")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Emodin-induced autophagy against cell apoptosis through the PI3K/AKT/mTOR pathway in human hepatocytes
Authors Zheng X, Yang S, Zhang R, Wang S, Li G, Zhou S
Received 12 February 2019
Accepted for publication 5 July 2019
Published 3 September 2019 Volume 2019:13 Pages 3171—3180
DOI https://doi.org/10.2147/DDDT.S204958
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Xiao-yuan Zheng1, Shi-ming Yang2, Rong Zhang1, Su-min Wang2, Guo-bing Li1, Shi-wen Zhou1
1Department of Pharmacy, Xinqiao Hospital, Army Medical University, Chongqing, People’s Republic of China; 2Department of Gastroenterology, Xinqiao Hospital, Army Medical University, Chongqing, People’s Republic of China
Correspondence: Shi-wen Zhou
Department of Pharmacy, Xinqiao Hospital, Army Medical University, Chongqing 400037, People’s Republic of China
Email [email protected]
Background: Emodin, a major component of Polygonum multiflorum (PM), has been reported to exert both protective and toxic effects in several cell types. However, the effects and underlying mechanisms of action of emodin in hepatic cells are still obscure.
Methods: The present study used the normal human liver cell line L02 to investigate the effects and mechanisms of emodin in hepatic cells. After treatment with emodin, L02 cells were examined for viability, apoptosis and autophagy with the Cell Counting Kit-8 (CCK-8), annexin V/PerCP staining and GFP-LC3 plasmid transfection. The expression of proteins including cleaved caspase-3, LC3B-I/II, p-PI3K, PI3K, p-AKT, AKT, p-mTOR, mTOR and actin was examined by using Western blot.
Results: Emodin significantly inhibited the viability of and induced apoptosis in L02 cells in a dose- and time-dependent manner. In addition, emodin increased the number of GFP-LC3 puncta in L02 cells and upregulated the expression of LC3B-II compared to those in control cells. Furthermore, emodin significantly decreased the expression of p-PI3K, p-AKT and p-mTOR in a dose-dependent manner compared to that in control cells without altering the expression of PI3K, AKT and mTOR. Notably, cotreatment with emodin and 3-methyladenine (3-MA) or rapamycin significantly increased and decreased the apoptosis rate of L02 cells, respectively, compared to that of cells treated with emodin alone.
Conclusion: In conclusion, emodin exhibited cytotoxicity in the L02 human hepatic cell line by promoting apoptosis, and it also induced autophagy through the suppression of the PI3K/AKT/mTOR signalling pathway. The autophagy could play a protective role following emodin treatment.
Keywords: Polygonum multiflorum, emodin, hepatoprotective, apoptosis, hepatotoxic, autophagy, PI3K, AKT, mTOR
Corrigendum for this paper has been published
Introduction
Polygonum multiflorum is a traditional Chinese herb that has been widely used as an anti-ageing treatment in China for centuries, but it has recently been reported to cause liver injury.1,2 Anthraquinones, stilbenes and tannins are thought to be the major active components of PM.3 Emodin is an anthraquinone derivative (Figure 1A) isolated from PM. Emodin exhibits a variety of therapeutic effects, such as anti-tumour,4 antioxidant,5 anti-inflammatory,6 and anti-virus activities.7 Despite its various biological properties, both protective activities and toxic effects were reported with emodin treatment in vitro and in vivo.8,9 Thus, for its safe clinical use, the underlying mechanism of action of emodin in hepatic cells still needs to be elucidated.
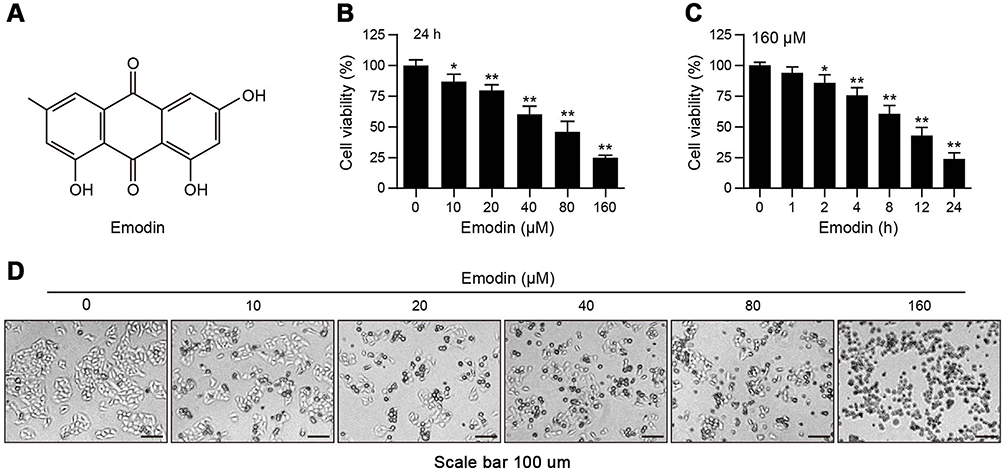 |
Figure 1 Emodin suppressed the proliferation of L02 cells. (A) The chemical structure of emodin. (B and C) L02 cells were treated with different concentrations of emodin for 24 h or with emodin (160 µM) for different time intervals. Cell viability was determined by CCK-8 assay. Data are presented as the means ± SDs for 3 independent experiments. *P<0.05 compared with the control group; **P<0.01 compared with the control group. (D) Representative photos depict the morphology of L02 cells exposed to various concentrations of emodin for 24 h. Scale bars: 100 μm. |
Macroautophagy (hereafter, autophagy) is a catabolic process that degrades cytoplasmic components within the lysosome and provides additional nutrients and energy to the cell.10 In addition, autophagy plays an essential role in quality control by selectively degrading long-lived proteins and organelles. Because of its anti-stress properties, autophagy is commonly thought to be an essential cytoprotective response to several stresses, including starvation, hypoxia and pathogen invasion. Conversely, it has also been reported that excessive autophagy in cells may promote caspase- and apoptosis-independent cell death.11 The dynamic process of autophagy is tightly regulated. At the initiation of autophagy, most signal pathways converge to the same mammalian target, rapamycin complex 1 (mTORC1, a complex containing TOR, raptor, GβL/mLST8, PRAS40 and DEPTOR), especially mTOR.12,13 MTOR is a serine/threonine protein kinase belonging to the phosphatidylinositol kinase-related kinase family. The activity of mTOR is suppressed under nutrient starvation, which induces autophagy. The phosphatidylinositol 3-kinase (PI3K)/AKT pathway is a pathway upstream of mTOR that plays a central role in cell survival and proliferation. Previous studies showed that emodin exerted a pro-apoptotic effect on K562 cells by suppressing the PI3K/AKT signalling pathway.14 Controversially, emodin attenuated autophagy in mouse C2C12 myoblasts through the phosphorylation of AKT.15 These results pose the questions of whether emodin had a role in autophagic activity in hepatic cells and what the mechanism of emodin treatment in hepatocytes was.
In this study, we investigated the effects and molecular mechanisms of emodin on L02 hepatic cells and sought to understand the hepatoprotective activities and hepatotoxic effects of emodin treatment. Our data demonstrated that emodin can inhibit L02 cell proliferation, trigger cell apoptosis, and induce autophagy in L02 cells through the PI3K/AKT/mTOR pathway. These results provide evidence for the rational use of emodin in the future.
Materials and methods
Cell lines and reagents
L02 cells were purchased from the Cell Bank of the Chinese Academy of Sciences and maintained in RPMI 1640 medium (Gibco, 31800022) supplemented with 10% foetal bovine serum (FBS; Gibco, 10100147), 1.5 g/L NaHCO3 (Gibco, 25080094), and 0.11 g/L sodium pyruvate (Gibco, 11360070) in 5% CO2 at 37°C. Emodin (E7881), 3-methyladenine (M9281) and rapamycin (V900930) were purchased from Sigma-Aldrich Chemical. The antibodies used were as follows: anti-cleaved caspase-3 (#9664), anti-LC3B-I/II (#3868), anti-phospho-AKT(Thr308) (#13038), anti-AKT (#4691), anti-phospho-mTOR(Ser2448) (#5536), anti-mTOR (#2983), and anti-actin (#8457), which were purchased from Cell Signalling Technology. Anti-phospho-PI3K(Tyr485) (sc-130211) and anti-PI3K (sc-365404) were purchased from Santa Cruz Biotechnology. Horseradish peroxidase-conjugated species-specific secondary antibodies (4741506, 4741806) were purchased from Kirkegaard and Perry Laboratories.
Cell viability (CCK-8) assay
Cells were seeded in 96-well plates at a density of 5,000 cells per well for 24 h, and 5 identical wells were prepared for each group. After the indicated treatments, the cells were treated with 10 µl of Cell Counting Kit-8 reagent (CCK-8) (Beyotime, C0038) for 1 h. The absorption values (optical density (OD)) were measured by a microplate reader at 450 nm. Cell viabilities were normalized to the control group according to the following formula: cell viability (%) = (ODblank - ODexperiment)/ODblank ×100%.
Cell morphology
To view the cell morphology, L02 cells were seeded in 6-well plates at a density of 5×104 cells per well for 24 h and treated with various concentrations of emodin for 24 h. The morphology of the L02 cells was observed under a light microscope (Diaphot 300, Nikon).
GFP-LC3 plasmid transfection and confocal microscopy
A total of 2×104 cells were plated on coverslips for 24 h. GFP-LC3 plasmid transfection was performed using Lipofectamine 3000 transfection reagent (Invitrogen, L3000) according to the manufacturer’s protocol. Plasmids encoding GFP-LC3 (Addgene, #22405) were a gift from Jayanta Debnath. After a 48 h incubation, the medium containing the transfection mixture was replaced with fresh complete medium and the cells were treated with emodin (80 µM) for 12 h or were not treated with emodin before being viewed by using a laser scanning confocal microscope (Zeiss, Germany). All images were analysed by ImageJ software (MD, USA).
Flow cytometry
After the cells were treated as indicated, the apoptosis rates were determined by flow cytometry using an Annexin V Apoptosis Detection Kit PerCP-eFluor (eBioscience, 88-8008-74) according to the manufacturer’s instructions.
Western blots
The cells were treated as indicated, harvested and then washed twice with ice-cold PBS for total protein extraction. The protein concentration was determined using an Enhanced BCA Protein Assay Kit (Beyotime Biotechnology, P0011, China). Equal amounts of protein were separated by 10% SDS-PAGE and subsequently transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% skim milk in TBST buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.1% Tween 20).Then, the membranes were incubated with primary antibodies at 4 °C overnight. After that, the membranes were washed 3 times with TBST buffer and incubated with the secondary antibodies at room temperature for 2 h. After the membranes were washed with TBST buffer 3 times, bands were observed and analysed with a charge-coupled device (CCD) system (Kodak, Image Station 2000 MM, Japan).
Statistical analysis
Statistical analyses were performed with SAS. Data were normalized to the control and analysed with one-sample t-tests. Statistical significance was indicated as follows: 0.01≤ *P<0.05; **P<0.01.
Results
Emodin suppressed the proliferation of L02 cells
To observe the inhibitory effects of emodin on hepatic cells, we first performed CCK-8 assays to investigate the effects of emodin on L02 cell growth. As shown in Figure 1B and C, emodin decreased the viability of L02 cells in a dose- and time-dependent manner. To directly observe alterations to L02 cells with emodin treatment, we viewed cells under an optical microscope with a camera. As shown in Figure 1D, the untreated L02 cells grew well, and the cellular structures were clear. After treatment with emodin for 24 h, we found increased cellular shrinkage and decreased cell density in a dose-dependent manner.
Emodin induced apoptosis in L02 cells through the activation of caspase-3
To examine the inhibitory effects of emodin on L02 cells, we assessed the effects of emodin on apoptosis in L02 cells by using an annexin V/PerCP staining assay. Cells treated with emodin exhibited increased apoptosis in a dose- and time-dependent manner (Figure 2A and B). In addition to the increase in apoptosis, following emodin treatment, the expression of cleavage/activation caspase-3 (c-caspase-3) was increased in a dose- and time-dependent manner compared to that observed without emodin treatment (Figure 2C–F). These results indicated that emodin could induce apoptosis in L02 cells by activating caspase-3.
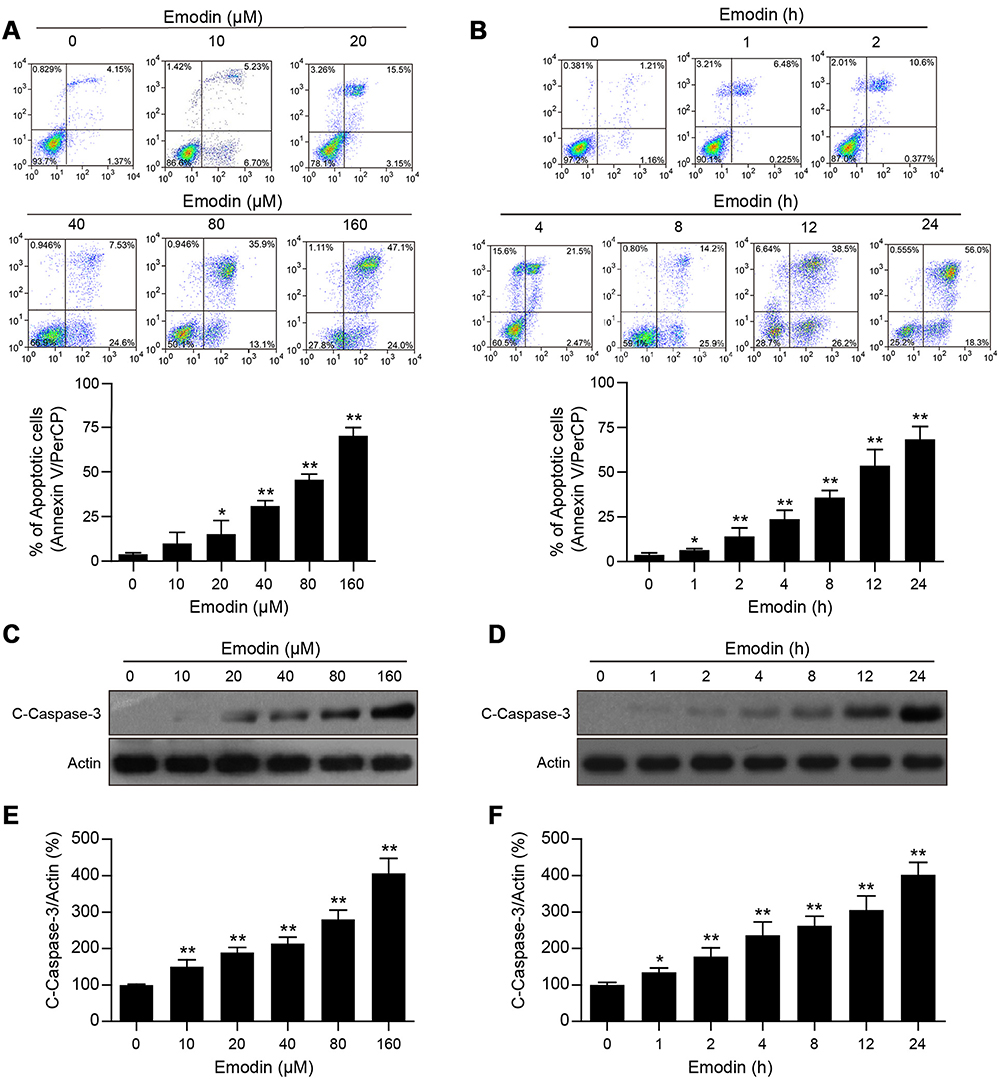 |
Figure 2 Emodin induced apoptosis in L02 cells through activating caspase-3. (A and B) L02 cells were treated with different concentrations of emodin for 24 h or with 160 µM emodin for different time intervals. Cell apoptosis was detected by flow cytometry using annexin V/PerCP staining. Representative flow cytometric images and statistical data are showed, and the data are presented as means ± SDs for 3 independent experiments. *P<0.05 compared with the control group; **P<0.01 compared with the control group. (C and D) Cells were treated as in (A) and (B), respectively, and then subjected to Western blot analysis to determine the expression of c-caspase-3. Actin was used as a loading control. (E) The intensities of c-caspase-3 in (C) and (D) normalized to actin were statistically analysed and represented as the mean ± SD for 3 independent experiments. *P<0.05 compared with the control group; **P<0.01 compared with the control group. (F) The intensities of c-caspase-3 in (D) normalized to actin were statistically analysed and represented as the mean ± SD for 3 independent experiments. *P<0.05 compared with the control group; **P<0.01 compared with the control group. |
Emodin induced autophagic alterations in L02 cells
Whether emodin plays a role in the autophagic activity of L02 cells remains unclear. Because the conversion of LC3B-I to LC3B-II is commonly used to evaluate autophagy,16 we first detected LC3B-I/II levels in L02 cells using Western blotting. As shown in Figure 3A–D, emodin treatment unexpectedly led to an increase in the expression of LC3B-II in a dose- and time-dependent manner, except for 160 μM emodin treatment for 24 h, and the expression of LC3B-I was largely unchanged compared to that in the untreated cells. Furthermore, L02 cells expressing GFP-LC3 were detected under a confocal laser scanning microscope used to observe autophagosomes. The treatment of cells with emodin (80 μM) for 12 h resulted in a significant increase in GFP-LC3 puncta formation compared to that in untreated cells, which indicated the accumulation of autophagosomes (Figure 3E and F).
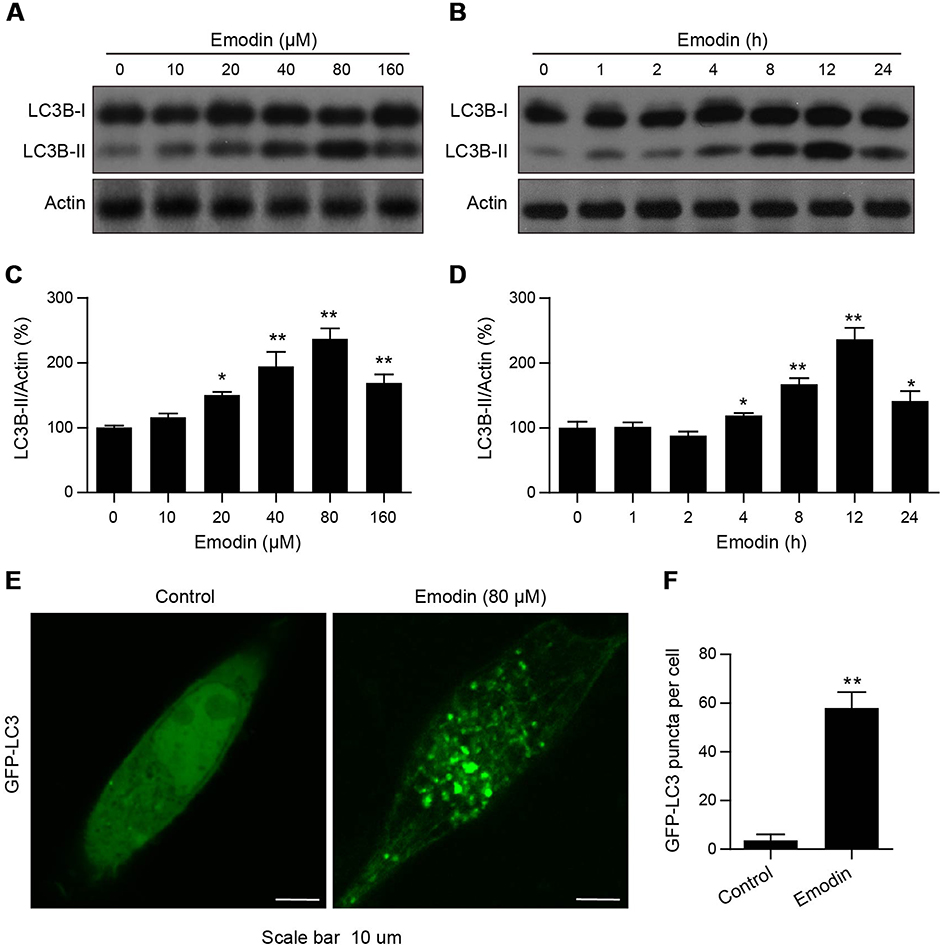 |
Figure 3 Emodin induced autophagic alterations in L02 cells. (A and B) Cells were treated with different concentrations of emodin for 24 h or with emodin (160 µM) for different time intervals. The expression of LC3B-I/II was analysed by Western blot. (C and D) The intensities of LC3B-II in (A and B) normalized to actin were statistically analysed and represented as the mean ± SD for 3 independent experiments. *P<0.05 compared with the control group; **P<0.01 compared with the control group. (E) GFP-LC3 expression in L02 cells treated with or without emodin (80 µM) for 12 h. GFP-LC3 puncta were examined using confocal microscopy; scale bars: 10 μm. (F) The average number of GFP-LC3 puncta per cell in (E) was quantitated from 3 independent experiments. Data are presented as the mean ± SD for 3 independent experiments. Thirty cells were analysed per treatment condition. **P<0.01 compared with the control group. |
Emodin inhibited the PI3K/AKT/mTOR pathway
To further understand the mechanism of emodin in autophagy, the levels of p-PI3K, PI3K, p-AKT, AKT, p-mTOR and mTOR were detected. Western blot analysis showed that emodin treatment resulted in a dose-dependent decrease in the expression of p-PI3K, p-AKT and p-mTOR in L02 cells compared to that observed in cells without emodin treatment; however, the level of p-mTOR was marginally elevated with 10 μM emodin treatment compared to that observed in cells without emodin treatment (Figure 4A and B). In addition, changes in the expression levels of PI3K, AKT and mTOR were not observed (Figure 4A and B). These results suggested that emodin might induce autophagy through the suppression of the PI3K/AKT/mTOR pathway.
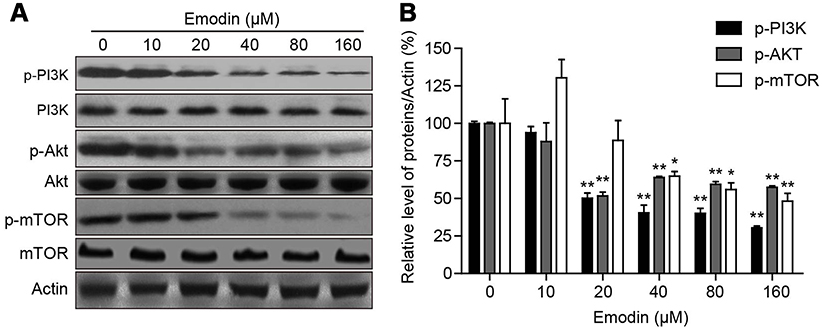 |
Figure 4 Emodin inhibited the PI3K/AKT/mTOR pathway. (A) L02 cells were treated with different concentrations of emodin for 24 h. The expression of p-PI3K(Tyr485), PI3K, p-AKT (Thr308), AKT, p-mTOR (Ser2448) and mTOR were analysed by Western blot. (B) The intensities of p-PI3K, p-AKT and p-mTOR in (A) normalized to actin were statistically analysed and represented as the mean ± SD for 3 independent experiments. *P<0.05 compared with the control group; **P<0.01 compared with the control group. |
Emodin-induced autophagy played a protective role in L02 cells
Autophagy has been reported to be both a protective and a lethal process in cells, so we investigated the role of emodin-induced autophagy by using 3-MA, an autophagy inhibitor that targets the initiation of autophagy.17 Cotreatment of L02 cells with emodin and 3-MA significantly reduced the expression of LC3B-II and cell viability compared with those in cells treated with emodin alone (Figure 5A–C). In addition, the apoptosis rates were significantly increased with emodin and 3-MA treatment compared with those after treatment with emodin alone (Figure 5D). The L02 cells were largely shrinkage with apoptotic morphologies under cotreatment with emodin and 3-MA (Figure 5E). Moreover, cotreatment of L02 cells with rapamycin (an autophagy inducer that targets mTOR) and emodin significantly increased the expression of LC3B-II and cell viability compared with those after treatment with emodin alone (Figure 5F–H) and markedly attenuated apoptosis compared with the apoptosis rates following treatment with emodin alone (Figure 5I). The L02 cells exhibited reduced shrinkage with rapamycin and emodin cotreatment than with emodin alone (Figure 5J). These data suggested that emodin-induced autophagy had a protective role in L02 cells.
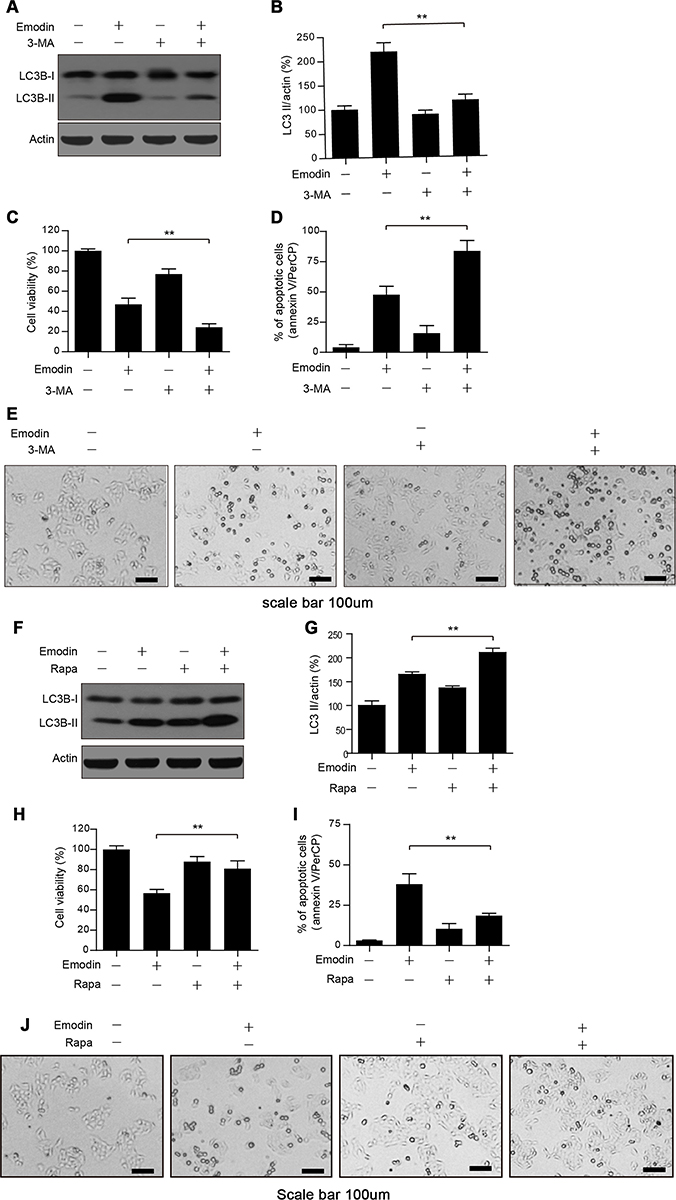 |
Figure 5 Emodin-induced autophagy played a protective role in L02 cells. (A) Cells were treated with or without emodin (40 µM) in the absence or presence of 3-MA (5 mM) for 24 h. The expression of LC3B-I/II was analysed by Western blot. (B) The intensities of LC3B-II in (A) normalized to actin were statistically analysed and represented as the mean ± SD for 3 independent experiments. **P<0.01. (C) Cell viability in (A) was determined by CCK-8 assay. Data are presented as the means ± SDs for 3 independent experiments. **P<0.01. (D) Cell apoptosis in (A) was detected by flow cytometry using annexin V/PerCP staining. Data are presented as the means ± SDs for 3 independent experiments. (E) Representative photos depicting the morphology of L02 cells treated in (A) Scale bars: 100 μm. (F) Cells were treated with or without emodin (40 µM) in the absence or presence of rapamycin (Rapa; 100 nM) for 24 h. The expression of LC3B-I/II was analysed by Western blot. (G) The intensities of LC3B-II in (F) normalized to actin were statistically analysed and are represented as the mean ± SD for 3 independent experiments. **P<0.01. (H) Cell viability in (F) was determined by CCK-8 assay. Data are presented as the means ± SDs for 3 independent experiments. **P<0.01. (I) Cell apoptosis in (F) was detected by flow cytometry using annexin V/PerCP staining. Data are presented as the means ± SDs for 3 independent experiments. (J) Representative photos depicting the morphology of L02 cells treated in (F). Scale bars: 100 μm. |
Discussion
In recent years, the role of traditional Chinese herbs in the treatment of multiple diseases has drawn much attention from scientists because of their high efficiencies and low toxicities. Emodin, an anthraquinone derivative isolated from Polygonum multiflorum, has gained increasing attention as a therapeutic agent. Leukaemia, cervical cancer, lung cancer, colorectal cancer and hepatocellular carcinoma cells were reported to be targets of emodin, with less toxicity for non-tumour cells reported in previous studies.18–22 Recently, the hepatotoxicity of emodin was reported to be ~30 μM.23 Consistent with this report, we found that emodin at concentrations above 20 μM had an obvious cytotoxic effect on the hepatic cell line L02 by promoting cell apoptosis. In addition to its anti-tumour activities, emodin protected against diabetic cardiomyopathy by regulating the AKT/GSK-3β signalling pathway,24 and it was also reported that emodin protected against cisplatin-induced apoptosis in rat renal tubular cells by activating autophagy through the modulation of the AMPK/mTOR pathway.25 Consistent with these reports, we also found that emodin could induce autophagy in the L02 hepatic cell line with the following evidence: (a) emodin treatment significantly upregulated the expression of LC3B-II in a dose- and time-dependent manner compared to that observed without emodin treatment and resulted in the significant accumulation of autophagosomes in L02 cells; (b) emodin caused the significant downregulation of the expression of p-PI3K, p-AKT and p-mTOR without any obvious changes in the expression of PI3K, AKT and mTOR in a dose-dependent manner compared to that observed without emodin treatment; and (c) the cotreatment of cells with emodin and 3-MA reduced the expression of LC3B-II, and the combinational treatment of cells with emodin (40 μM, a non-saturating concentration that induces autophagy) and rapamycin enhanced the expression of LC3B-II compared to that observed following treatment with emodin alone.
Autophagy is commonly considered an anti-apoptotic process that maintains the health of mammalian cells, probably by eliminating damaged organelles such as mitochondria,26 the endoplasmic reticulum,27 and part of the nucleus.28 In addition, autophagy is also thought to be a kind of programmed cell death with excessive engulfment and degradation of the cytoplasm, although the mechanism of this is still largely unclear, and this classification is still controversial.29 In the present study, both the autophagy inhibitor 3-MA and the autophagy inducer rapamycin were recruited to understand the role of emodin-induced autophagy in hepatocytes. The combination of emodin with 3-MA significantly promoted apoptosis in L02 cells compared to that observed in cells treated with emodin alone. Furthermore, emodin and rapamycin cotreatment markedly attenuated apoptosis in L02 cells compared with that observed following emodin treatment alone. These results suggested that emodin-induced autophagy has a protective role against cellular apoptosis, especially in at low/medium dose (10–80 μM). Notably, with a high dose (160 μM) of emodin, the expression of LC3B-II was reduced compared to that observed with 80 μM emodin treatment. This could partly be due to the caspase-mediated cleavage of essential autophagy proteins, which produced conditions in which autophagy could not cope with cellular stresses.30 Additionally, the protective effect of emodin-induced autophagy in hepatic cells could be reinforced by rapamycin, illustrating that mTOR might be a potential therapeutic target for liver protection in the future. More attention needs to be paid to emodin for its future clinical usage because, although emodin showed hepatoprotective effects in some reports, it exerted hepatotoxic effects in a dose- and time-dependent manner in our present study.
In summary, we demonstrated that emodin induced L02 cell apoptosis through upregulating the expression of cleaved caspase-3 and induced L02 cells autophagy through attenuating the phosphorylation of PI3K, AKT and mTOR. Autophagy could play a protective role against cell apoptosis in hepatocytes (Figure 6). These findings improved the understanding of the hepatoprotective activities and hepatotoxic effects of emodin treatment.
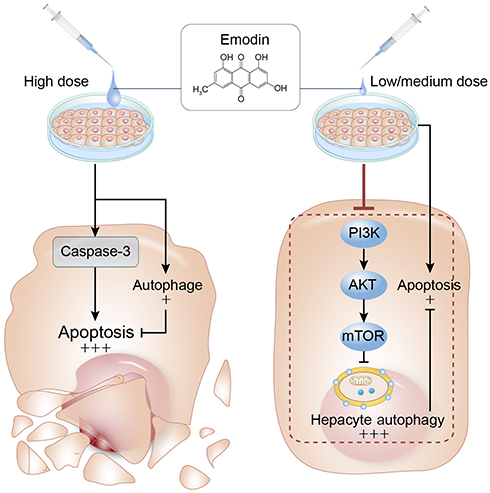 |
Figure 6 Summary of the current study. At low/medium concentrations, emodin-induced autophagy hinders apoptosis and maintains L02 cell viability, while high concentrations of emodin induces a marginal autophagy which hardly cope apoptosis and leads cell death. Arrows and black words part represent activation of target molecules or stimulation of production, and blunt lines and grey words part indicate target inhibition. |
Abbreviation list
3-MA, 3-methyladenine; AMPK, adenosine 5ʹ-monophosphate (AMP)-activated protein kinase; DEPTOR, DEP domain-containing mTOR-interacting protein; GFP, green fluorescent protein; LC3, microtubule-associated protein 1A/1B-light chain 3; PRAS40, proline-rich AKT substrate of 40 kDa; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Acknowledgments
This work was partially supported by the Natural Science Foundation of Chongqing (cstc2018jscx-msyX0172), and the authors would like to thank the Central Laboratory of Xinqiao Hospital at the Army Medical University.
Author contributions
The study was conceived and designed by Shi-wen Zhou, Shi-ming Yang and Xiao-yuan Zheng. Technical support was provided by Rong Zhang, Guo-bing Li and Su-min Wang. The manuscript was drafted by Xiao-yuan Zheng and revised by Shi-wen Zhou and Shi-ming Yang. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Park GJH, Mann SP, Ngu MC. Acute hepatitis induced by Shou‐Wu‐Pian, a herbal product derived from polygonum multiflorum. J Gastroenterol Hepatol. 2001;16(1):115–117.
2. Mazzanti G, Battinelli L, Daniele C, et al. New case of acute hepatitis following the consumption of Shou Wu Pian, a Chinese herbal product derived from Polygonum multiflorum. Ann Intern Med. 2004;140:7. doi:10.7326/0003-4819-140-7-200404060-00042-w3
3. Ma J, Zheng L, He YS, Li HJ. Hepatotoxic assessment of polygoni multiflori radix extract and toxicokinetic study of stilbene glucoside and anthraquinones in rats. J Ethnopharmacol. 2015;162:61–68. doi:10.1016/j.jep.2014.12.045
4. Shrimali D, Shanmugam MK, Kumar AP, et al. Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 2013;341(2):139–149. doi:10.1016/j.canlet.2013.08.023
5. Ahmad W, Zaidi SMA, Mujeeb M, Ansari SH, Ahmad S. HPLC and HPTLC methods by design for quantitative characterization and in vitro anti-oxidant activity of polyherbal formulation containing rheum emodi. J Chromatogr Sci. 2014;52(8):911–918. doi:10.1093/chromsci/bmt123
6. Chang C-H, Lin -C-C, Yang -J-J, Namba T, Hattori M. Anti-inflammatory effects of emodin from ventilago leiocarpa. Am J Chin Med (Gard City N Y). 1996;24(02):139–142. doi:10.1142/S0192415X96000189
7. Zhang HM, Wang F, Qiu Y, et al. Emodin inhibits coxsackievirus B3 replication via multiple signalling cascades leading to suppression of translation. Biochem J. 2016;473(4):473–485. doi:10.1042/BJ20150419
8. Yin X, Gong X, Jiang R, et al. Emodin ameliorated lipopolysaccharide-induced fulminant hepatic failure by blockade of TLR4/MD2 complex expression in D-galactosamine-sensitized mice. Int Immunopharmacol. 2014;23(1):66–72. doi:10.1016/j.intimp.2014.08.018
9. Liu X, Liu Y, Qu Y, Cheng M, Xiao H. Metabolomic profiling of emodin-induced cytotoxicity in human liver cells and mechanistic study. Toxicol Res (Camb). 2015;4(4):948–955. doi:10.1039/C4TX00246F
10. Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75. doi:10.1016/j.cell.2014.02.049
11. Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19(1):87–95. doi:10.1038/cdd.2011.146
12. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–1295. doi:10.1016/j.febslet.2010.01.017
13. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi:10.1016/j.cell.2012.03.017
14. Chun-Guang W, Liang Z, Yong LL, et al. Emodin exerts an antiapoptotic effect on human chronic myelocytic leukemia K562 cell lines by targeting the PTEN/PI3K-AKT signaling pathway and deleting BCR-ABL. Integr Cancer Ther. 2016;16(4):526–539. doi:10.1177/1534735416664784
15. Chen D, Liu J, Lu L, et al. Emodin attenuates TNF-alpha-induced apoptosis and autophagy in mouse C2C12 myoblasts though the phosphorylation of Akt. Int Immunopharmacol. 2016;34:107–113. doi:10.1016/j.intimp.2016.02.023
16. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222.
17. Li X, Xu HL, Liu YX, An N, Zhao S, Bao JK. Autophagy modulation as a target for anticancer drug discovery. Acta Pharmacol Sin. 2013;34(5):612–624. doi:10.1038/aps.2013.23
18. Pooja T, Karunagaran D. Emodin suppresses Wnt signaling in human colorectal cancer cells SW480 and SW620. Eur J Pharmacol. 2014;742:55–64. doi:10.1016/j.ejphar.2014.08.028
19. Yaoxian W, Hui Y, Yunyan Z, Yanqin L, Xin G, Xiaoke W. Emodin induces apoptosis of human cervical cancer hela cells via intrinsic mitochondrial and extrinsic death receptor pathway. Cancer Cell Int. 2013;13(1):1. doi:10.1186/1475-2867-13-71
20. Yu JQ, Bao W, Lei JC. Emodin regulates apoptotic pathway in human liver cancer cells. Phytother Res. 2013;27(2):251–257. doi:10.1002/ptr.4703
21. Ok S, Kim S-M, Kim C, et al. Emodin inhibits invasion and migration of prostate and lung cancer cells by downregulating the expression of chemokine receptor CXCR4. Immunopharmacol Immunotoxicol. 2012;34(5):768–778. doi:10.3109/08923973.2012.654494
22. Chen Y, Li J, Hu J, et al. Emodin enhances ATRA-induced differentiation and induces apoptosis in acute myeloid leukemia cells. Int J Oncol. 2014;45(5):2076–2084. doi:10.3892/ijo.2014.2610
23. Li CL, Ma J, Zheng L, Li HJ, Li P. Determination of emodin in L-02 cells and cell culture media with liquid chromatography-mass spectrometry: application to a cellular toxicokinetic study. J Pharm Biomed Anal. 2012;71:71–78. doi:10.1016/j.jpba.2012.07.031
24. Wu Z, Chen Q, Ke D, Li G, Deng W. Emodin protects against diabetic cardiomyopathy by regulating the AKT/GSK-3beta signaling pathway in the rat model. Molecules. 2014;19(9):14782–14793. doi:10.3390/molecules190914782
25. Liu H, Gu LB, Tu Y, Hu H, Huang YR, Sun W. Emodin ameliorates cisplatin-induced apoptosis of rat renal tubular cells in vitro by activating autophagy. Acta Pharmacol Sin. 2016;37(2):235–245. doi:10.1038/aps.2015.114
26. Li Q, Zhang T, Wang J, et al. Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem Biophys Res Commun. 2014;444(2):182–188. doi:10.1016/j.bbrc.2014.01.032
27. Lipatova Z, Segev N, Hochstrasser M. A role for macro-ER-phagy in ER quality control. PLoS Genet. 2015;11(7):e1005390. doi:10.1371/journal.pgen.1005390
28. Dou Z, Xu C, Donahue G, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527(7576):105. doi:10.1038/nature15724
29. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–1010. doi:10.1038/nrm2529
30. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. doi:10.1038/nrm3735
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.