73 m2; 0.2-mg/kg groups only) occurred by Week 13 and remained stable until the end of the study, indicating possible normalization of renal function in these patients. The eGFR of patients in the 0.2-mg/kg groups with all other CKD stages was maintained throughout, thus indicating that renal function remained stable in these treatment groups. The numbers in this CKD subset analysis were small; thus, the results should be interpreted with care. However, the apparent stability demonstrated in renal function was mirrored by the A/Cr data, which although highly variable, also showed no significant changes by Week 53 or between the 0.2-mg/kg groups in median values.
Mean baseline plasma Gb3 values were only slightly elevated and did not significantly differ in changes from baseline between the two 0.2-mg/kg treatment groups. Males typically exhibit higher baseline plasma Gb3 values than females; thus, their expected reductions posttreatment are greater. In this study, the slightly greater decrease observed by Week 53 in the 0.2-mg/kg weekly group is difficult to interpret because mean baseline plasma Gb3 levels in both groups were relatively low, and numbers of males and females in each group differed. Although not analyzed in this study, globotriaosylsphingosine (lysoGb3), a deacylated Gb3, has been introduced as a promising new biomarker for monitoring the course of FD.33 It is reported to have greater sensitivity and reliability than plasma or urine Gb3;34 thus, future reports analyzing LysoGb3 in FD, rather than Gb3, are likely to be of interest.
This study was not powered to detect differences in secondary endpoints between the treatment groups, and as such, these results should be interpreted with caution; however, they are consistent with the primary LVMI endpoint in that no significant changes were observed from baseline by the end of the study within or between the 0.2-mg/kg EOW and weekly groups.
Agalsidase alfa was generally found to be safe and well tolerated. The percentages of patients who experienced AEs such as infusion-related reactions and who developed anti–agalsidase alfa antibodies were similar to previous experience.15 The results suggest that fewer patients in the 0.2-mg/kg weekly group experienced severe or life-threatening AEs or SAEs, and increasing dosing frequency from EOW to weekly did not seem to result in more patients developing antibodies nor changes in the safety profile. Furthermore, there was no apparent impact on treatment effect or safety from being anti–agalsidase alfa positive.
Evaluation of the 0.4-mg/kg group was exploratory only and not planned for comparison with the two 0.2-mg/kg treatment groups, but the efficacy and safety results for this group were generally consistent with those of the 0.2-mg/kg groups. No new safety concerns were introduced by this increased dosage.
As is often the case in rare disease studies, the total number of patients in this study was small, especially given the echocardiography-based primary endpoint; this fact, along with the relatively short-term study, limits the conclusions that can be drawn. A longer period of follow-up may be needed to confirm the apparent stabilization in cardiac status that was observed. To evaluate left ventricular mass, we chose echocardiography as a standardized, readily available, reproducible, and noninvasive technique.35 However, the limitation of echocardiography is the inability to detect fibrosis. Gadolinium-enhanced cardiovascular magnetic resonance can detect fibrosis, but it has other limitations (eg, toxicity in advanced renal failure, the impossibility of examining patients with implanted ferromagnetic materials, and less readily available technology).36
FD manifestations have been reported to occur later, progress at a slower rate, and vary in degree of severity in females compared with males.10,37–39 Thus, the greater number of male patients and patients >45 years old, plus the inclusion of patients with NYHA Class III in the 0.2-mg/kg EOW group, could suggest a somewhat greater disease burden at the start in this group compared with the 0.2-mg/kg weekly group. However, these baseline differences were not statistically significant; furthermore, mean age and LVMI did not significantly differ between the two groups at baseline.
This study was not designed to assess genotype; the lack of mutation analyses for all patients meant that the proportion of patients with later onset cardiac variant FD, and thus potential differences in treatment efficacy with agalsidase alfa for classic and cardiac variant FD, is unknown.40 Also, although further detailed information on antiproteinuric therapy with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers throughout the study might be useful for evaluating proteinuria and albuminuria, the variability found in baseline A/Cr ratios may still hamper interpretation. Furthermore, the inclusion of patients with different stages of CKD, including some with end-stage renal disease, limits the extent of renal conclusions that can be drawn for the overall study population.
Conclusion
Overall, this study found no differences in efficacy outcomes or safety when the frequency of agalsidase alfa enzyme replacement therapy was increased from the approved dose of 0.2 mg/kg EOW to the same dose administered weekly. Furthermore, the exploratory evaluation of 0.4 mg/kg weekly showed no significant deviation from these results. The study data herein support the current approved dosing regimen.
Acknowledgments
The authors thank all the patients who participated in this study, which was funded by Shire. Medical writing assistance (supported financially by Shire) was provided by Annie Rowe and Tina Rose (Excel Scientific Solutions, Horsham, UK) during the preparation of the manuscript.
Disclosure
LG was the principal investigator for the current study with Shire and has received travel funding from Genzyme and Shire. OG has received research support from Actelion, Amicus, Genzyme Corp, Pfizer-Protalix Biotherapeutics, and Shire; payments for consultancy from Actelion, Pfizer-Protalix Biotherapeutics, and Shire; and payments for speaker bureaus from Actelion, Genzyme, and Shire. MH has received consultancy payments from Shire; his institution has received grant support from Protalix, Amicus, and Genzyme. IK has received speaking fees and travel funding from Genzyme and Shire. AL is a member of the Fabry Registry Board of Advisors, which is sponsored by Genzyme. KN has received research support and travel grants from Amicus, Genzyme, and Shire and speaker honoraria from Genzyme and Shire. RS was the principal investigator for a study with Shire, has received travel funding from Shire, and has attended Shire’s Fabry Advisory Board meetings. BV has received speaking fees and travel grants from Genzyme and Shire and is a member of the Fabry Registry Board of Advisors, which is sponsored by Genzyme. PC and AW are employees of, and hold stock options in, Shire. MK, JK, JM, and DGR declare no conflicts of interest in this work. The authors report no other conflicts of interest in this work.
References
Online Mendelian Inheritance in Man OMIM®. Fabry Disease. 2012–13. Available from: http://www.omim.org/entry/301500. Accessed December 4, 2014. | ||
Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J Mol Biol. 2004;337(2):319–335. | ||
MacDermot KD, Holmes A, Miners AH. Natural history of Fabry disease in affected males and obligate carrier females. J Inherit Metab Dis. 2001;24(suppl 2):13–14. [discussion 11–12]. | ||
MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001;38(11):750–760. | ||
Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34(3):236–242. | ||
Mehta A, Clarke JT, Giugliani R, et al; FOS Investigators. Natural course of Fabry disease: changing pattern of causes of death in FOS – Fabry Outcome Survey. J Med Genet. 2009;46(8):548–552. | ||
West M, Nicholls K, Mehta A, et al. Agalsidase alfa and kidney dysfunction in Fabry disease. J Am Soc Nephrol. 2009;20(5):1132–1139. | ||
Branton MH, Schiffmann R, Sabnis SG, et al. Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). 2002;81(2):122–138. | ||
Schiffmann R, Warnock DG, Banikazemi M, et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009;24(7):2102–2111. | ||
Kampmann C, Linhart A, Baehner F, et al. Onset and progression of the Anderson-Fabry disease related cardiomyopathy. Int J Cardiol. 2008;130(3):367–373. | ||
Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke. 2009;40(3):788–794. | ||
Kampmann C, Wiethoff CM, Perrot A, Beck M, Dietz R, Osterziel KJ. The heart in Anderson Fabry disease. Z Kardiol. 2002;91(10):786–795. | ||
de Simone G, Daniels SR, Devereux RB, et al. Left ventricular mass and body size in normotensive children and adults: assessment of allometric relations and impact of overweight. J Am Coll Cardiol. 1992;20(5):1251–1260. | ||
Hughes DA, Elliott PM, Shah J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008;94(2):153–158. | ||
Schiffmann R, Kopp JB, Austin HA 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743–2749. | ||
Breunig F, Weidemann F, Strotmann J, Knoll A, Wanner C. Clinical benefit of enzyme replacement therapy in Fabry disease. Kidney Int. 2006;69(7):1216–1221. | ||
Mehta A, Beck M, Elliott P, et al; Fabry Outcome Survey investigators. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009;374(9706):1986–1996. | ||
Ramaswami U, Wendt S, Pintos-Morell G, et al. Enzyme replacement therapy with agalsidase alfa in children with Fabry disease. Acta Paediatr. 2007;96(1):122–127. | ||
Schiffmann R, Martin RA, Reimschisel T, et al. Four-year prospective clinical trial of agalsidase alfa in children with Fabry disease. J Pediatr. 2010;156(5):832–837, 837e831. | ||
Kampmann C, Linhart A, Devereux RB, Schiffmann R. Effect of agalsidase alfa replacement therapy on Fabry disease-related hypertrophic cardiomyopathy: a 12- to 36-month, retrospective, blinded echocardiographic pooled analysis. Clin Ther. 2009;31(9):1966–1976. | ||
Clarke JT, West ML, Bultas J, Schiffmann R. The pharmacology of multiple regimens of agalsidase alfa enzyme replacement therapy for Fabry disease. Genet Med. 2007;9(8):504–509. | ||
Lang RM, Bierig M, Devereux RB, et al; Chamber Quantification Writing Group; American Society of Echocardiography’s Guidelines and Standards Committee; European Association of Echocardiography. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18(12):1440–1463. | ||
Northridge DB, Grant S, Ford I, et al. Novel exercise protocol suitable for use on a treadmill or a bicycle ergometer. Br Heart J. 1990;64(5):313–316. | ||
ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166(1):111–117. | ||
Rector TS, Kubo SH, Cohn JN. Validity of the Minnesota living with heart failure questionnaire as a measure of therapeutic response to enalapril or placebo. Am J Cardiol. 1993;71(12):1106–1107. | ||
American Heart Association. Classification of Functional Capacity and Objective Assessment; 1994. Available from: http://my.americanheart.org/professional/StatementsGuidelines/ByPublicationDate/PreviousYears/Classification-of-Functional-Capacity-and-Objective-Assessment_UCM_423811_Article.jsp. Accessed December 10, 2013. | ||
National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002;39(2: suppl 1):S1–S266. | ||
Zeidner KM, Desnick RJ, Ioannou YA. Quantitative determination of globotriaosylceramide by immunodetection of glycolipid-bound recombinant verotoxin B subunit. Anal Biochem. 1999;267(1):104–113. | ||
Palmieri V, Dahlöf B, DeQuattro V, et al. Reliability of echocardiographic assessment of left ventricular structure and function: the PRESERVE study. Prospective randomized study evaluating regression of ventricular enlargement. J Am Coll Cardiol. 1999;34(5):1625–1632. | ||
Weidemann F, Linhart A, Monserrat L, Strotmann J. Cardiac challenges in patients with Fabry disease. Int J Cardiol. 2010;141(1):3–10. | ||
Casanova C, Celli BR, Barria P, et al; Six Minute Walk Distance Project (ALAT). The 6-min walk distance in healthy subjects: reference standards from seven countries. Eur Respir J. 2011;37(1):150–156. | ||
Chetta A, Zanini A, Pisi G, et al. Reference values for the 6-min walk test in healthy subjects 20–50 years old. Respir Med. 2006;100(9):1573–1578. | ||
Aerts JM, Groener JE, Kuiper S, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008;105(8):2812–2817. | ||
Liao HC, Huang YH, Chen YJ, et al. Plasma globotriaosylsphingosine (lysoGb3) could be a biomarker for Fabry disease with a Chinese hotspot late-onset mutation (IVS4+919G>A). Clin Chim Acta. 2013;426:114–120. | ||
Yousef Z, Elliott PM, Cecchi F, et al. Left ventricular hypertrophy in Fabry disease: a practical approach to diagnosis. Eur Heart J. 2013;34(11):802–808. | ||
Moon JC, Sachdev B, Elkington AG, et al. Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J. 2003;24(23):2151–2155. | ||
Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase a deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw Hill; 2001:3733–3774. | ||
Whybra C, Kampmann C, Willers I, et al. Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001;24(7):715–724. | ||
Wilcox WR, Oliveira JP, Hopkin RJ, et al; Fabry Registry. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93(2):112–128. | ||
Serra W, Fagnani S, Ardissino D, Gherli T. Late-onset cardiac variant of Fabry disease responsive to short-term treatment with agalsidase alpha. J Clin Exp Cardiol. 2010;1(2):109. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.