")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Enhanced oral bioavailability of fluvastatin by using nanosuspensions containing cyclodextrin
Received 15 June 2018
Accepted for publication 3 September 2018
Published 23 October 2018 Volume 2018:12 Pages 3491—3499
DOI https://doi.org/10.2147/DDDT.S177316
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Jun Li,1,2,* Min Yang,2,* Wen-Rong Xu1
1School of Medical Science and Laboratory Medicine, Jiangsu University, Zhenjiang, Jiangsu 212013, China; 2Department of Intensive Care Unit, Chest Hospital Affiliated to Shanghai Jiaotong University, Shanghai 200030, China
*These authors contributed equally to this work
Background: In this study, fluvastatin (FVT) nanosuspensions containing cyclodextrin were developed to improve oral bioavailability.
Methods: FVT nanosuspensions containing cyclodextrin were prepared by a high pressure homogenization technique. The nanosuspensions system was then characterized by transmission electron microscopy (TEM), particle size, differential scanning calorimetry (DSC) and powder X-ray diffractometry (PXRD). In addition, in vitro drug release properties, pharmacokinetics and pharmacodynamics were also investigated in detail.
Results: After lyophilization, the nanosuspensions could be redispersed gently and with a narrow particle size distribution, but the particle size has no obvious change. The powder X-ray diffraction and differential scanning calorimetry of FVT nanosuspensions showed that FVT existed in amorphous form in nanosuspensions. In vitro release, FVT nanosuspensions have sustained-release properties. Meanwhile, FVT nanosuspensions could significantly modify the pharmacokinetic profile and increase the bioavailability of FVT by more than 2.4-fold in comparison with the FVT capsules group. In vivo irritation test showed that there was almost no evidence of hemorrhagic mucosal erosion and intestinal villus destruction in rat gastric mucosa.
Conclusion: The combination of nanocrystallization and cyclodextrin complexation techniques is a new attempt to formulate poorly water-soluble FVT.
Keywords: fluvastatin, HP-β-CD, nanosuspensions, bioavailability, irritation test
Introduction
Fluvastatin (FVT) belongs to the statin family and is considered as the first line of defense against hyperlipidemia.1 It inhibits cholesterol formation in the liver by inhibiting HMG-CoA reductase enzyme.2 Most of the statins have been developed for immediate release preparations. However, FVT is a poorly water-soluble drug with a short half-life (1–3 hours) and can be metabolized by cytochrome P3A in intestine gut and liver.3 The recommended dose is 20 or 40 mg daily, 1 hour is required to reach peak plasma concentration, and the volume of distribution of FVT is 330 L. The oral bioavailability of FVT is as low as 30% due to the slow dissolution rate in the intestinal tract and obvious first-pass effect.4 In addition, considering the long-term use of FVT, it is an essential requirement to enhance FVT bioavailability and sustain its release to lower both the dose and the frequency, hence improving patient tolerability. Clinically, the increase in low-density lipoproteins (LDLs) in plasma concentration leads to vascular stenosis, which carries the risk of atherosclerosis, coronary artery disease, and plague and has life-threatening consequences.5 Therefore, it is urgent for researchers to improve the existing drugs. Recent strategies have been used to solve problems related to low bioavailability and poor solubility of FVT. These include nanostructured lipid carriers,6 solid dispersion,7 nanoparticles,8,9 and microspheres.10 In the past few years, the use of particle size reduction method to form stable nanoparticles has proved an effective strategy to solve this thorny problem. Nanoscale suspensions can be defined as liquid dispersions consisting of solid drug nanoparticles, which are stabilized by polymers and/or surfactants.11 According to the Noyes–Whitney equation,12 the reduction in particle size leads to a significant increase in the dissolution rate of API, which in turn can lead to a significant increase in bioavailability.13,14
In addition, nanosuspensions containing cyclodextrin can offset the shortcoming of cyclodextrin-loading technology because surfactants play a role in stabilizing drug nanocrystals.15,16 In this study, we attempted to prepare FVT nanosuspensions containing cyclodextrin by using high-pressure homogenization method. The nanosuspensions’ system was then characterized by transmission electron microscopy (TEM), particle size, differential scanning calorimetry (DSC), and powder X-ray diffraction (PXRD). In addition, in vitro drug release properties, pharmacokinetics, and pharmacodynamics were investigated in detail. The combination of nanocrystallization and cyclodextrin complexation techniques is a new attempt to formulate poorly water-soluble FVT.
Materials and methods
Materials
FVT was sourced from Kang Baotai Fine Chemical Co., Ltd. (Hubei, China). The commercially available FVT capsule (Lescol® (Fluvastatin), 40 mg/capsule) was purchased from Beijing Novatis Pharmaceutical Co., Ltd., (Beijing, China) 2-Hydroxypropyl-β-cyclodextrin (HP-β-CD), poloxamer 407, and other reagents were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Double-distilled water from a Milli-Q system (EMD Millipore, Billerica, MA, USA) was used throughout the experiment.
Preparation of FVT/HP-β-CD nanosuspensions
FVT/HP-β-CD nanosuspensions were prepared by the high-pressure homogenization technique. Typically, FVT and HP-β-CD (weight ratio =10:15 mg) were dissolved in ethanol (50%, 5 mL) and, then, the solvent was removed under reduced pressure using a rotatory evaporator until no residual ethanol could be detected. Subsequently, 20% of poloxamer 407 aqueous solution (5 mL) was introduced into the dried FVT/HP-β-CD system. The system was subjected to an AH-1500 high shear homogenizer (ATS Engineering Limited Co., Ltd., Suzhou, China) with 2,200 bar for 12 cycles to form nanosuspensions. FVT/HP-β-CD nanosuspensions were lyophilized using trays that were frozen in deep freezer (BCD-228D11SY; Hisense Ronshen, Guangdong, China) at −80°C for 24 hours. The trays were then transferred to the vacuum adapter of lyophilizer (Genesis Pilot; SP Scientific, Warminster, PA, NY, USA). The solvent was sublimed under a pressure of 0.01 mbar for 48 hours (Figure 1A).
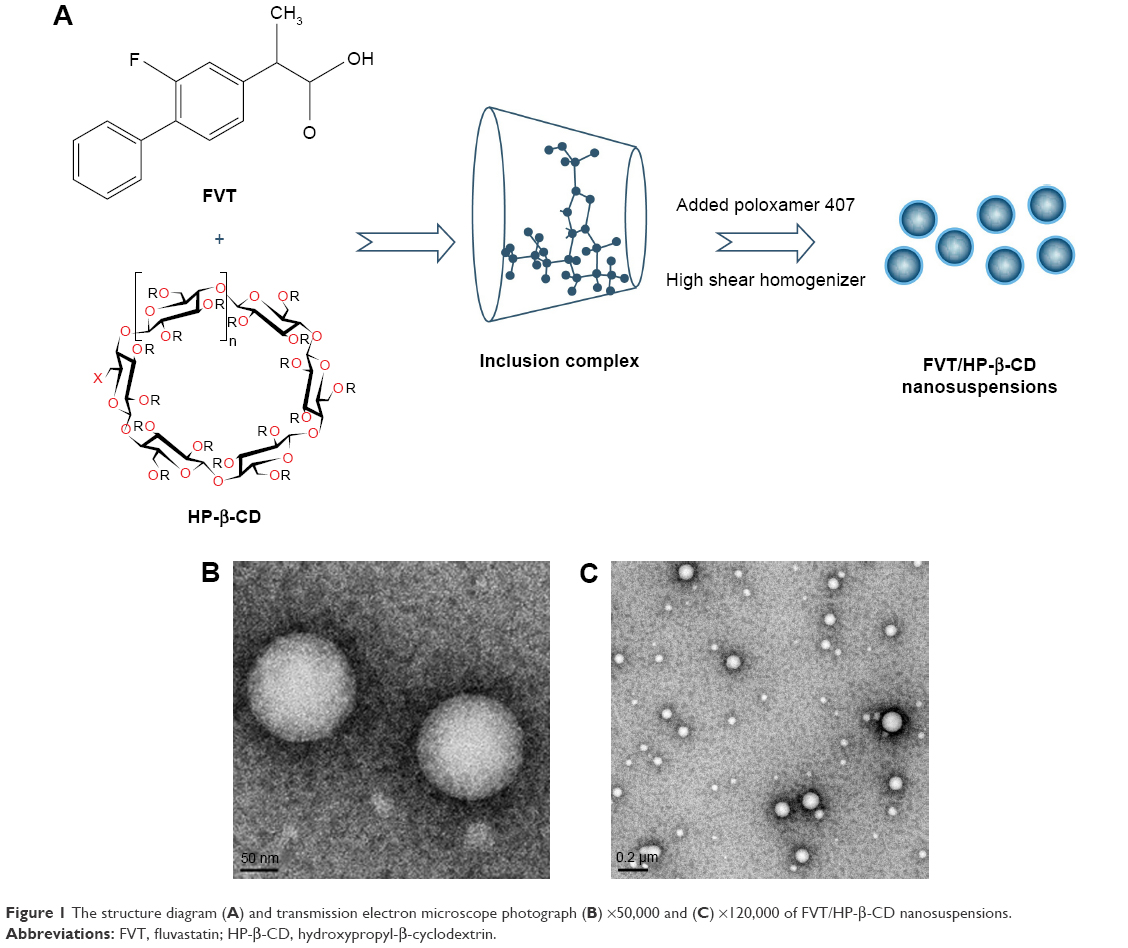 | Figure 1 The structure diagram (A) and transmission electron microscope photograph (B) ×50,000 and (C) ×120,000 of FVT/HP-β-CD nanosuspensions. |
Effect of pH on the stability of nanosuspensions
Nanosuspensions containing poloxamer 407 were prepared in an acidic medium of pH 3 and in an alkaline medium of pH 10. The results of particle size analysis and nanosuspension stability were compared with those of nanosuspensions prepared in neutral distilled water (pH 6.5).
Characterization
Particle size and zeta potential analysis
The lyophilized FVT/HP-β-CD nanosuspensions powder was redispersed with PBS (pH 7.4) before measurement (0.5 mg/mL). The particle size and zeta potential were measured by dynamic light scattering and electrophoretic mobility, respectively, at 25°C (Zetasizer Nano ZS 3000 SH; Malvern Instruments, Malvern, UK).
TEM
The external morphology of FVT/HP-β-CD nanosuspensions was determined by TEM (CM120; Philips, Amsterdam, Netherlands). A drop of freshly prepared FVT/HP-β-CD nanosuspensions was dripped onto the surface of a copper grid and air-dried. Because of the poor conductivity of organic samples, negative staining with a drop of 2% aqueous sodium phosphotungstate for contrast enhancement was performed 2 minutes before TEM measurements.17
DSC and PXRD
DSC and PXRD were used to determine the crystal form of FVT dispersed in the HP-β-CD nanosuspensions. DSC analysis was performed using a DSC8000 differential scanning calorimeter. Accurately weighed samples were placed in aluminum pans sealed with a lid. Al2O3 was used as the reference. During the scanning process, a heating rate of 5°C/minute was applied in the temperature range from 20°C to 150°C. PXRD studies were performed using the Phillips X-ray diffractometer and Cu-Kα radiation. The samples were scanned over a 2θ range of 0°–90° at a rate of 0.05°/second. The freeze-drying procedure was performed by a freeze dryer to practically determine the formulation of FVT/HP-β-CD nanosuspensions.
Stability studies
FVT/HP-β-CD nanosuspensions powder (FVT 10 mg) was stored at 25°C for 3 months before the polydispersity index (PDI) and zeta potential were determined. All measurements were performed in triplicate.
In vitro release
Analysis of the release characteristics of FVT/HP-β-CD nanosuspensions powder was carried out according to the procedure outlined in a previous study.18 Briefly, 100 mg of FVT/HP-β-CD nanosuspensions powder was introduced into a dialysis bag (molecular weight cut-off =10 kDa) and resuspended in 500 mL of PBS, pH 7.4. These samples were placed in a shaker with constant agitation at 60 rpm and kept in an incubator at 37°C. At scheduled time points (0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 hours), the samples were centrifuged and 2 mL of the supernatant was removed and replaced with 2 mL of fresh PBS. Then, the samples were filtered through a 0.45 μm syringe filter and LC-MS/MS analysis was carried out. Commercial FVT capsules were used as control.
Pharmacokinetics
All in vivo experimental protocols were approved by the animal care committee of the Faculty of Medicine, Jiangsu University Animal Center. In all studies of animals, the procedures were in line with the National Institutes of Health guide for the care and use of laboratory animals. Twelve Wistar rats were used in vivo to observe the changes in pharmacokinetic parameters of FVT after oral administration. All rats were randomly divided into two groups and administrated commercial FVT capsules (prepared by dispersing commercial FVT capsules in 0.5% sodium carboxymethylcellulose solution and having it sonicated for 10 minutes) and FVT/HP-β-CD nanosuspensions powder by intragastric administration (10 mg/kg dose). Blood samples (0.5 mL) were collected from the caudal vein into heparinized tubes at 0, 0.5, 1, 2, 4, 6, 12, 16, 24, and 48 hours after intragastric administration. Blood was immediately centrifuged for plasma at 4,000× g for 10 minutes. Plasma samples were collected at −70°C and kept frozen until analysis.
HPLC analysis condition
The quantification of FVT in samples obtained from in vitro and in vivo studies was performed on the LC-MS/MS system (API3000) based on a validated method described previously.19,20
In short, 100 μL of plasma was collected at each time of sampling, and the same volume of simvastatin, used as an internal standard with concentration (50 ng/mL), was dissolved in acetonitrile to precipitate the plasma proteins. The supernatant was transferred into an automatic sampler bottle, and 20 μL of sample was injected into LC-MS/MS. Separation was performed on an Advanced Chromatography Technologies 3C18 column (50 × 4.5 mm) with a mobile phase of ammonium acetate (1 mM, pH 4.0) and acetonitrile (40:60) at a flow rate of 1.2 mL/minute. Using the line equation of the standard curve plotted in the range of 0.25–20 ng/mL and considering interday SD and intraday SD, the peak area of FVT in each sample was analyzed.
In vivo irritation test
Wistar rats were used to evaluate the oral tolerance of commercial FVT capsules and FVT/HP-β-CD nanosuspensions. The rats were randomly divided into three groups with three rats in each. Provided with water, they were fasted for 12 hours before euthanasia. FVT capsules and FVT/HP-β-CD nanosuspensions (10 mg/kg) were orally administered to rats in two groups once a day for 7 days. The control group received an equal amount of saline. Two hours after the last administration, the rats were euthanized and the tissue specimens (5 mm of small intestine) were fixed with 4% formaldehyde and embedded in paraffin for histopathological evaluation.
Statistical analysis
Each experiment was repeated at least three times, and mean ± SD (standard error) was calculated from the representative experiments. Statistically significant differences were determined using two-tailed Student’s t-test (Version 10.0; SPSS Inc., Chicago, IL, USA). The P-values for significance were set at 0.05.
Results and discussion
Characterization of FVT/HP-β-CD nanosuspensions
In this study, FVT/HP-β-CD nanosuspensions with an average particle size of 132.4±6.9 nm were prepared. After lyophilization, the nanosuspensions could be redispersed gently, but the particle size had no obvious change. FVT/HP-β-CD nanosuspensions had a narrow particle size distribution. Figure 1B and C shows the TEM of FVT/HP-β-CD nanosuspensions obtained by drying at room temperature (TEM). These particle size data and TEM images indicated that nanoscale particles would be expected to improve their dissolution behavior and bioavailability.21 In the preliminary experiment, we found that it was difficult to obtain nanosuspensions less than 200 nm. The addition of surfactants (poloxamer series) greatly reduced the particle size of FVT/HP-β-CD nanosuspensions. Therefore, poloxamer 407 was used in the preparation process of FVT/HP-β-CD nanosuspensions. The particle size, together with the PDI, of nanosuspensions decreased as the dosage of poloxamer 407 increased. In this system, the encapsulation technology of cyclodextrin can greatly improved the solubility of drugs (≈0.3–4.2 mg/mL). Preparation of FVT/HP-β-CD nanosuspensions could either improve the dissolution of FVT or reduce the binding by intestinal-free mucins, which helped improve the bioavailability. In addition, the zeta potential of nanosuspensions indicated that there were many negative charges on their surface, resulting in good physical stability of the whole system (Table 1). The zeta potential reflected the charge of particles to a certain extent and had a great influence on the stability of the system. The influence of nanoparticles on the zeta potential was mainly from its material and particle size. When the material was fixed, the influence of particle size on the surface charge density included the following: 1) the smaller the particle size, the larger the surface area, the lower the surface charge density, and the smaller the potential; 2) the particle size affected the viscosity of the system, which affected the thickness of the diffusion layer; and the more the diffusion layer compressed, the farther the sliding surface was from the neutral region, the higher the potential was; and 3) the particle size affected the mass and motion of the particles in the electric field. In this study, the zeta potential absolute value of nanosuspensions was about 20, which had a stabilizing effect on the system.
 | Table 1 The characteristics data of FVT/HP-β-CD nanosuspensions at room temperatures (n=3) |
Effect of pH on stability of nanosuspensions
In an attempt to enhance the physical stability of FVT/HP-β-CD nanosuspensions using poloxamer 407 as stabilizers, the effect of pH was investigated. Nanosuspensions were prepared in an acidic medium of pH 3 and in an alkaline medium of pH 10, and results were compared with those prepared in neutral distilled water (pH 6.5, 132.4±6.9 nm). In general, the results indicated that the nanosuspensions exhibited a decrease in their mean size in the acidic medium (121.5±5.5 nm) and alkaline medium (111.2±3.3 nm). Best stability was recorded in the alkaline medium where stability persisted for up to 3 days with an almost constant mean particle size. Improved stability was attributed to the adsorption of hydroxyl ions of nanosuspensions as indicated by the negative sign of zeta potential giving rise to a certain degree of between dispersed particles.
DSC and PXRD
DSC was used to investigate the melting and crystallization behaviors of the crystalline material of FVT/HP-β-CD nanosuspensions. A sharp melting peak of FVT at 69.6°C (Figure 2, a) indicated its crystalline nature. The DSC thermal behavior of the blank nanosuspensions (Figure 2, b) and physical mixture (FVT + blank nanosuspensions) were chosen as a reference. The temperature of the melting peak of FVT in the nanosuspensions (Figure 2, c) was 67.5°C, signifying that FVT and other components interacted with each other while heating. During the preparation of nanosuspensions, FVT and HP-β-CD were dissolved in a mixture of solvents, which were subsequently evaporated, allowing homogeneous dispersion of the drug in the materials (Figure 2, d). Therefore, the disappearance of the specific peak in FVT/HP-β-CD nanosuspensions revealed that FVT existed in the nanosuspensions in an uncrystallized form rather than in a crystallized form.
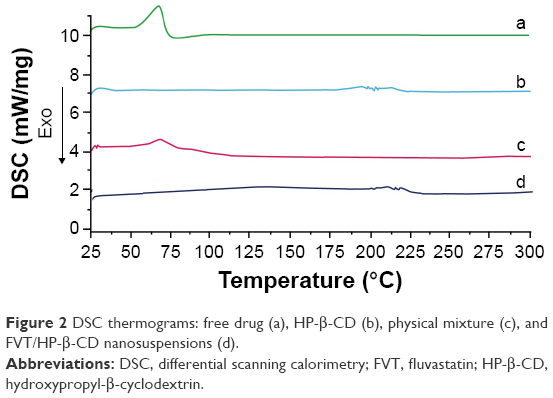 | Figure 2 DSC thermograms: free drug (a), HP-β-CD (b), physical mixture (c), and FVT/HP-β-CD nanosuspensions (d). |
The PXRD spectra for free FVT, blank nanosuspensions, the physical mixture of FVT and the blank nanosuspensions, and the FVT/HP-β-CD nanosuspensions are shown in Figure 3. FVT showed characteristic intense peaks between the 2θ of 2 and 15 (Figure 3, a and c). However, in the cases of nanosuspensions (Figure 3, d), the intensity of peaks was decreased, showing the amorphous nature of the drug after entrapment into nanosuspensions by freeze-drying.
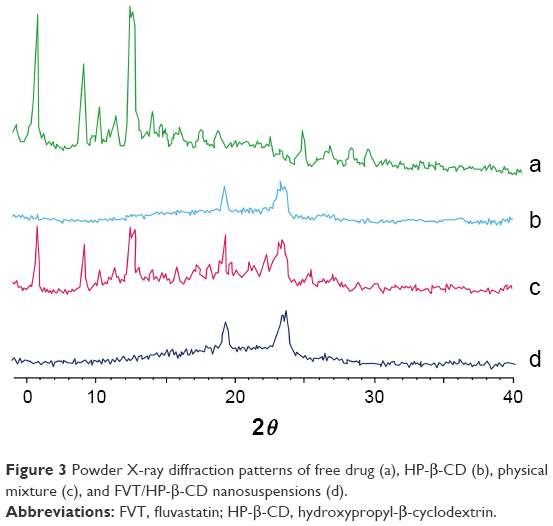 | Figure 3 Powder X-ray diffraction patterns of free drug (a), HP-β-CD (b), physical mixture (c), and FVT/HP-β-CD nanosuspensions (d). |
Stability studies
One week of investigation into nanosuspension stability showed that slight sedimentation on particles would take place upon storage in ambient conditions. After lyophilization, FVT/HP-β-CD nanosuspensions exhibited good stability in a period of 3 months. No significant change in physical appearance (re-dispersed) and particle aggregation was observed. All the indexes did not change obviously during the observation period (Table 2).
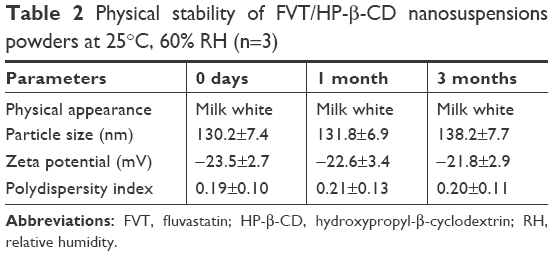 | Table 2 Physical stability of FVT/HP-β-CD nanosuspensions powders at 25°C, 60% RH (n=3) |
In vitro release
FVT is a poorly water-soluble drug with a short half-life in vivo. Entrapping it with biodegradable polymers to supply a sustained-release effect was an effective way to lengthen its half-life and enhance its therapeutic efficacy. In order to develop a long-term release system, it is essential to understand the mechanism and dynamics of its release. Figure 4 illustrates the release profiles of commercial FVT capsules and FVT/HP-β-CD nanosuspensions. On the whole, in vitro release data showed that FVT capsules were released faster than FVT/HP-β-CD nanosuspensions; almost 95% of FVT was released from capsules after 2 hours. In contrast, about 30% of the total FVT was released from the HP-β-CD nanosuspension with an initial burst release in the first 2 hours. The rapidly released FVT was unencapsulated or loosely attached to the HP-β-CD surface and was thus easily extracted at the early stage. After 8 hours, the cumulative proportion of FVT released from the HP-β-CD nanosuspension was 60%. In the next few hours, there was a typical plateau period. At that time, the amount of remaining drug was quite limited and the release was slow. When the plateau period was extended to 24 hours, the cumulative drug release percentage of HP-β-CD nanosuspension increased to 72%. It is worth noting that there was no significant change in the release behavior of the sample after 3 months of stability observation. The kinetics of in vitro release was analyzed according to the zero-order, first-order, and diffusion-controlled release mechanisms. After calculation, the in vitro drug-release kinetic model of FVT in the release medium fit well with the Higuchi equation: Q=6.254t1/2–1.321 (r=0.996).
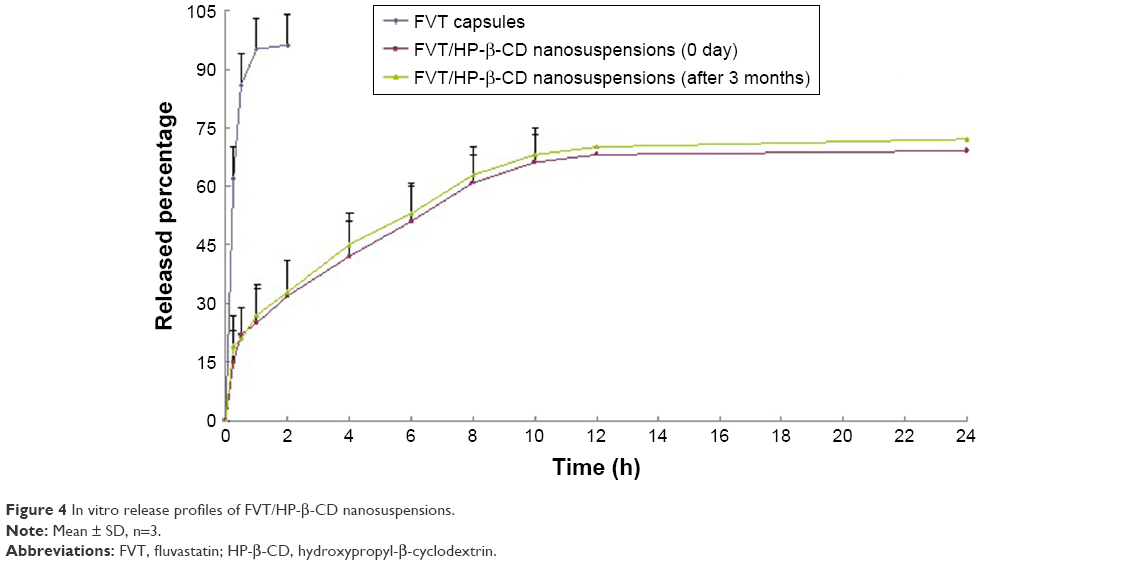 | Figure 4 In vitro release profiles of FVT/HP-β-CD nanosuspensions. |
Pharmacokinetics
The time plots of plasma concentration in rats after intragastric administration of test formulations (FVT/HP-β-CD nanosuspensions and commercial FVT capsules) are shown in Figure 5, and the pharmacokinetic parameters are tabulated in Table 3. The Tmax was 2 hours and the Cmax was 303.7 ng/mL after intragastric administration of FVT capsules. However, the time to achieve maximum concentration of FVT was delayed by 2 hours in the form of HP-β-CD nanosuspensions. The Cmax of FVT/HP-β-CD nanosuspensions was 562.9 ng/mL, significantly (P<0.05) higher than that obtained with the FVT capsules. Meanwhile, the concentration in the plasma of FVT/HP-β-CD nanosuspensions from 4 to 16 hours was remarkably higher than in rats administered with FVT capsules. Twenty-four hours after intragastric administration, the concentration of FVT in nanosuspensions was still 65 ng/mL, whereas the concentration of FVT in capsule was undetectable. The AUC0–∞ of FVT/HP-β-CD nanosuspensions was 6,952.4 ng hour/mL, 2.85-fold higher than AUC0–∞ of 2,436.5 ng hour/mL for FVT capsules, clearly displaying the performance superiority of nanosuspensions over capsules.
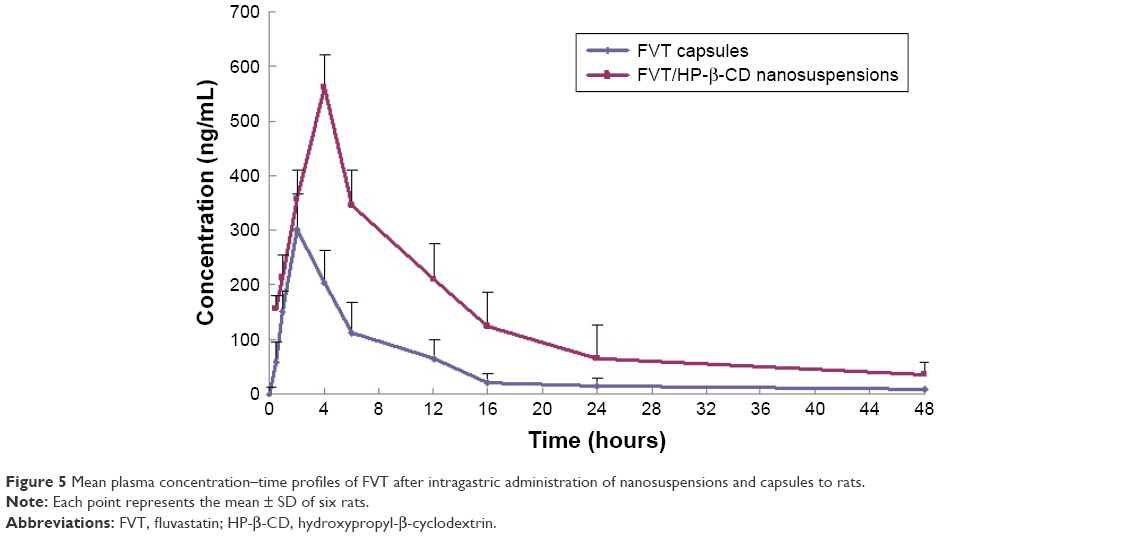 | Figure 5 Mean plasma concentration–time profiles of FVT after intragastric administration of nanosuspensions and capsules to rats. |
 | Table 3 Pharmacokinetic parameters of FVT after intragastric administration of nanosuspensions and capsules to rats (each point represents the mean ± SD of 6 rats) |
Nanosuspensions have shown potential in enhancing the oral bioavailability of poorly soluble drugs. Similarly, the ability of cyclodextrin in promoting drug absorption has been verified. However, there are relatively few reports on the combination of the two methods. Nanosuspensions containing FVT/HP-β-CD possess high drug dispersity and additional formulation advantages. Therefore, mechanisms that help enhance the bioavailability of FVT were proposed: 1) the nanosuspensions of FVT/HP-β-CD dramatically improved the dissolution of the drug and 2) the excipients involved in nanosuspensions significantly enhanced the intestinal permeability through interaction with biomembrane and antidrug efflux effect.22
In vivo irritation test
The long-term irritation effect of FVT/HP-β-CD nanosuspensions after oral administration was observed in rats. Figure 6 shows the histopathology of the rat gastric mucosa treated with various preparations to observe their effect on tissue integrity and cell structure. There was almost no evidence of hemorrhagic mucosal erosion or intestinal villus destruction in rat gastric mucosa. The results showed that the preparation had good biocompatibility with rat gastric mucosa.
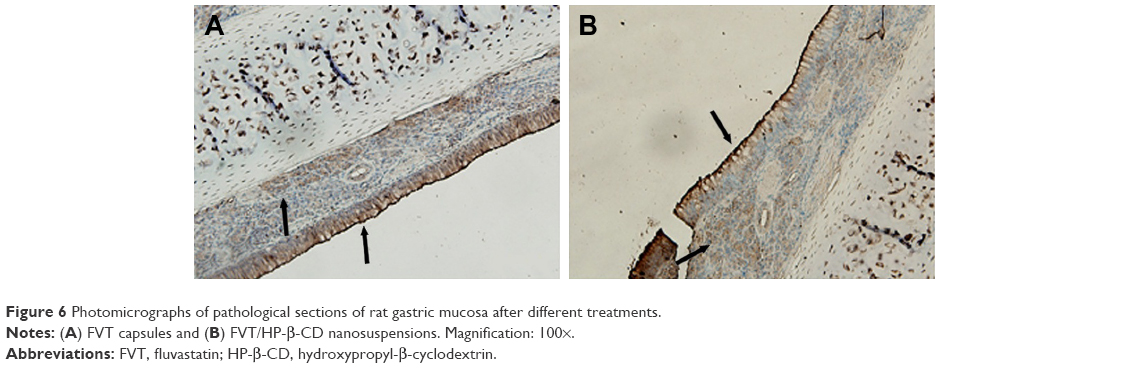 | Figure 6 Photomicrographs of pathological sections of rat gastric mucosa after different treatments. |
Conclusion
In this study, FVT nanosuspensions containing cyclodextrin were developed by a high-pressure homogenization technique to improve oral bioavailability. After lyophilization, the nanosuspensions could be redispersed gently with a narrow particle size distribution, but the particle size had no obvious change. The PXRD and DSC of FVT nanosuspensions showed that FVT existed in amorphous form in nanosuspensions. In vitro release showed sustained-release properties of FVT nanosuspensions. Meanwhile, FVT nanosuspensions could significantly modify the pharmacokinetic profile and increase the bioavailability of FVT by more than 2.4-fold in comparison with the FVT capsules’ group. In vivo irritation test showed that there was almost no evidence of hemorrhagic mucosal erosion or intestinal villus destruction in rat gastric mucosa.
Disclosure
The authors report no conflicts of interest in this work.
References
Yano M, Ikeda M, Abe K, et al. Oxidative stress induces anti-hepatitis C virus status via the activation of extracellular signal-regulated kinase. Hepatology. 2009;50(3):678–688. | ||
Kah J, Wüstenberg A, Keller AD, et al. Selective induction of apoptosis by HMG-CoA reductase inhibitors in hepatoma cells and dependence on p53 expression. Oncol Rep. 2012;28(3):1077–1083. | ||
Mahley RW, Bersot TP. Drug therapy for hypercholesterolemia and dys-lipidemia. In: Gilman AG, Goodman LS, Rall TW, Murad F, editors. The Pharmacological Basis of Therapeutics. 11th ed. New Delhi: McGraw Hill Medical Publishing Div; 2006:933–966. | ||
Lee CK, Choi JS, Bang JS. Effects of Fluvastatin on the Pharmacokinetics of Repaglinide: Possible Role of CYP3A4 and P-glycoprotein Inhibition by Fluvastatin. Korean J Physiol Pharmacol. 2013;17(3):245–251. | ||
Lopes-Virella MF, Virella GT. U.S. Patent and Trademark Office. Washington DC: 8,568,995 U.S. Patent No. 2013. | ||
El-Helw AR, Fahmy UA. Improvement of fluvastatin bioavailability by loading on nanostructured lipid carriers. Int J Nanomedicine. 2015;10:5797–5804. | ||
Papageorgiou GZ, Papadimitriou S, Karavas E, Georgarakis E, Docoslis A, Bikiaris D. Improvement in chemical and physical stability of fluvastatin drug through hydrogen bonding interactions with different polymer matrices. Curr Drug Deliv. 2009;6(1):101–112. | ||
Bikiaris DN, Papageorgiou GZ, Papadimitriou SA, Karavas E, Avgoustakis K. Novel biodegradable polyester poly(propylene succinate): synthesis and application in the preparation of solid dispersions and nanoparticles of a water-soluble drug. AAPS PharmSciTech. 2009;10(1):138–146. | ||
Amoli-Diva M, Pourghazi K, Mashhadizadeh MH. Magnetic pH-responsive poly(methacrylic acid-co-acrylic acid)-co-polyvinylpyrrolidone magnetic nano-carrier for controlled delivery of fluvastatin. Mater Sci Eng C Mater Biol Appl. 2015;47:281–289. | ||
Yasunami N, Ayukawa Y, Furuhashi A, et al. Acceleration of hard and soft tissue healing in the oral cavity by a single transmucosal injection of fluvastatin-impregnated poly (lactic-co-glycolic acid) microspheres. An in vitro and rodent in vivo study. Biomed Mater. 2015;11(1):015001. | ||
Patravale VB, Date AA, Kulkarni RM. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56(7):827–840. | ||
Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19(12):930–934. | ||
Scholz A, Abrahamsson B, Diebold SM, et al. Influence of hydrodynamics and particle size on the absorption of felodipine in labradors. Pharm Res. 2002;19(1):42–46. | ||
Hintz R, Johnson K. The effect of particle size distribution on dissolution rate and oral absorption. Int J Pharm. 1989;51(1):9–17. | ||
Liu P, Rong X, Laru J, et al. Nanosuspensions of poorly soluble drugs: preparation and development by wet milling. Int J Pharm. 2011;411(1–2):215–222. | ||
Tuomela A, Liu P, Puranen J, et al. Brinzolamide nanocrystal formulations for ophthalmic delivery: reduction of elevated intraocular pressure in vivo. Int J Pharm. 2014;467(1–2):34–41. | ||
Su Z, Liang Y, Yao Y, Wang T, Zhang N. Polymeric complex micelles based on the double-hydrazone linkage and dual drug-loading strategy for pH-sensitive docetaxel delivery. J Mater Chem B. 2016;4(6):1122–1133. | ||
Balzus B, Colombo M, Sahle FF, Zoubari G, Staufenbiel S, Bodmeier R. Comparison of different in vitro release methods used to investigate nanocarriers intended for dermal application. Int J Pharm. 2016;513(1–2):247–254. | ||
Gonzalez O, Iriarte G, Rico E, et al. LC-MS/MS method for the determination of several drugs used in combined cardiovascular therapy in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878(28):2685–2692. | ||
di Pietro G, Coelho EB, Geleilete TM, Marques MP, Lanchote VL. Chiral evaluation of fluvastatin in human plasma by high-performance liquid chromatography electrospray mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;832(2):256–261. | ||
Mosharraf M, Nyström C. The effect of particle size and shape on the surface specific dissolution rate of microsized practically insoluble drugs. Int J Pharm. 1995;122(1–2):35–47. | ||
Kumar L, Reddy MS, Shirodkar RK, Pai GK, Krishna VT, Verma R. Preparation and characterisation of fluconazole vaginal films for the treatment of vaginal candidiasis. Indian J Pharm Sci. 2013;75(5):585–590. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.