")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Evaluation of glycine-bearing celecoxib derivatives as a colon-specific mutual prodrug acting on nuclear factor-κB, an anti-inflammatory target
Authors Lee S, Lee Y, Kim W, Nam J, Jeong S, Yoo J , Kim M, Moon HR, Jung Y
Received 13 May 2015
Accepted for publication 7 July 2015
Published 7 August 2015 Volume 2015:9 Pages 4227—4237
DOI https://doi.org/10.2147/DDDT.S88543
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Sunyoung Lee,1,* Yonghyun Lee,1,2,* Wooseong Kim,1 Joon Nam,1 Seongkeun Jeong,1 Jin-Wook Yoo,1 Min-Soo Kim,1 Hyung Ryong Moon,1 Yunjin Jung1
1College of Pharmacy, Pusan National University, Busan, 2Bio-Nanomedicine Lab, Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon, South Korea
*These authors contributed equally to this work
Abstract: In an inflammatory state where HOCl is generated, glycine readily reacts with HOCl to produce glycine chloramine, an anti-inflammatory oxidant. Colonic delivery of celecoxib elicits anticolitic effects in a trinitrobenzene sulfonic acid-induced rat colitis model. Glycine-bearing celecoxib derivatives were prepared and evaluated as a colon-specific mutual prodrug acting on nuclear factor-κB (NFκB), an anticolitic target. Glycylcelecoxib (GC), N-glycylaspart-1-ylcelecoxib (N-GA1C), and C-glycylaspart-1-ylcelecoxib (C-GA1C) were synthesized and their structures identified using infrared and proton nuclear magnetic resonance spectrometer. The celecoxib derivatives were chemically stable in pH 6.8 and 1.2 buffers. GC and C-GA1C were resistant to degradation in the small intestinal contents, while N-GA1C was substantially cleaved to release celecoxib. In contrast, all the celecoxib derivatives were degraded to liberate celecoxib in the cecal content. These results suggest that GC and C-GA1C could be delivered to and liberate celecoxib and glycine in the large intestine. In human colon carcinoma HCT116 and murine macrophage RAW264.7 cells, combined celecoxib–glycine chloramine treatment additively suppressed the production of proinflammatory NFκB target gene products. Collectively, our data suggest that C-GA1C is a potential colon-specific mutual prodrug acting against NFκB.
Keywords: colon-specific drug delivery, mutual prodrug, celecoxib, glycine chloramine, nuclear factor kappa-B
Introduction
Proinflammatory mediators, including proinflammatory cytokines, are upregulated in the colon mucosa and epithelial cells of inflammatory bowel disease (IBD) patients.1 In response to many proinflammatory cytokines, including TNFα, the transcription factor nuclear factor-κB (NFκB) is activated in many types of cells in the large intestine, leading to expression of its target genes involved in immunity and inflammation, thereby playing a central role in the pathological progress of IBD.2 For this reason, NFκB has been considered a therapeutic target for the development of anti-IBD agents.3,4
Chloramines of such amino acids as glycine and taurine, which are generated by reaction with a strong oxidant, HOCl, produced by the neutrophil enzyme myeloperoxidase in the inflamed tissues, have an inhibitory effect on NFκB activity.5,6 Chloramines of amino acids retain the oxidizing capability of HOCl, but are much less reactive and more selective for thiols and methionine residues in amino acids in a variety of functional proteins,5,7 which could affect signaling pathways in cells. In fact, the oxidation of methionine in NFκB interferes with its biological activity, leading to inhibition of NFκB pathways. Such inhibition provides a mechanism whereby production of neutrophil oxidants could dampen the inflammatory response.8,9
Although the therapeutic effectiveness and safety of celecoxib, a selective COX-2 inhibitor, in IBD are disputed,10–14 colon-targeted delivery of celecoxib is suggested to be a pharmaceutical strategy to settle the controversy by tackling potential troublesome issues raised by long-term use of celecoxib for the treatment of IBD.15,16 A recent paper demonstrated that colonic delivery of celecoxib improves its anticolitic effects as well as cardiovascular toxicity, which may be achieved by increasing the therapeutic concentration at the target site and decreasing the systemic absorption of celecoxib.16
To improve further the therapeutic efficacy of celecoxib, we designed a colon-specific mutual prodrug of celecoxib in which aspart-4-yl glycine and glycine were used not only for a colon-specific carrier but also for a therapeutic moiety. Glycine in the carrier moiety should be converted to glycine chloramine in the inflamed colon where HOCl is generated.8 In this study, glycine-bearing celecoxib derivatives were synthesized and their colon specificity and pharmacologic mutuality against an anti-inflammatory target (NFκB) assessed in vitro.
Materials and methods
Materials
Human colon carcinoma HCT116 cells (American Type Culture Collection, Manassas, VA, USA) were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and penicillin–streptomycin (HyClone™; GE Healthcare Bio-Sciences Corp, Piscataway, NJ, USA). Lipopolysaccharides (LPSs), 1,1′-carbonyldiimidazole (CDI), N-benzoyl-glycine, and N-(2-hydroxybenzoyl)-glycine were obtained from Sigma-Aldrich Co (St Louis, MO, USA). Recombinant human TNFα was obtained from R&D Systems Inc (Minneapolis, MN, USA). N-(t-Boc)-glycine, 1-benzyl N-(t-Boc)-aspartate, and 4-benzyl N-(t-Boc)-aspartate were purchased from Tokyo Chemical Industry (Tokyo, Japan). 5-Aminosalicyloylglycine was prepared as described in our previous paper.17 Celecoxib was ether-extracted from Celebrex capsules (Pfizer Inc, New York, NY, USA). Solvents for proton nuclear magnetic resonance (1H-NMR) and high-performance liquid chromatography (HPLC) were obtained from EMD Millipore (Billerica, MA, USA). All other chemicals were reagent grade, commercially available products. Infrared (IR) spectra were recorded on a Varian Fourier-transform IR spectrophotometer (Varian Medical Systems, Palo Alto, CA, USA). 1H-NMR spectra were taken on a Varian AS 500 spectrometer, and the chemical shifts are in parts per million downfield from tetramethylsilane. The HPLC system consisted of a model 306 pump, a 117 variable ultraviolet detector, a model 234 autoinjector, and a Model 805 manometric module from Gilson Inc (Middleton, WI, USA). A symmetry column (R18; Waters, Milford, MA, USA) (250×4.6 mm) with a guard column (Waters, 3.9×20 mm) was used. The animal protocol used in this study was reviewed and approved by the Pusan National University Institutional Animal Care and Use Committee on ethical procedures and scientific care.
HPLC analysis
Samples prepared were filtered through a membrane filter (0.45 μm). The filtrate (20 μL) was injected on a symmetry C18 column (Waters), which was eluted with a mobile phase at a flow rate of 1 mL/min. The mobile phase consisted of 60% acetonitrile in 0.067 M phosphate buffer (pH 4.5) containing 0.1% trifluoroacetic acid, which was filtered through a 0.45 μm membrane filter before use. The eluate was monitored at 273 nm. The detection limit was about 0.2 μg/mL under our experimental conditions. Accuracy and relative standard deviations were 98.7% and 0.43%, respectively. Gilson Trilution® LC software was used for the data analysis. The retention times of celecoxib, aspart-1-ylcelecoxib (A1C), glycylcelecoxib (GC), C-glycylaspart-1-ylcelecoxib (C-GA1C), and N-glycylaspart-1-ylcelecoxib (N-GA1C) were 10.68, 3.11, 5.95, 3.16, and 2.43 minutes, respectively. HPLC analysis of aromatic acids released from glycine-conjugated aromatic acids in the cecal contents was performed as described in previous papers.17,18
Synthesis of glycine-bearing celecoxib derivatives
Preparation of 2-amino-N-(4-(5-p-tolyl-3-(trifluoromethyl)-1H-pyrazol-1-yl)phenylsulfonyl)acetamide (glycylcelecoxib)
N-(t-Boc)-glycine (0.872 g, 4.978 mmol) was dissolved in 30 mL acetonitrile and added to CDI (0.950 g, 5.8 mmol) in portions with stirring at room temperature (RT) for 10 minutes. The reaction mixture was added to celecoxib (0.5 g, 1.31 mmol [1]) dissolved in triethylamine (4.3 mL, 33 mmol) and stirred for 4 hours at 55°C. The residue obtained by evaporation was dissolved in 40 mL ethyl acetate/ether (1:3) and washed with 5% NaHCO3 and subsequently dried over anhydrous Na2SO4. The solvent was removed by flash evaporation, and N-(t-Boc)-glycine celecoxib (2) in the oily residue was precipitated by the addition of ether. Compound 2 was deprotected in 1 M HCl/acetic acid (10 mg/mL) to yield the final product: 2-amino-N-(4-(5-p-tolyl-3-(trifluoromethyl)-1H-pyrazol-1-yl)phenylsulfonyl)acetamide (GC [3]). Yield: 79%; mp: 189°C–194°C; IR (Nujol): νmax (cm−1) =1,720 (C=O, SO2NHCO); 1H-NMR (dimethyl sulfoxide [DMSO]-d6): d =2.27 (s, 3H), 3.29 (m, 2H), 7.12 (s, 1H), 7.13–7.21 (m, 4H), 7.33 (d, 2H, J =8.5 Hz), 7.80 (d, 2H, J =9.0 Hz) C19H17F3N4O3S (438); calculated: C, 52.05; H, 3.88; N, 12.78; found: C, 52.10; H, 3.89; N, 12.75.
Preparation of 1-(l-aspart-1-yl)aminosulfonyl-4-[5-(4-methylphenyl)-3-(trifluoromethyl) pyrazol-1-yl]benzene (aspart-1-ylcelecoxib)
A1C was synthesized as described in a previous paper.15 Briefly, 4-benzyl N-(t-Boc)-aspartic acid (1.61 g, 4.98 mmol, 1) was dissolved in 30 mL acetonitrile followed by the addition of CDI (0.95 g, 5.80 mmol), which was stirred at RT for 10 minutes. Celecoxib [1] (0.5 g, 1.31 mmol) and triethylamine (0.3 mL) were added to the reaction mixture and reacted at 55°C for 4 hours. After the removal of solvents by evaporation, the residue was dissolved in 40 mL ethyl acetate/ether (1:3), washed with 5% NaHCO3, and subsequently dried over anhydrous Na2SO4, which was subjected to flash evaporation to obtain compound 4. Compound 4 was deprotected with 1 M HCl/acetic acid (10 mg/mL) at RT for 3 hours, yielding compound 5, and subsequently 1 M NaOH (5 ml) at RT for 2 hours followed by acidification to obtain the final product 1-(l-aspart-1-yl)aminosulfonyl-4-[5-(4-methylphenyl)-3-(trifluoromethyl) pyrazol-1-yl]benzene (A1C, [6]) as white precipitate: physical data, including melting point, IR (Nujol), 1H-NMR (DMSO-d6), and elemental analysis, are identical with those in the previous paper.15
Preparation of 1-(N-glycyl-l-aspart-1-yl)aminosulfonyl-4-[5-(4-methylphenyl)-3-(trifluoromethyl) pyrazol-1-yl]benzene (N-GA1C)
N-(t-Boc)-glycine (0.33 g, 1.9 mmol) was dissolved in 12 mL acetonitrile followed by the addition of CDI (0.36 g, 2.21 mmol), which was stirred at RT for 10 minutes. Compound 5 (0.30 g, 0.5 mmol) and triethylamine (1.64 mL, 12.6 mmol) were added to the reaction mixture for 4 hours at 55°C. After the removal of solvents by evaporation, the residue was dissolved in 20 mL ethyl acetate/ether (1:3), washed with 5% NaHCO3, and subsequently dried over anhydrous Na2SO4. The solvent was removed by flash evaporation to obtain compound 7. Compound 7 was deprotected in 1 M HCl/acetic acid (10 mg/mL) at RT for 3 hours and subsequently 1 M NaOH (5 ml) for 2 hours, followed by acidification to yield the final product 1-(N-glycyl-l-aspart-1-yl)aminosulfonyl-4-[5-(4-methylphenyl)-3-(trifluoromethyl) pyrazol-1-yl]benzene (N-GA1C [8]) as white precipitate. Yield: 27%; mp: 154°C–158°C; IR (Nujol): νmax (cm−1) =1,700 (C=O, SO2NHCO), 1,668 (C=O, amide), 1,596 (C=O, carboxylic) 1H-NMR (DMSO-d6): δ =2.30 (s, 3H), 2.35 (dd, 1H, J =7.5, 15.5 Hz), 2.61 (dd, 1H, J =5.5, 12.5 Hz), 4.02 (t, 2H, J =5.0 Hz), 4.4 (m, 1H), 7.13 (s, 1H), 7.14–7.21 (m, 4H), 7.51 (d, 2H, J =8.5 Hz), 7.85 (d, 2H, J =8.0 Hz); C23H22F3N5O6S (553.12); calculated: C, 49.91; H, 4.01; N, 12.65; found: C, 49.77; H, 3.97; N, 12.51.
Preparation of 1-[(2S)-2-(amino)-3-[(carboxymethyl)carbamoyl]propanoyl]amino sulfonyl-4-[5-(4-methylphenyl)-3(trifluoromethyl)pyrazol-1-yl]benzene (C-GA1C)
The benzyl group of 4-benzyl N-(t-Boc)-aspart-1-yl celecoxib (4) was removed in 1 M NaOH solution to yield compound 9. Compound 9 (0.29 g, 0.5 mmol) was dissolved in 12 mL acetonitrile followed by the addition of CDI (0.09 g, 0.55 mmol), which was stirred at RT for 10 minutes. O-benzylglycine (0.33 g, 2 mmol) and triethylamine (0.41 mL, 3.15 mmol) were added to the reaction mixture and stirred at 50°C for 6 hours. After removal of the solvent by evaporation, the residue was dissolved in 20 mL ethyl acetate/ether (1:3), washed with 0.1 M HCl, and subsequently dried over anhydrous Na2SO4, which was subjected to flash evaporation to obtain compound 10. Compound 10 was deprotected in 1 M HCl/acetic acid (10 mg/mL) at RT for 3 hours and subsequently 1 M NaOH for 2 hours followed by acidification to obtain the final product 1-[(2S)-2-(amino)-3-[(carboxymethyl)carbamoyl]propanoyl]aminosulfonyl-4-[5-(4-methylphenyl)-3 (trifluoromethyl)pyrazol-1-yl]benzene (C-GA1C [11]) as white precipitate. Yield: 20%; mp: 140°C–143°C; IR (Nujol): νmax (cm−1) =1,715 (C=O, SO2NHCO), 1,679 (C=O, amide), 1,595 (C=O, carboxylic); 1H-NMR (DMSO-d6): δ =2.29 (s, 3H), 1.96 (dd, 1H, J =8, 16 Hz), 2.3 (dd, 1H, J =4, 15 Hz), 3.76 (m, 2H), 4.0 (m, 1H), 7.16 (s, 1H), 7.15–7.21 (m, 4H), 7.51 (d, 2H, J =9.0 Hz), 7.85 (d, 2H, J =8.5 Hz); C23H22F3N5O6S (553.12); calculated: C, 49.91; H, 4.01; N, 12.65; found: C, 49.81; H, 3.99; N, 12.54.
Apparent partition coefficient
To each solution of celecoxib derivatives (10 mL, 500 μM) in pH 6.8 isotonic phosphate buffer presaturated with 1-octanol, 10 mL of 1-octanol presaturated with the phosphate buffer was added. The mixture was shaken for 1 hour and left at 37°C for 3 hours. The concentration of each celecoxib derivative in the aqueous phase was analyzed by HPLC. The apparent partition coefficients were calculated by employing the equation (Co–Cw)/Cw, where Co and Cw represent the initial and equilibrium concentration of the drug in the aqueous phase, respectively.
Chemical stability
Each solution of celecoxib derivatives in pH 1.2 hydrochloric acid buffer or in pH 6.8 isotonic phosphate buffer (500 μM) was incubated at 37°C for 10 hours. At a predetermined time interval, a 20 μL portion of each solution was removed and the concentrations of celecoxib and celecoxib derivatives analyzed by HPLC.
Incubation of amino acid-conjugated derivatives in suspensions of gastrointestinal tract contents
Male Sprague Dawley rats (250–260 g; Samtako, Osan, South Korea) were killed by carbon dioxide and a midline incision was made. The contents of proximal small intestine, distal small intestine, and cecum were collected separately and suspended in pH 6.8 isotonic phosphate buffer to 20% (w/v). In a microtube, each celecoxib derivative in the buffer (0.5 mL, 1 mM) was added to the suspension (0.5 mL) and incubated at 37°C under nitrogen (for incubation in the cecal contents). At an appropriate time interval, the samples were extracted with ethyl acetate (0.5 mL) followed by centrifugation at 6,000 g for 5 minutes. Methanol (1.0 mL) was added to the residue obtained from evaporation of the organic layer (0.1 mL), vortexed, and centrifuged at 20,000 g at 4°C for 10 minutes. The concentration of celecoxib in a 20 μL portion of the supernatant was determined by HPLC. For analysis of benzoic acid, salicylic acid, and 5-aminosalicylic acid, samples were centrifuged at 6,000 g for 3 minutes, skipping the extraction process. Methanol (0.9 mL) was added to the supernatant (0.1 mL), vortexed, and centrifuged at 20,000 g at 4°C for 10 minutes. The concentration of the aromatic acids in the supernatants (20 μL) was analyzed by HPLC.
Western blot
Cells were lysed to obtain whole-cell lysates as described previously.19 Cell lysates were electrophoretically separated using 7.5% or 10% gels. Proteins were transferred to nitrocellulose membranes (Protran; Schleicher and Schuell BioScience Inc, Keene, NH, USA). COX-2 and iNOS proteins in cell lysates and tissue homogenates were detected using a monoclonal anti-COX-2 antibody (Cell Signaling Technology Inc, Danvers, MA, USA) and anti-iNOS (NOS-2) antibody (Santa Cruz Biotechnology Inc, Dallas, TX, USA). Signals were visualized using the SuperSignal chemiluminescence substrate (Thermo Fisher Scientific, Waltham, MA, USA). Experiments were performed in duplicate, and equivalent loading was confirmed by probing the blots anti-α-tubulin antibody (Santa Cruz Biotechnology).
Luciferase assay
Cells were plated in six-well plates to be 50%–60% confluent on the day of transfection with either NFκB-dependent luciferase plasmid (0.5 μg, a gift from Dr. M. Birrer, National Cancer Institute/National Institute of Health, USA). FuGene (Hoffman-La Roche Ltd, Basel, Switzerland) was used as a transfection reagent. At 24 hours posttransfection, cells were treated with TNFα (10 ng/mL) in the presence of the reagent at the indicated concentrations in the figure legends. Cells were lysed 6 hours later, and luciferase activities were measured and normalized to cytomegalovirus Renilla luciferase activities using a Dual Luciferase Reporter Assay System (Promega Corporation, Fitchburg, WI, USA).
IL-8 ELISA
For analysis of IL-8 (neutrophil chemotactic factor) in cell-culture supernatants, HCT116 cells were treated with TNFα in the presence of various concentrations of drugs, and an appropriate volume of the supernatants was subjected to IL-8 enzyme-linked immunosorbent assay (ELISA; R&D Systems).
Statistical analysis
The results are expressed as means ± standard error of the mean. One-way analysis of variance followed by Tukey’s honest significant difference test was used to test for differences between data. Differences with P<0.05 were considered significant. XLStat® software (Addinsoft Inc, New York, NY, USA) was used for the statistical analysis.
Results
Design and synthesis of potential colon-specific mutual prodrugs of celecoxib
Colon-targeted celecoxib is more effective than conventional celecoxib in ameliorating rat colitis.16 To improve further the therapeutic efficacy of colon-targeted celecoxib, glycine was selected as a colon-specific carrier for colon-targeted delivery of celecoxib. In the inflamed colon, glycine is very likely converted to glycine chloramine, which has anti-inflammatory activity.8 Glycine was conjugated to celecoxib directly or via aspartic acid to yield colon-specific mutual prodrugs. As shown in Figure 1A and B, A1C (6) and three glycine-bearing celecoxib derivatives – GC (3), C-GA1C (11), and N-GA1C (8) – were synthesized. Formation of the final products was confirmed from the spectral data of IR, 1H-NMR, and elemental analysis.
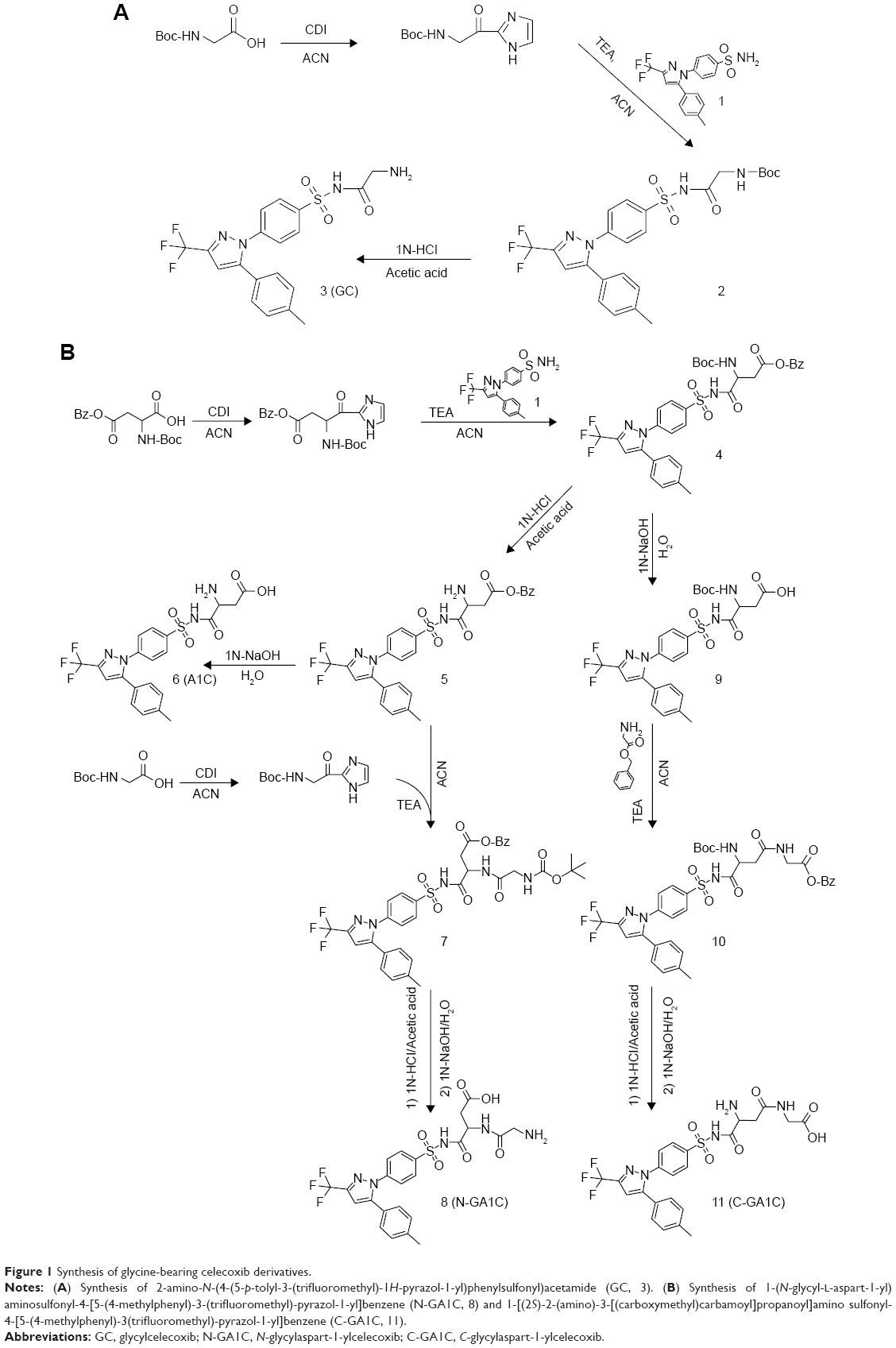 | Figure 1 Synthesis of glycine-bearing celecoxib derivatives. |
Chemical stability and apparent partition coefficient
Each celecoxib derivative was incubated in pH 1.2 and pH 6.8 buffer solutions, which represent the pH of the stomach and small intestine, respectively. Neither the concentration of the celecoxib derivatives changed nor was celecoxib detected during 10 hours’ incubation at 37°C. Apparent partition coefficients of celecoxib, GC, N-GA1C, and C-GA1C in 1-octanol/isotonic phosphate buffer were 4.86, 1.2, −0.31, and 0.34, respectively. These results indicate that celecoxib derivatives might be chemically stable during the transit through the upper intestine, and conjugation of celecoxib with the amino acids lowered partition coefficients.
Incubation of the celecoxib derivatives in the contents of small intestine and cecum
For a prodrug to be colon-specific, the prodrug should be chemically and enzymatically stable during the transit of the upper gastrointestinal tract (stomach and small intestine) and be deconjugated to liberate the active agent at the large intestine.20 To test this in vitro, glycine-bearing celecoxib derivatives were incubated with the small intestinal contents or cecal contents of rats, and changes in levels of celecoxib and the celecoxib derivatives were monitored. As shown in Figure 2A, GC and C-GA1C were resistant to degradation in the proximal and distal small intestinal contents, while N-GA1C was substantially cleaved to release celecoxib. In contrast, all the celecoxib derivatives were degraded to liberate celecoxib in the cecal content (Figure 2B). The celecoxib-release rates in the cecal contents were in the order of N-GA1C > C-GA1C > GC, which corresponded to the disappearance rates of the derivatives. Celecoxib released from N-GA1C, C-GA1C, and GC was 100% (4 hours after incubation), 65%, and 30% of the dose, respectively, 24 hours after incubation. To examine whether the cecal conversion of the derivatives to celecoxib took place by microbial enzymes, the same experiments were performed using autoclaved cecal contents. Autoclaving of cecal contents inactivates microbial enzymes that participate in colonic metabolisms.21 In contrast, no change in the levels of the celecoxib derivatives was observed in the autoclaved cecal contents throughout the experimental period (10 hours). These results suggest that GC and C-GA1C administered orally could be delivered to (without significant loss in the upper intestine) and liberate celecoxib in the large intestine.
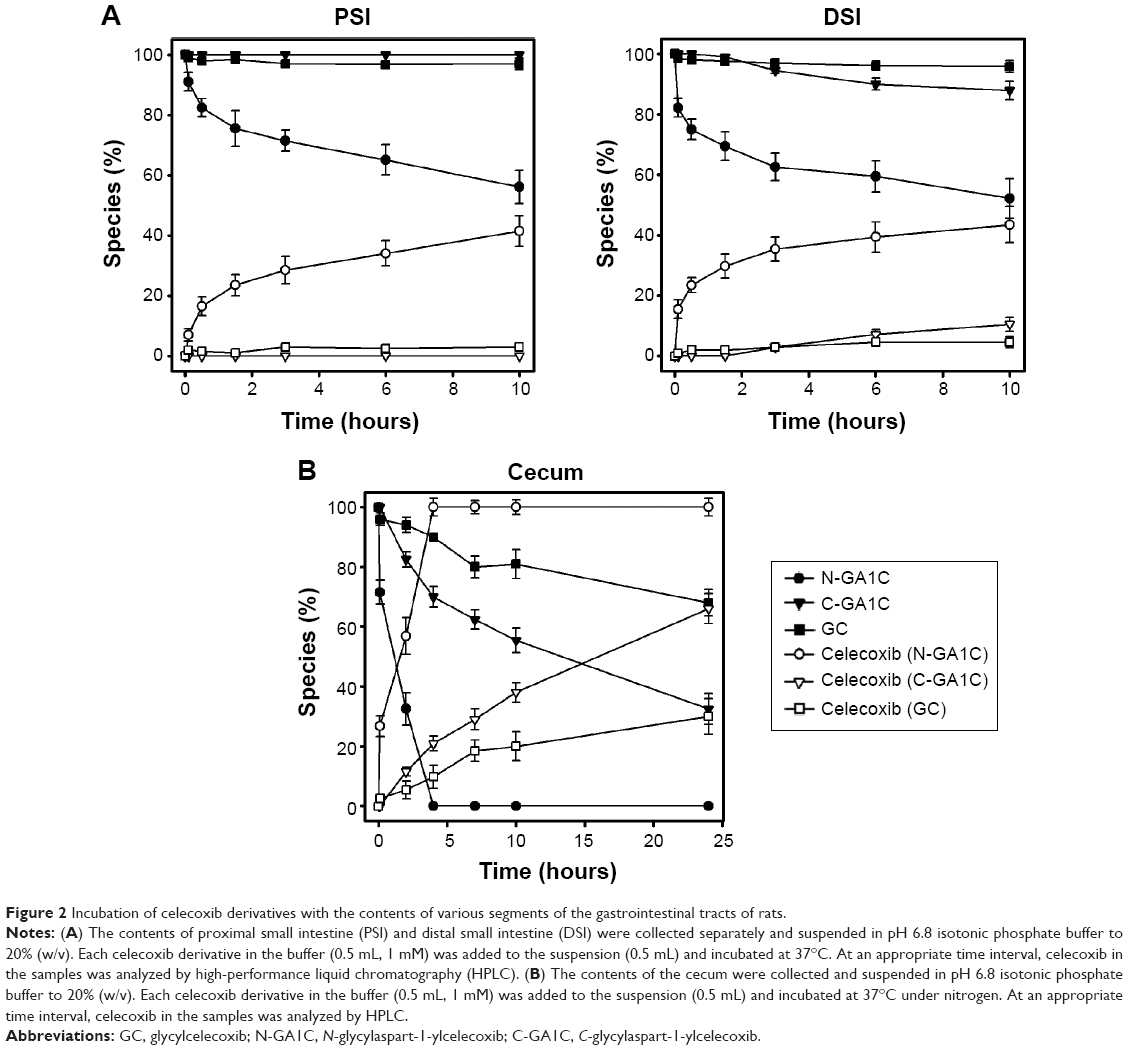 | Figure 2 Incubation of celecoxib derivatives with the contents of various segments of the gastrointestinal tracts of rats. |
C-GA1C likely liberates glycine in the cecal contents
Since the celecoxib derivatives were designed as a mutual prodrug liberating celecoxib and glycine to act together against inflammation, it is important whether such celecoxib derivatives provide glycine along with celecoxib. For GC, it is certain that the derivative delivered to the large intestine should release glycine as well as celecoxib. For C-GA1C, this is not the case. To examine whether C-GA1C can release glycine, additional experiments were performed to speculate on a degradation pathway of C-GA1C in the cecal contents. A1C, the celecoxib derivative obtained when glycine is cleaved off C-GA1C and N-GA1C, was incubated in the cecal contents, and its degradation and celecoxib release were compared to those of C-GA1C and N-GA1C. As shown in Figure 3A, the rates of the derivatives’ degradation corresponded to those of celecoxib production, and the order of the rates was N-GA1C > A1C > C-GA1C. A1C production was also monitored during incubation of N-GA1C and C-GA1C with the cecal contents. A1C was not detected in the cecal contents. These results suggest that N-GA1C and C-GA1C undergo a one-step degradation pathway to release celecoxib without conversion to A1C, likely being cleaved between the dipeptides C-(aspart-4-yl)glycine or N-glycylaspartic acid and celecoxib. According to the speculated degradation pathway of C-GA1C, it is possible that C-(aspart-4-yl)glycine, the carrier of C-GA1C, is released without further degradation to glycine and aspartic acid. To examine the possibility that the amide bond in the dipeptide carrier is susceptible to colonic microbial enzymes, aromatic acid–glycine conjugates that have the same linkage type as C-(aspart-4-yl)glycine, N-benzoylglycine, N-salicyloylglycine, and N-(5-aminosalicyloyl)glycine were subjected to release experiments in the cecal contents. As shown in Figure 3B, aromatic acid–glycine conjugates were susceptible to cecal degradation, leading to release of the aromatic acids. Therefore, it is likely that N-(aspart-4-yl)glycine released from C-GA1C in the large intestine is cleaved to liberate glycine.
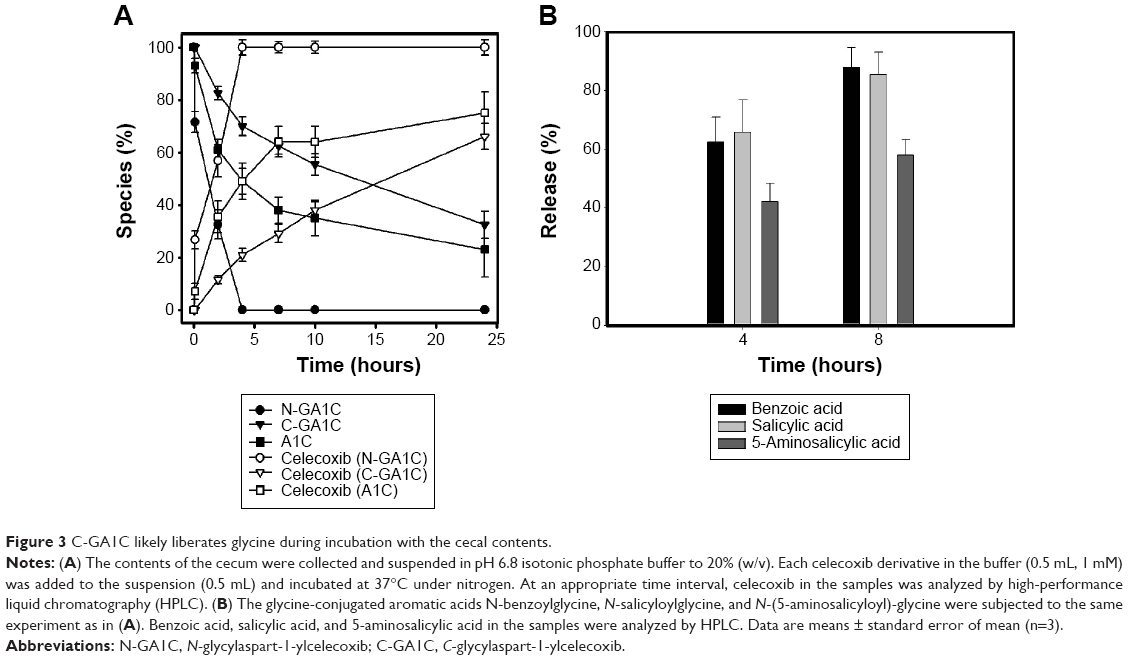 | Figure 3 C-GA1C likely liberates glycine during incubation with the cecal contents. |
Glycine chloramine and celecoxib elicit an additive inhibitory effect against NFκB
Our data suggest that C-GA1C and GC delivered to the target site could liberate celecoxib and glycine. Since HOCl is generated in the inflamed colon and reacts with taurine to produce taurine chloramine, glycine liberated from C-GA1C is very likely changed to glycine chloramine.8 Celecoxib as well as glycine chloramine inhibits NFκB, a transcription factor involved in the regulation of inflammation and immunity,5,22,23 in various types of cells. NFκB activity is elevated in the inflamed intestine, playing an important role in the progression of chronic intestinal inflammation.4 It was hypothesized that glycine and celecoxib may elicit an additive activity by acting together on the common target – NFκB. To test this hypothesis, we first examined whether glycine chloramine inhibited NFκB in human colon carcinoma HCT116 cells and murine macrophage RAW264.7 cells. In our previous paper, inhibitory effect of celecoxib on NFκB was demonstrated in the cell lines.16 Since colon-specific prodrugs of celecoxib can produce more than 100 μM celecoxib to the large intestine,15 cell experiments were performed between 10 and 100 μM. Human colon carcinoma HCT116 cells were transfected with an NFκB-dependent luciferase plasmid followed by treatment with TNFα in the presence of glycine chloramine and/or celecoxib. As shown in Figure 4A, TNFα induced luciferase up to 18.5-fold, glycine chloramine attenuated TNF-mediated induction of the luciferase, and combined celecoxib–glycine chloramine treatment showed an additive inhibitory effect on the expression of NFκB-dependent luciferase. To confirm this, cells were treated with TNFα (for HCT116 cells) or LPS (for RAW264.7 cells) in the presence of glycine chloramine and/or celecoxib, and secretion of IL-8, a chemoattractant for neutrophil, and induction of COX-2 and iNOS protein were monitored. IL-8, COX-2 and iNOS are NFκB target genes products involved in gut inflammation.19 As shown in Figure 4B, consistent with the luciferase results, various concentrations of glycine chloramine suppressed TNFα secretion of IL-8 and LPS induction of COX-2 and iNOS proteins. Combined celecoxib–glycine chloramine treatment elicited an additive inhibitory effect on production of the NFκB target gene products. The inhibitory effects of celecoxib on NFκB are shown in Figure S1.
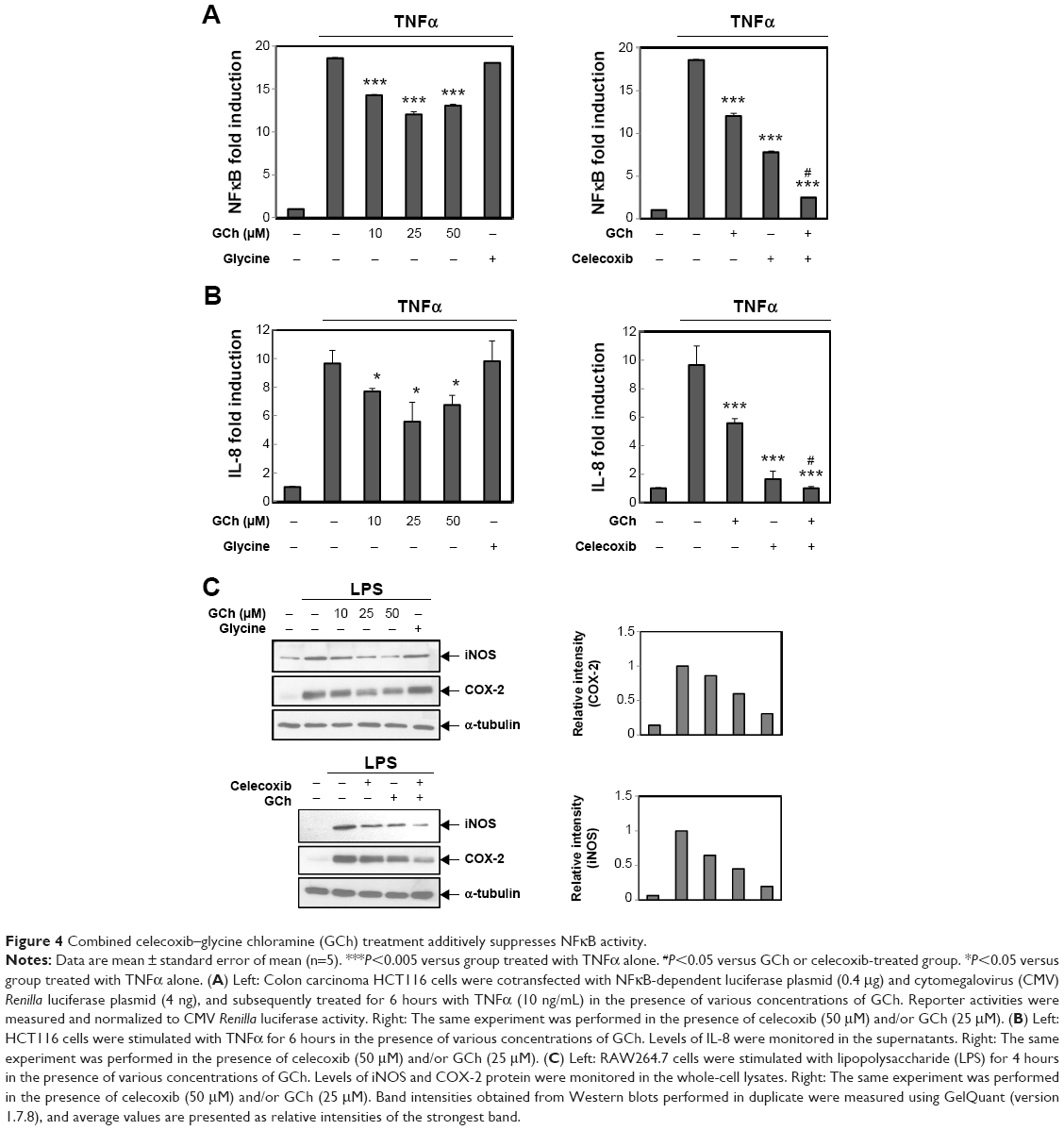 | Figure 4 Combined celecoxib–glycine chloramine (GCh) treatment additively suppresses NFκB activity. |
Discussion
Glycine-bearing celecoxib derivatives were designed and prepared as a colon-specific mutual prodrug acting on NFκB. The derivatives were chemically stable in pH conditions representing the stomach and intestine, and had lower partition coefficients than celecoxib. GC and C-GA1C were resistant to degradation in the small intestinal contents, while N-GA1C liberated a substantial amount of celecoxib. In contrast, all the derivatives generated celecoxib in the cecal contents. In human colon carcinoma cells, celecoxib or glycine chloramine inhibited NFκB activity, and combined treatment with the two agents elicited an additive inhibitory activity.
The (bio)chemical stability of glycine-bearing celecoxib derivatives in the contents of the gastrointestinal tract suggests that GC and C-GA1C but not N-GA1C are appropriate for colonic delivery of celecoxib. N-GA1C is very likely to release a substantial amount of celecoxib before reaching the large intestine. Based on lower partition coefficients and greater cecal conversion of C-GA1C to celecoxib, C-GA1C may be more efficient than GC in increasing therapeutic availability of celecoxib in the large intestine, which depends on the restriction of systemic absorption and colonic conversion rate of a colon-specific prodrug.20
Since glycine in the cecal contents was not able to be quantified directly, celecoxib released from GC and C-GA1C was considered to reflect glycine release. Celecoxib release from GC, in which glycine is directly conjugated to celecoxib, should come along with glycine. However, for C-GA1C, in which aspartic acid acts as a linker between celecoxib and glycine, it is possible that C-aspart-4-ylglycine is cleaved off C-GA1C to release celecoxib without further degradation to glycine and aspartic acid. Actually, our data showing that A1C was not detected during incubation of C-GA1C and conversion of A1C to celecoxib occurred faster than that of C-GA1C in the cecal contents suggest that C-GA1C could be degraded to liberate celecoxib and C-aspart-4-ylglycine. Nonetheless, this may not cause a problem in generating glycine in the large intestine, since C-aspart-4-ylglycine is likely susceptible to cecal metabolism, resulting in glycine release. This is supported by our data showing that glycine-conjugated aromatic acids with the same bond type as C-aspart-4-ylglycine were efficiently metabolized to release aromatic acids and glycine in the cecal contents. Although one-step degradation of C-GA1C is suggested, it is also possible that C-GA1C undergoes a sequential degradative pathway liberating A1C and glycine. No detection of A1C may be ascribed to faster conversion of A1C to celecoxib than that of C-GA1C to A1C (Figure 3A). Since this degradation pathway surely produces glycine, no further experiment was performed to clarify this possibility.
Consistent with previous papers,5,16 either glycine chloramine or celecoxib inhibited NFκB in colon carcinoma cells. Moreover, the two molecules elicited an additive inhibitory effect on NFκB. Considering that NFκB is a major drug target for the treatment of colitis, C-GA1C may act as a mutual prodrug against colitis. This argument is supported by the in vitro results showing potential colon targetability and a previous paper strongly suggesting conversion of glycine to glycine chloramine in the inflamed colon.8 Furthermore, concentrations of glycine and celecoxib used for cell-based experiments may be pharmacologically relevant, given that colon-specific prodrugs of celecoxib produces more than 100 μM celecoxib in the large intestine.15 However, in vivo experiments are needed to explicate the therapeutic feasibility of the prodrugs. Taken together, our data suggest that C-GA1C is a potential colon-specific mutual prodrug acting against colitis.
Acknowledgment
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology 2012R1A1A2003641).
Disclosure
The authors report no conflicts of interest in this work.
References
Sanchez-Munoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008;14(27):4280–4288. | ||
Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. 2002;2(10):725–734. | ||
Atreya I, Atreya R, Neurath MF. NF-κB in inflammatory bowel disease. J Intern Med. 2008;263(6):591–596. | ||
Neurath MF, Becker C, Barbulescu K. Role of NF-κB in immune and inflammatory responses in the gut. Gut. 1998;43(6):856–860. | ||
Midwinter RG, Cheah FC, Moskovitz J, Vissers MC, Winterbourn CC. IκB is a sensitive target for oxidation by cell-permeable chloramines: inhibition of NF-κB activity by glycine chloramine through methionine oxidation. Biochem J. 2006;396(1):71–78. | ||
Barua M, Liu Y, Quinn MR. Taurine chloramine inhibits inducible nitric oxide synthase and TNF-α gene expression in activated alveolar macrophages: decreased NF-κB activation and IκB kinase activity. J Immunol. 2001;167(4):2275–2281. | ||
Kanayama A, Inoue J, Sugita-Konishi Y, Shimizu M, Miyamoto Y. Oxidation of IκBα at methionine 45 is one cause of taurine chloramine-induced inhibition of NF-κB activation. J Biol Chem. 2002;277(27):24049–24056. | ||
Kim H, Jeon H, Kong H, et al. A molecular mechanism for the anti-inflammatory effect of taurine-conjugated 5-aminosalicylic acid in inflamed colon. Mol Pharmacol. 2006;69(4):1405–1412. | ||
Kim C, Cha YN. Production of reactive oxygen and nitrogen species in phagocytes is regulated by taurine chloramine. Adv Exp Med Biol. 2009;643:463–472. | ||
Cuzzocrea S, Mazzon E, Serraino I, et al. Celecoxib, a selective cyclo-oxygenase-2 inhibitor reduces the severity of experimental colitis induced by dinitrobenzene sulfonic acid in rats. Eur J Pharmacol. 2001;431(1):91–102. | ||
El-Medany A, Mahgoub A, Mustafa A, Arafa M, Morsi M. The effects of selective cyclooxygenase-2 inhibitors, celecoxib and rofecoxib, on experimental colitis induced by acetic acid in rats. Eur J Pharmacol. 2005;507(1–3):291–299. | ||
Hegazi RA, Mady HH, Melhem MF, Sepulveda AR, Mohi M, Kandil HM. Celecoxib and rofecoxib potentiate chronic colitis and premalignant changes in interleukin 10 knockout mice. Inflamm Bowel Dis. 2003;9(4):230–236. | ||
Mahadevan U, Loftus EV Jr, Tremaine WJ, Sandborn WJ. Safety of selective cyclooxygenase-2 inhibitors in inflammatory bowel disease. Am J Gastroenterol. 2002;97(4):910–914. | ||
Sandborn WJ, Stenson WF, Brynskov J, et al. Safety of celecoxib in patients with ulcerative colitis in remission: a randomized, placebo-controlled, pilot study. Clin Gastroenterol Hepatol. 2006;4(2):203–211. | ||
Lee Y, Kim J, Kim H, et al. N-succinylaspart-1-yl celecoxib is a potential colon-specific prodrug of celecoxib with improved therapeutic properties. J Pharm Sci. 2012;101(5):1831–1842. | ||
Lee Y, Kim W, Hong S, et al. Colon-targeted celecoxib ameliorates TNBS-induced rat colitis: a potential pharmacologic mechanism and therapeutic advantages. Eur J Pharmacol. 2014;726:49–56. | ||
Jung YJ, Lee JS, Kim YM. Synthesis and in vitro/in vivo evaluation of 5-aminosalicyl-glycine as a colon-specific prodrug of 5-aminosalicylic acid. J Pharm Sci. 2000;89(5):594–602. | ||
Kong H, Kim H, Do H, et al. Structural effects of N-aromatic acyl-amino acid conjugates on their deconjugation in the cecal contents of rats: implication in design of a colon-specific prodrug with controlled conversion rate at the target site. Biopharm Drug Dispos. 2011;32(6): 343–354. | ||
Hong S, Yum S, Yoo HJ, et al. Colon-targeted cell-permeable NFκB inhibitory peptide is orally active against experimental colitis. Mol Pharm. 2012;9(5):1310–1319. | ||
Jung Y, Kim YM. What should be considered on design of a colon-specific prodrug? Expert Opin Drug Deliv. 2010;7(2):245–258. | ||
Kim H, Kong H, Choi B, et al. Metabolic and pharmacological properties of rutin, a dietary quercetin glycoside, for treatment of inflammatory bowel disease. Pharm Res. 2005;22(9):1499–1509. | ||
Funakoshi-Tago M, Shimizu T, Tago K, et al. Celecoxib potently inhibits TNFα-induced nuclear translocation and activation of NF-κB. Biochem Pharmacol. 2008;76(5):662–671. | ||
Shishodia S, Koul D, Aggarwal BB. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates TNF-induced NF-κB activation through inhibition of activation of IκBα kinase and Akt in human non-small cell lung carcinoma: correlation with suppression of COX-2 synthesis. J Immunol. 2004;173(3):2011–2022. |
Supplementary material
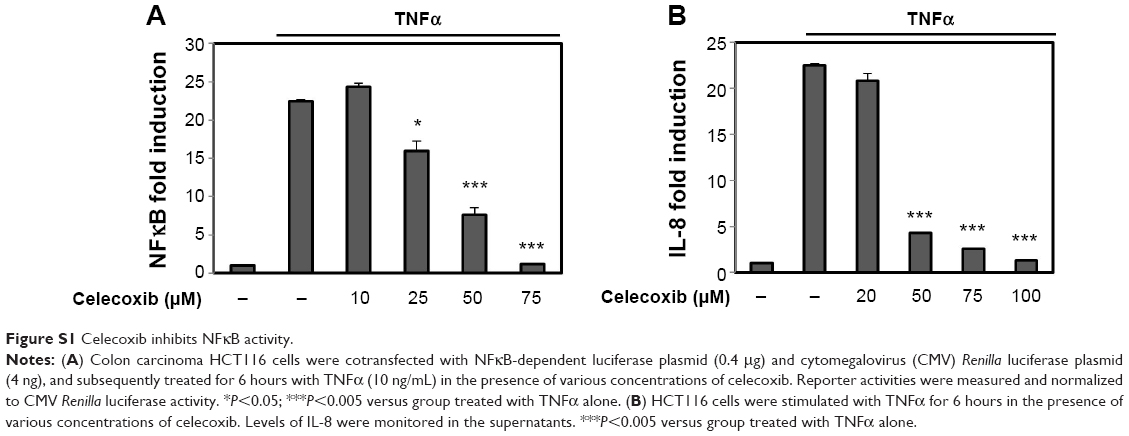 | Figure S1 Celecoxib inhibits NFκB activity. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.