")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Evaluation of effects of various drugs on platelet functions using phorbol 12-myristate 13-acetate-induced megakaryocytic human erythroid leukemia cells
Authors Tada T, Aki K, Oboshi W, Kawazoe K, Yasui T, Hosoi E
Received 27 June 2016
Accepted for publication 9 August 2016
Published 26 September 2016 Volume 2016:10 Pages 3099—3107
DOI https://doi.org/10.2147/DDDT.S115910
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Tomoki Tada,1 Kensaku Aki,2 Wataru Oboshi,1,3 Kazuyoshi Kawazoe,4 Toshiyuki Yasui,5 Eiji Hosoi2
1Subdivision of Biomedical Laboratory Sciences, Graduate School of Health Sciences, Tokushima University, 2Department of Cells and Immunity Analytics, Institute of Biomedical Sciences, Tokushima University Graduate School, Tokushima, 3Department of Medical Technology, Kagawa Prefectural University of Health Sciences, Kagawa, 4Department of Clinical Pharmacy Practice Pedagogy, Institute of Biomedical Sciences, 5Department of Reproductive and Menopausal Medicine, Institute of Biomedical Sciences, Tokushima University Graduate School, Tokushima, Japan
Background: The hyperfunction and activation of platelets have been strongly implicated in the development and recurrence of arterial occlusive disease, and various antiplatelet drugs are used to treat and prevent such diseases. New antiplatelet drugs and many other drugs have been developed, but some drugs may have adverse effects on platelet functions.
Objective: The aim of this study was to establish an evaluation method for evaluating the effect and adverse effect of various drugs on platelet functions.
Materials and methods: Human erythroid leukemia (HEL) cells were used after megakaryocytic differentiation with phorbol 12-myristate 13-acetate as an alternative to platelets. Drugs were evaluated by changes in intracellular Ca2+ concentration ([Ca2+]i) mobilization in Fura2-loaded phorbol 12-myristate 13-acetate-induced HEL cells. Aspirin and cilostazol were selected as antiplatelet drugs and ibuprofen and sodium valproate as other drugs.
Results: There was a positive correlation between [Ca2+]i and platelet aggregation induced by thrombin. Aspirin (5.6–560 µM) and cilostazol (5–10 µM) significantly inhibited thrombin-induced increases in [Ca2+]i in a concentration-dependent manner. On the other hand, ibuprofen (8–200 µM) and sodium valproate (50–1,000 µg/mL) also significantly inhibited thrombin-induced increases in [Ca2+]i in a concentration-dependent manner. Furthermore, the interaction effects of the simultaneous combined use of aspirin and ibuprofen or sodium valproate were evaluated. When the inhibitory effect of aspirin was higher than that of ibuprofen, the effect of aspirin was reduced, whereas when the inhibitory effect of aspirin was lower than that of ibuprofen, the effect of ibuprofen was reduced. The combination of aspirin and sodium valproate synergistically inhibited thrombin-induced [Ca2+]i.
Conclusion: It is possible to induce HEL cells to differentiate into megakaryocytes, which are a useful model for the study of platelet functions, and the quantification of the inhibition of thrombin-induced increases in [Ca2+]i is applicable to the evaluation of the effects of various drugs on platelets.
Keywords: platelets, intracellular Ca2+ concentration ([Ca2+]i), aspirin, cilostazol, ibuprofen, sodium valproate
Corrigendum for this paper has been published
Introduction
Platelets play an important role in hemostasis through adhesion and aggregation, and these functions are influenced by diseases and drugs. For example, the hyperfunction and activation of platelets have been strongly implicated in the development and recurrence of arterial occlusive disease.1–4 In the mechanisms responsible for platelet activation, it is important to regulate Ca2+ mobilization through the activation of phospholipase C5 and the production of thromboxane A2 (TXA2), which strongly induces platelet cohesion.6 Different substances have the ability to activate platelets in vivo, with thrombin being considered important in diseases such as thrombosis.7,8 On the other hand, various antiplatelet drugs are used in the treatment and prevention of these diseases.9 New antiplatelet drugs and many other drugs have also been developed, but some of the drugs may have adverse effects on platelet functions. Therefore, the effects and adverse effects of various drugs on platelets need to be examined in more detail.
Platelet activation is regulated through intracellular Ca2+, which plays a pivotal role as a second messenger in the platelet aggregation mechanism.10,11 In order to clarify their state, it is important to evaluate changes in intracellular Ca2+ concentrations ([Ca2+]i) in platelets. Furthermore, changes in [Ca2+]i induced by thrombin have been observed in human erythroid leukemia (HEL) cells and platelets, and HEL has been shown to cause Ca2+ mobilization induced by thrombin.12 HEL cells are progenitor cells that have the characteristics of a megakaryocyte/platelet lineage such as platelet membrane glycoprotein IIb/IIIa, glycoprotein Ib, and TXA2 receptor and may differentiate into megakaryocyte-like cells. HEL cells have become a useful model for the study of platelet functions,13–15 since anyone can study platelet functions using HEL cells anytime/anywhere without sampling blood.
In the present study, attempts were made to improve the evaluation method for evaluating the effects of various drugs (aspirin and cilostazol, antiplatelet drugs; ibuprofen, an anti-inflammatory drug that has been reported to inhibit cyclooxygenase-1 [COX-1] in the same as aspirin; and sodium valproate, an anticonvulsant that has been reported to cause impairment of platelet16) on platelet functions using phorbol 12-myristate 13-acetate (PMA)-induced megakaryocytic HEL cells as an alternative to platelets.
Materials and methods
Ethics statement
This study was approved by the ethics committee of the University of Tokushima Hospital (approval number 2072). Written informed consent was obtained from all volunteers, and all participants signed consent forms approved by the ethics committee.
Chemicals
PMA was purchased from Sigma-Aldrich Co. (St Louis, MO, USA), and thrombin was from Mochida Pharmaceutical Co., Ltd. (Tokyo, Japan). Aspirin (acetylsalicylic acid) was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan), and cilostazol was provided from Otsuka Pharmaceutical Co., Ltd. (Tokyo, Japan). Ibuprofen and sodium valproate were purchased from Sigma Chemicals (Perth, Australia). Fura2-AM was purchased from Dojindo Laboratories (Kumamoto, Japan). Dimethyl sulfoxide was purchased from Kanto Chemical Industry Co., Ltd (Tokyo, Japan). Triton X-100 was purchased from Wako Pure Chemical Industries, Ltd., acid citrate dextrose (ACD) was purchased from Terumo (Tokyo, Japan), and ethylene glycol bis(2-aminoethyl ether)-N, N, N′, N′-tetraacetic acid (EGTA) was from Sigma-Aldrich Co.
Platelet preparation
Human blood plasma and platelets were collected from five healthy adult volunteers. Blood collected from humans was mixed with ACD (blood:ACD =6:1) and centrifuged at 200× g at room temperature for 15 minutes. Supernatant platelet-rich plasma (PRP) was incubated with Fura2-AM (3 μL Fura2-AM in 1 mL PRP) at 37°C for 50 minutes in the dark. After being incubated, ACD at 15% of total volume of PRP was added, and the mixture was centrifuged at 700× g for 10 minutes. After removal of the supernatant, the platelet pellet was washed twice with 5 mL HEPES buffer (145 mM NaCl, 5 mM KCl, 1 mM MgSO4, 10 mM HEPES, 5 mM glucose, and pH 7.4)/750 μL ACD (700× g for 6 minutes). The platelet count was adjusted with HEPES buffer to 2×108/mL, and changes in [Ca2+]i and platelet aggregation intensity were measured.
Cell culture
HEL cells were used as platelet model cells.17 HEL cells were maintained in RPMI-1640 (Wako Pure Chemical Industries, Ltd.) supplemented with 10% fetal bovine serum (Biosera LTD, East Sussex, UK), 100 μg/mL penicillin G, and 100 μg/mL streptomycin at 37°C in a humidified atmosphere of 5% CO2.
Megakaryocytic differentiation of HEL cells and cell processing
Untreated HEL cells and PMA-induced HEL cells were subcultured at a density of 2×105 cells/mL. In order to induce megakaryocytic differentiation, cells were treated with 100 nM PMA (final concentration of 0.1% dimethyl sulfoxide). After being treated for 4 days, a large number of untreated HEL cells had attached to the bottom of the culture bottle. On the other hand, most PMA-induced HEL cells had attached to the bottom of the culture bottle. The supernatants of both culture bottles were removed and washed in HEPES buffer (145 mM NaCl, 5 mM KCl, 1 mM MgSO4, 10 mM HEPES, 5 mM glucose, 1 mM CaCl2, and pH 7.4) or phosphate-buffered saline (PBS) twice. Adherent cells were detached form the culture bottle by treatment with 1 mM EDTA/4Na-PBS at 37°C for 10 minutes and centrifuged at 260× g at room temperature for 5 minutes. After removal of the supernatant, cell pellets were washed three times with HEPES buffer or PBS (at 260× g for 5 minutes) and then suspended in 1 mL of HEPES buffer or PBS.
Flow cytometry analysis of surface CD41 expression
In order to quantify surface CD41 expression on untreated HEL cells and PMA-induced HEL cells, 100 μL of cells suspended in PBS (1×106 cells/mL) were incubated with 10 μL of fluorescein isothiocyanate (FITC)-conjugated anti-CD41 (Mouse Monoclonal Anti-Human CD41-FITC, Clone 5B12; DAKO Denmark A/S, Glostrup, Denmark) at 4°C for 30 minutes in the dark. A negative control was then used under the same conditions of FITC-conjugated mouse IgG isotype mAb. After the reaction had been stopped by the addition of 2,000 μL of PBS, cells were centrifuged at 260× g at room temperature for 5 minutes, and the supernatant was removed. Cells were resuspended in PBS and washed twice with PBS (at 260× g for 5 minutes). After the last wash, cells were resuspended in 500 μL of PBS and analyzed using flow cytometry (Beckman Coulter, CA, USA).
Measurement of thrombin-induced [Ca2+]i in HEL cells and human platelets
In order to measure [Ca2+]i, untreated HEL cells or PMA-induced HEL cells suspended in 1 mL of HEPES buffer were incubated with 2 μL of Fura2-AM at 37°C for 60 minutes in the dark, washed, and resuspended in the HEPES buffer at 1×106 cells/mL. The cell suspension in a volume of 480 μL, after a 50-second preincubation at 37°C, was treated with 10 μL of the different drugs being tested (antiplatelet drugs: aspirin and cilostazol, anti-inflammatory drug: ibuprofen, anticonvulsant drug: sodium valproate, and simultaneous combined use: aspirin and ibuprofen or sodium valproate) for 6 minutes. The suspended cells were stimulated with 20 μL of thrombin at 0.8–1.0 U/mL for 100 seconds, resulting in [Ca2+]i of ~200 nM, after which 100 μL of 1% Triton X-100, and then 100 μL of 100 mM EGTA were added. Fluorescence was measured at excitation wavelengths of 340 nm and 380 nm and an emission wavelength of 510 nm, and platelet aggregation was measured by light scattering after 3 minutes at an excitation wavelength of 380 nm and an emission wavelength of 400 nm using a fluorescence spectrophotometer (F-2500; Hitachi Ltd., Tokyo, Japan). [Ca2+]i and its aggregation were calculated with intracellular Ca2+ measurement software (Hitachi Ltd.) using the formula of Grynkiewicz et al.18
Statistical analysis
Data were expressed as the mean ± standard deviation. The significance of differences was analyzed using Dunnett’s test or Student’s t-test (Excel Statistics, Tokyo, Japan) and set at P<0.05.
Results
Relationship between [Ca2+]i in human platelets and platelet aggregation
Figure 1 shows the relationship between [Ca2+]i in human platelets and platelet aggregation intensity following a stimulation with thrombin (0.01–0.08 U/mL). [Ca2+]i strongly correlated with platelet aggregation intensity (r=0.946, P<0.01).
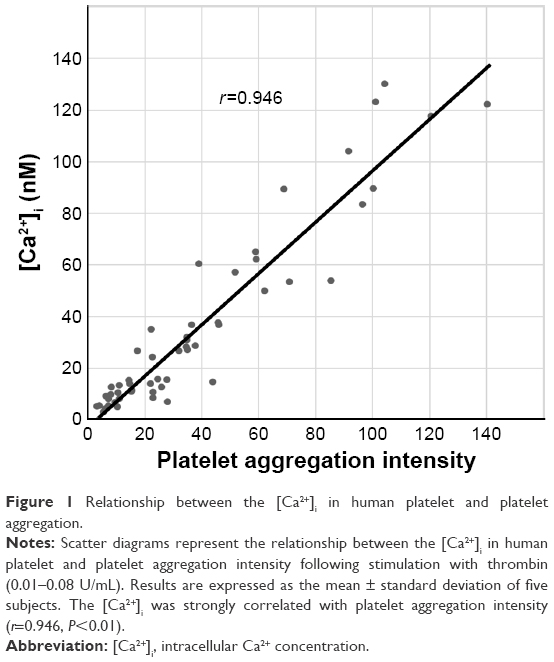 | Figure 1 Relationship between the [Ca2+]i in human platelet and platelet aggregation. |
Proliferative state and morphological changes in PMA-induced HEL cells
HEL cells consisted of small floating cells with a high nucleus/cytoplasm ratio, including cells with adhesive ability. As shown in Figure 2, adherent HEL cells also consisted of small cells with a high nucleus/cytoplasm ratio (Figure 2A). The proliferation of HEL cells treated for 4 days with PMA was suppressed, and distinct morphological changes were observed in microscopic and flow cytometric analyses; most of the small cells became larger megakaryocyte-like cells with positive multinuclear and periodic acid–Schiff staining. These cells adhered strongly to the bottom of the culture bottle (Figure 2B). Furthermore, untreated HEL cells were mainly a small cell population of gate I (Figure 2C). On the other hand, PMA-induced HEL cells were divided into three gates based on size by cell flow cytometry (Figure 2D). The expression of the surface CD41 antigen, a platelet marker, in each group was confirmed. The expression of the surface CD41 antigen was stronger on PMA-induced HEL cells than that on untreated HEL cells. Its strong expression was observed on large cells in gate II (Table 1).
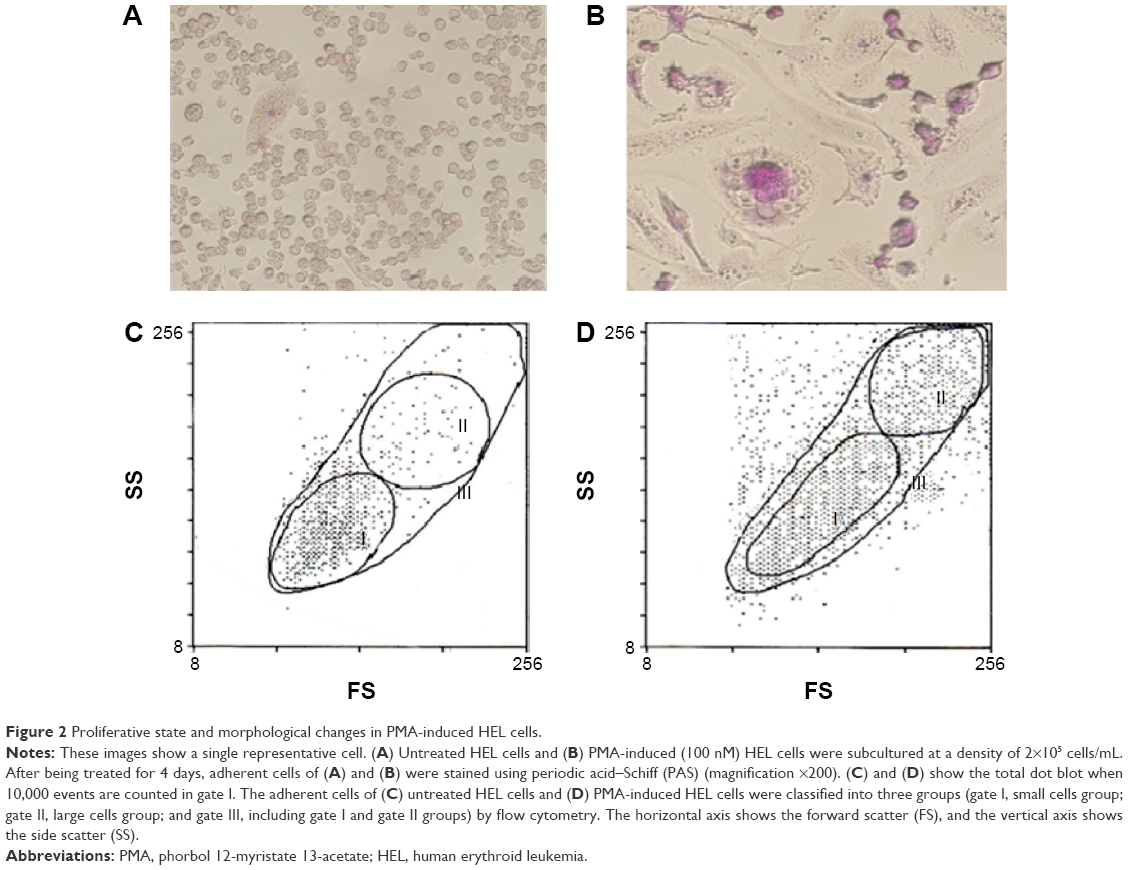 | Figure 2 Proliferative state and morphological changes in PMA-induced HEL cells. |
 | Table 1 Comparison of surface CD41 expression on untreated HEL cells and PMA-induced HEL cells after 4 days of subculturing by flow cytometry |
Effects of antiplatelet agents on [Ca2+]i induced by thrombin in untreated HEL cells or PMA-induced HEL cells
Figure 3 shows the effects of aspirin and cilostazol on thrombin-induced Ca2+ release in untreated HEL cells and PMA-induced HEL cells. Aspirin (5.6–560 μM) caused the concentration-dependent inhibition of increases in [Ca2+]i in both HEL cell types. Increases in [Ca2+]i were significantly inhibited in PMA-induced HEL cells at all concentrations tested (Figure 3A). On the other hand, cilostazol at 1–10 μM also caused the concentration-dependent inhibition of increases in [Ca2+]i in both HEL cell types, and this inhibition was stronger in PMA-induced HEL cells than in untreated HEL cells (Figure 3B).
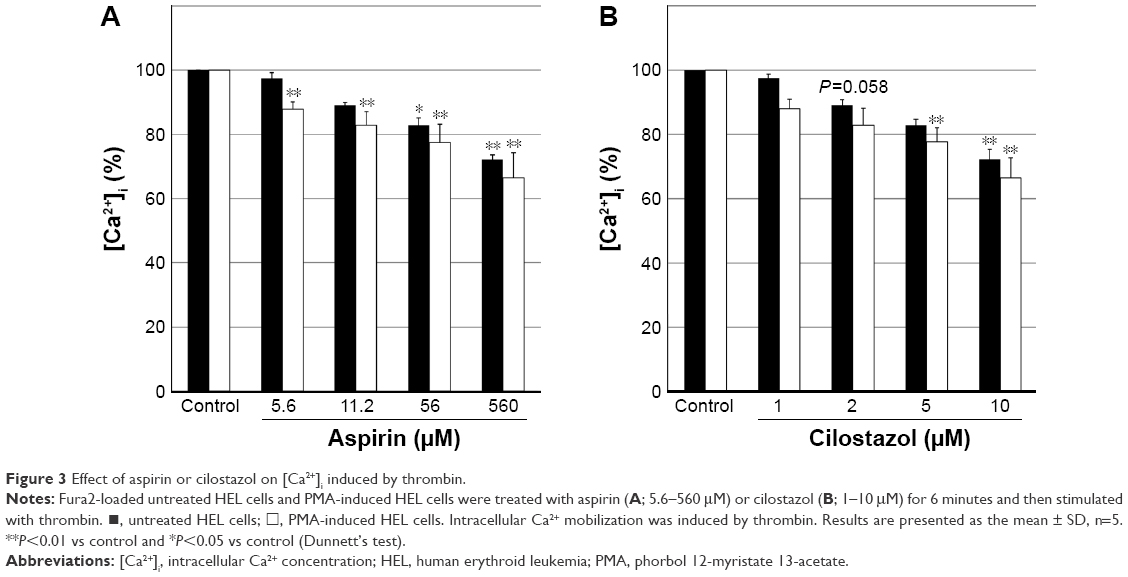 | Figure 3 Effect of aspirin or cilostazol on [Ca2+]i induced by thrombin. |
Effects of ibuprofen and sodium valproate on [Ca2+]i increases induced by thrombin in untreated HEL cells or PMA-induced HEL cells
Figure 4 shows the effects of ibuprofen and sodium valproate on thrombin-induced Ca2+ release in untreated HEL cells and PMA-induced HEL cells. Ibuprofen at 0.8–200 μM caused the concentration-dependent inhibition of increases in [Ca2+]i in both HEL cell types. Increases in [Ca2+]i were significantly inhibited in PMA-induced HEL cells at 8–200 μM (Figure 4A). Similarly, sodium valproate at 50–1,000 μg/mL also caused the concentration-dependent inhibition of increases in [Ca2+]i in both HEL cell types, with increases in [Ca2+]i being significantly inhibited in PMA-induced HEL cells at all concentrations tested (Figure 4B).
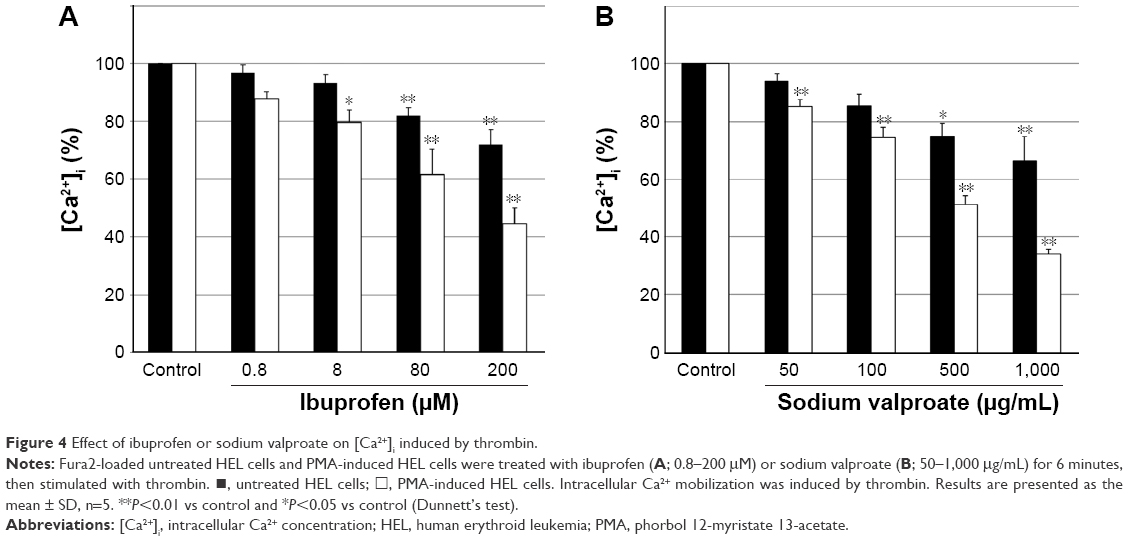 | Figure 4 Effect of ibuprofen or sodium valproate on [Ca2+]i induced by thrombin. |
Interaction effects of the simultaneous combined use of drugs on [Ca2+]i induced by thrombin in PMA-induced HEL cells
The combination experiment using PMA-induced HEL cells was carried out, since the effects and adverse effects of drugs could be widely evaluated by using PMA-induced HEL cells. Figure 5 shows the interaction effects of the simultaneous combined use of aspirin and ibuprofen or sodium valproate in PMA-induced HEL cells. As shown in Figure 5A, the simultaneous combination of 0.8 μM or 8 μM ibuprofen significantly reduced the inhibitory effects of aspirin on [Ca2+]i increases induced by thrombin, whereas the inhibitory effects of the simultaneous combination of 80 μM or 200 μM ibuprofen were significantly suppressed (Figure 5B). On the other hand, 50 μg/mL and 100 μg/mL of sodium valproate significantly increased the inhibitory effects of aspirin on [Ca2+]i increases induced by thrombin (Figure 5C).
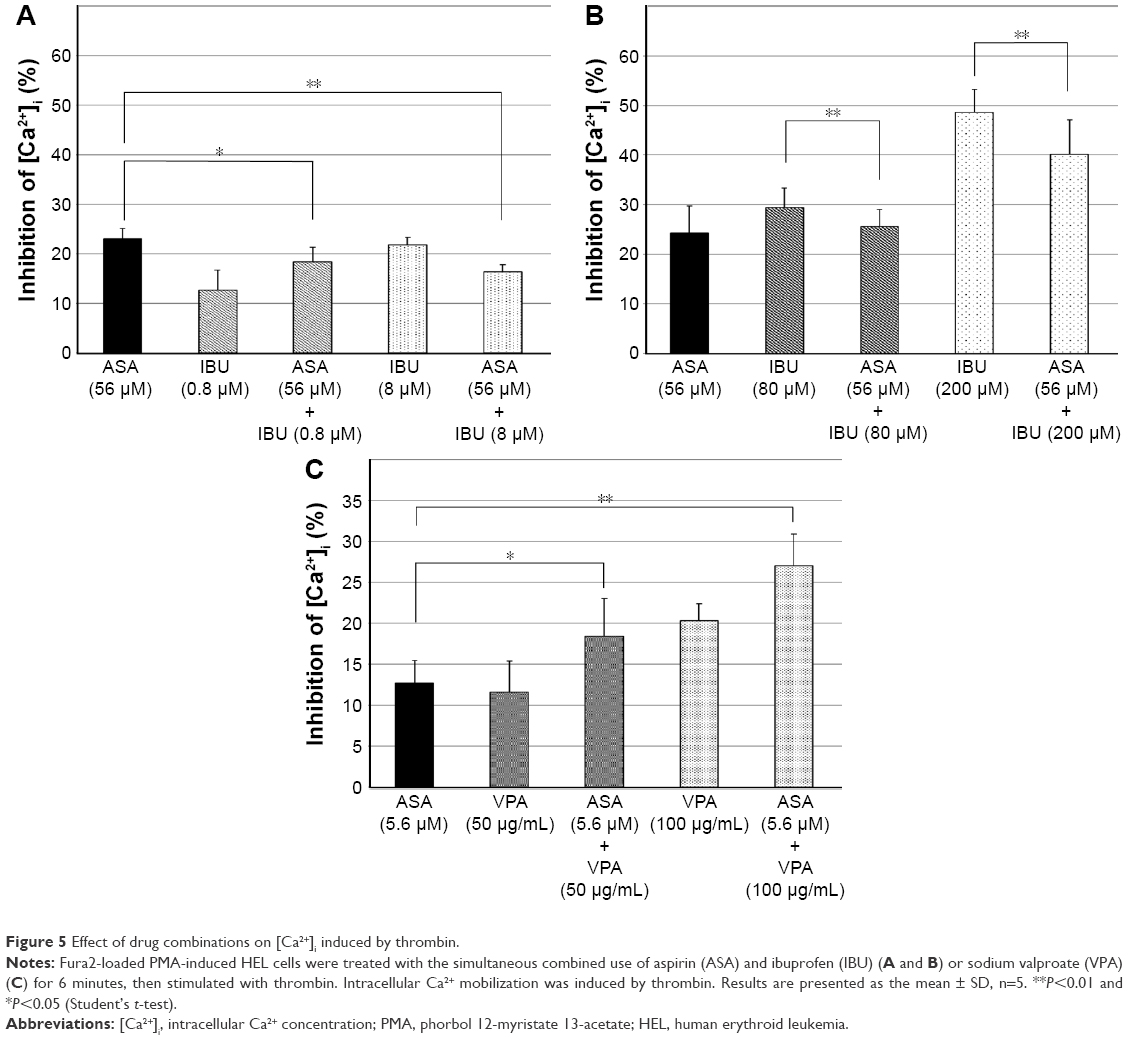 | Figure 5 Effect of drug combinations on [Ca2+]i induced by thrombin. |
Discussion
The activation and hyperfunction of platelets have recently been strongly implicated in the development and recurrence of arterial occlusive diseases, and various antiplatelet drugs are used in the treatment and prevention of these diseases, and new antiplatelet drugs and many other drugs have been developed. Therefore, the evaluation of platelet functions is of importance, particularly the effects and adverse effects of various drugs.
Platelet functions are currently assessed based on platelet aggregation with light transmission aggregometry. This is one of the methods used to measure platelet functions and has been routinely used to measure platelet aggregation activity, but such platelet aggregation is merely a secondary reaction. Platelets are activated by some factors, with thrombin being of importance, and previous studies have detected activated platelets in the blood of patients with progressive arterial thrombosis.7,8 Platelet aggregation is induced by intracellular Ca2+, which plays an important role as a second messenger in platelets. Therefore, in order to assess platelet functions, changes in [Ca2+]i need to be examined. It revealed that a positive correlation exists between [Ca2+]i and platelet aggregation induced by thrombin. Furthermore, changes in [Ca2+]i induced by thrombin were observed in HEL cells and platelets. HEL cells are progenitor cells with the ability to differentiate into megakaryocyte-like cells, and, thus, have served as a useful model for the study of platelet functions. Attempts were made to induce the differentiation of HEL cells into megakaryocyte-like cells using PMA. Within 4 days of the PMA treatment, most of the small HEL cells showed some features of megakaryocytic differentiation, including increased cell size, adhesive force, periodic acid–Schiff-positive, polyploidization of the nucleus, and CD41 expression. These results indicate that PMA-induced HEL cells differentiated into megakaryocyte-like cells. Thus, it was attempted to evaluate the effects of antiplatelet drugs and other drugs using PMA-induced megakaryocytic HEL cells as an alternative to platelets.
Aspirin and cilostazol were selected as antiplatelet drugs, and ibuprofen and sodium valproate have been reported to have adverse effects on platelet functions. Aspirin causes the irreversible inactivation of cyclooxygenase activity by acetylating Ser-530 (also referred to as Ser-529) of COX-1, thereby inhibiting TXA2 production and suppressing platelet function through the inhibition of [Ca2+]i in platelets.19–21 On the other hand, cilostazol is a cyclic AMP (cAMP) phosphodiesterase III inhibitor, inhibiting phosphodiesterase activity and suppressing [Ca2+]i by inhibiting cAMP in platelets.22–25 Although the mechanisms of action of aspirin and cilostazol differ, these drugs control platelet function by inhibiting [Ca2+]i in platelets. In the present study, aspirin (5.6–560 μM) inhibited thrombin-induced increases in [Ca2+]i in a concentration-dependent manner in untreated HEL cells and PMA-induced HEL cells. Thrombin-induced increases in [Ca2+]i in PMA-induced HEL cells were significantly inhibited by all concentrations of aspirin. Cilostazol (1–10 μM) also inhibited thrombin-induced increases in [Ca2+]i in a concentration-dependent manner in untreated HEL cells and PMA-induced HEL cells, with these increases being significantly inhibited by 10 μM or 5 μM and 10 μM cilostazol, respectively. On the other hand, ibuprofen is an anti-inflammatory drug that inhibits COX-1 in the same as aspirin, but the action sites of aspirin and ibuprofen are different. Ibuprofen inhibits COX-1 reversibly by forming a salt bridge with Arg-12026 and also inhibits the generation of TXA2, which is a platelet aggregation promoter.27 Ibuprofen (0.8–200 μM) inhibited thrombin-induced increases in [Ca2+]i in a concentration-dependent manner in untreated HEL cells and PMA-induced HEL cells, and these increases in untreated HEL cells and PMA-induced HEL cells were significantly inhibited by 80 μM and 200 μM or 8–200 μM ibuprofen, respectively. Sodium valproate is used in the treatment of epilepsy and bipolar disorder. Sodium valproate is used in the treatment of epilepsy and bipolar disorder, but hematologic side effect has been reported in sodium valproate therapy. In bipolar disorder, Suzuki et al28–30 reported that sodium valproate inhibited serotonin- or thrombin-induced [Ca2+]i increase in human platelets. It inhibited thrombin-induced increases in [Ca2+]i in a concentration-dependent manner in untreated HEL cells and PMA-induced HEL cells. Sodium valproate (50–1,000 μg/mL) also inhibited thrombin-induced increases in [Ca2+]i in a concentration-dependent manner in untreated HEL cells and PMA-induced HEL cells. Thrombin-induced increases in [Ca2+]i in PMA-induced HEL cells were significantly inhibited by all concentrations of sodium valproate. Recently, MacDonald and Wei31 reported a possible adverse interaction between aspirin and ibuprofen. Ouellet et al32 also reported reactions on platelet aggregation with the combination of aspirin and ibuprofen. Thus, it was attempted to evaluate the effects of interaction in thrombin-induced increases in [Ca2+]i in combination with aspirin and ibuprofen. In this study, high-dose aspirin (56 μM) was selected in order to clarify the interaction of ibuprofen. In combination with aspirin and ibuprofen, when the inhibitory effect of aspirin is higher than that of ibuprofen, the effect of aspirin was reduced, whereas when the inhibitory effect of aspirin is lower than that of ibuprofen, the effect of ibuprofen was reduced. The quantitative relationship between aspirin and ibuprofen has not yet been elucidated in detail. On the other hand, low-dose aspirin (5.6 μM) was selected in combination of sodium valproate. The combination of aspirin and sodium valproate synergistically inhibited thrombin-induced [Ca2+]i. The increase in this inhibition rate is considered to occur because the action sites of aspirin and sodium valproate are different. Gidal et al33 demonstrated that sodium valproate-induced platelet dysfunctions involve alterations in the arachidonic acid cascade (AA cascade). Kis et al34 also reported the lower activity of the AA cascade and decreased production of TXA2 in the platelets of patients receiving sodium valproate therapy. These findings suggest that sodium valproate inhibits the production of TXA2 in the AA cascade. However, the mechanism of action of sodium valproate on the platelet AA cascade has not yet been elucidated in detail. Furthermore, to the best of our knowledge, the mechanism of action of sodium valproate on intracellular Ca2+ in platelets currently remains unclear; therefore, further studies are warranted.
Conclusion
The results of the present study indicate that the effects and adverse effects of drug combinations may be evaluated based on the inhibition of increases in [Ca2+]i induced by thrombin in PMA-induced HEL cells. In conclusion, it is possible to induce HEL cells to differentiate into megakaryocytes, which are a useful model for the study of platelet functions, and the quantification of inhibitory effects on thrombin-induced increases in [Ca2+]i is applicable to the evaluation of the effects of various drugs on platelets. Furthermore, this system of evaluation using these platelet model cells may contribute to the selection of appropriate drugs and doses for the control of platelet functions without using platelets, and its use is expected in the future in this field.
Acknowledgment
This work was supported by a grant-in-aid for Challenging Exploratory Research from the Japanese Ministry of Education, Culture, Sports, Science, and Technology, grant number 26670282.
Disclosure
The authors report no conflicts of interest in this work.
References
Huo Y, Schober A, Forlow SB, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9(1):61–67. | ||
Massberg S, Brand K, Grüner S, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196(7):887–896. | ||
Massberg S, Vogt F, Dickfeld T, Brand K, Page S, Gawaz M. Activated platelets trigger an inflammatory response and enhance migration of aortic smooth muscle cells. Thromb Res. 2003;110(4):187–194. | ||
Wu KK. Platelet activation mechanisms and markers in arterial thrombosis. J Intern Med. 1996;239:17–34. | ||
Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1(1):11–21. | ||
Jackson SP, Nesbitt WS, Kulkarni S. Signaling events underlying thrombus formation. J Thromb Haemost. 2003;1(7):1602–1612. | ||
Jang IK, Gold HK, Ziskind AA, Leinbach RC, Fallon JT, Collen D. Prevention of platelet-rich arterial thrombosis by selective thrombin inhibition. Circulation. 1990;81(1):219–225. | ||
Winters JK, Santro SA, Miletich JP, Eisenberg PR. Relative importance of thrombin compared with plasmin-mediated platelet activation in response to plasminogen activation with streptokinase. Circulation. 1991;84(4):1552–1560. | ||
Laine M, Paganelli F, Bonello L. P2Y12-ADP receptor antagonists: days of future and past. World J Cardiol. 2016;8(5):327–332. | ||
Rink TJ, Hallam TJ. What turns platelets on? Trends Biochem Sci. 1984;9:215–219. | ||
Johnson PC, Ware JA, Cliveden PB, Smith M, Dvorak AM, Salzman EW. Measurement of ionized calcium in blood platelets with the photoprotein aequorin. Comparison with Quin 2. J Biol Chem. 1985;260(4):2069–2076. | ||
Brass LF, Manning DR, Williams AG, Woolkalis MJ, Poncz M. Receptor and G protein-mediated responses to thrombin in HEL cells. J Biol Chem. 1991;266(2):958–965. | ||
Tabilio A, Rosa JP, Testa U, et al. Expression of platelet membrane glycoproteins and alpha-granule proteins by a human erythroleukemia cell line (HEL). EMBO J. 1984;3(2):453–459. | ||
Nakajima M, Yamamoto M, Ushikubi F, Okuma M, Fujiwara M, Narumiya S. Expression of thromboxane A2 receptor in cultured human erythroleukemia cells and its induction by 12-O-tetradecanoylphorbol-13-acetate. Biochem Biophys Res Commun. 1989;158(3):958–965. | ||
Long MW, Heffner CH, Williams JL, Peters C, Prochownik EV. Regulation of megakaryocyte phenotype in human erythroleukemia cells. J Clin Invest. 1990;85(4):1072–1084. | ||
Vasudev K, Keown P, Gibb I, McAllister-Williams RH. Hematological effects of valproate in psychiatric patients: what are the risk factors? J Clin Psychopharmacol. 2010;30(3):282–285. | ||
Martin P, Papayannopoulou T. HEL cells: a new human erythroleukemia cell line with spontaneous and induced globin expression. Science. 1982;216(4551):1233–1235. | ||
Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–3450. | ||
Lecomte M, Laneville O, Ji C, DeWitt DL, Smith WL. Acetylation of human prostaglandin endoperoxide synthase-2(cyclooxygenase-2) by aspirin. J Biol Chem. 1994;269(18):13207–13215. | ||
Gordon JM, Person JD. Effects of sulphinpyrazone and aspirin on prostaglandin I2(prostacyclin) synthesis by endothelial cells. Br J Pharmacol. 1978;64(4):481–483. | ||
Patrono C. Aspirin as an antiplatelet drug. N Engl J Med. 1994;330:1287–1294. | ||
Umekawa H, Tanaka T, Kimura Y, Hidaka H. Purification of cyclic adenosine monophosphaterase from human platelets using new-inhibitor sepharose chromatography. Biochem Pharmacol. 1984;33:3339–3344. | ||
Sudo T, Tadhibana K, Toda K, et al. Potent effects of novel anti-platelet aggregatory cilostamide analogues on recombinant cyclic nucleotide phosphodiesterase isozyme activity. Biochem Pharmacol. 2000;59(4):347–356. | ||
Igawa T, Tani T, Chijiwa T, et al. Potentiation of anti-platelet aggregation activity of cilostazol with vascular endothelial cells. Thromb Res. 1990;57:617–623. | ||
Minami N, Suzuki Y, Yamamoto M, et al. Inhibition of shear stress-induced platelet aggregation by cilostazol, a specific inhibitor of cGMP-inhibited phosphodiesterase, in vitro and ex vivo. Life Sci. 1997;61(25):383–389. | ||
Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367(6460):243–249. | ||
McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96(1):272–277. | ||
Kusumi I, Suzuki K, Sasaki Y, Kameda K, Koyama T. Treatment response in depressed patients with enhanced Ca mobilization stimulated by serotonin. Neuropsychopharmacology. 2000;23(6):690–696. | ||
Suzuki K, Kusumi I, Akimoto T, Sasaki Y, Koyama T. Effects of lithium and valproate on agonist-induced platelet intracellular calcium mobilization: relevance to myosin light chain kinase. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):67–72. | ||
Akimoto T, Kusumi I, Suzuki K, Masui T, Koyama T. Effects of valproate on serotonin-induced intracellular calcium mobilization in human platelets. J Psychiatry Neurosci. 2007;32(1):17–22. | ||
MacDonald TM, Wei L. Is there an interaction between the cardiovascular protective effects of low-dose aspirin and ibuprofen? Basic Clin Pharmacol Toxicol. 2006;98(3):275–280. | ||
Ouellet M, Riendeau D, Percival MD. A high level of cyclooxygenase-2 inhibitor selectivity is associated with a reduced interference of platelet cyclooxygenase-1 inactivation by aspirin. Proc Natl Acad Sci U S A. 2001;98(25):14583–14588. | ||
Gidal B, Spencer N, Maly M, et al. Valproate-mediated disturbances of hemostasis: relationship to dose and plasma concentration. Neurology. 1994;44(8):1418–1422. | ||
Kis B, Szupera Z, Mezei Z, Gecse A, Telegdy G, Vécsei L. Valproate treatment and platelet function: the role of arachidonate metabolites. Epilepsia. 1999;40(3):307–310. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.