")
Back to Archived Journals » Cell Health and Cytoskeleton » Volume 7
Emerging roles for the pH-sensing G protein-coupled receptors in response to acidotic stress
Authors Sanderlin E, Justus C, Krewson E, Yang L
Received 19 November 2014
Accepted for publication 15 January 2015
Published 2 March 2015 Volume 2015:7 Pages 99—109
DOI https://doi.org/10.2147/CHC.S60508
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Denis Wirtz
Edward J Sanderlin,1 Calvin R Justus,1 Elizabeth A Krewson,2 Li V Yang1,2
1Department of Internal Medicine, Brody School of Medicine, East Carolina University, Greenville, NC, USA; 2Department of Anatomy and Cell Biology, Brody School of Medicine, East Carolina University, Greenville, NC, USA
Abstract: Protons (hydrogen ions) are the simplest form of ions universally produced by cellular metabolism including aerobic respiration and glycolysis. Export of protons out of cells by a number of acid transporters is essential to maintain a stable intracellular pH that is critical for normal cell function. Acid products in the tissue interstitium are removed by blood perfusion and excreted from the body through the respiratory and renal systems. However, the pH homeostasis in tissues is frequently disrupted in many pathophysiologic conditions such as in ischemic tissues and tumors where protons are overproduced and blood perfusion is compromised. Consequently, accumulation of protons causes acidosis in the affected tissue. Although acidosis has profound effects on cell function and disease progression, little is known about the molecular mechanisms by which cells sense and respond to acidotic stress. Recently a family of pH-sensing G protein-coupled receptors (GPCRs), including GPR4, GPR65 (TDAG8), and GPR68 (OGR1), has been identified and characterized. These GPCRs can be activated by extracellular acidic pH through the protonation of histidine residues of the receptors. Upon activation by acidosis the pH-sensing GPCRs can transduce several downstream G protein pathways such as the Gs, Gq/11, and G12/13 pathways to regulate cell behavior. Studies have revealed the biological roles of the pH-sensing GPCRs in the immune, cardiovascular, respiratory, renal, skeletal, endocrine, and nervous systems, as well as the involvement of these receptors in a variety of pathological conditions such as cancer, inflammation, pain, and cardiovascular disease. As GPCRs are important drug targets, small molecule modulators of the pH-sensing GPCRs are being developed and evaluated for potential therapeutic applications in disease treatment.
Keywords: acidosis, GPCR, GPR4, GPR65 (TDAG8), GPR68 (OGR1)
Introduction
Cellular metabolism produces acid as a byproduct. Metabolism of each glucose molecule by glycolysis generates two pyruvate molecules. Under anaerobic conditions the metabolism of pyruvate results in the production of the glycolytic end product lactic acid, which has a pKa of 3.9. Lactic acid is deprotonated at the carboxyl group and results in one lactate ion and one proton at the physiological pH. Under aerobic conditions pyruvate is converted into acetyl-CoA and CO2 in the mitochondria. CO2 in water forms a chemical equilibrium of carbonic acid and bicarbonate, an important physiological pH buffering system. The body must maintain suitable pH for proper physiological functions. Some regulatory mechanisms to control systemic pH are respiration, renal excretion, bone buffering, and metabolism.1–4 The respiratory system can buffer the blood by excreting carbonic acid as CO2 while the kidney responds to decreased circulatory pH by excreting protons and electrolytes to stabilize the physiological pH. Bone buffering helps maintain systemic pH by Ca2+ reabsorption and mineral dissolution. Collectively, it is clear that several biological systems require tight regulation to maintain pH for normal physiological functions. Cells utilize vast varieties of acid-base transporters for proper pH homeostasis within each biological context.5–8 Some such transporters are H+-ATPase, Na+/H+ exchanger, Na+-dependent HCO3–/C1– exchanger, Na+-independent anion exchanger, and monocarboxylate transporters. Cells can also maintain short-term pH homeostasis of the intracellular pH by rapid H+ consuming mechanisms. Some such mechanisms utilize metabolic conversions that move acids from the cytosol into organelles. Despite these cellular mechanisms that tightly maintain proper pH homeostasis, there are many diseases whereby pH homeostasis is disrupted. These pathological conditions are characterized by either local or systemic acidosis. Systemic acidosis can occur from respiratory, renal, and metabolic diseases and septic shock.1–4,9 Additionally, local acidosis is characterized in ischemic tissues, tumors, and chronically inflamed conditions such as in asthma and arthritis caused by deregulated metabolism and hypoxia.10–15
Acidosis is a stress for the cell. The ability of the cell to sense and modulate activity for adaptation to the stressful environment is critical. There are several mechanisms whereby cells sense acidosis and modulate cellular functions to facilitate adaptation. Cells can detect extracellular pH changes by acid sensing ion channels (ASICs) and transient receptor potential (TRP) channels.16 Apart from ASIC and TRP channels, extracellular acidic pH was shown to stimulate inositol polyphosphate formation and calcium efflux.17,18 This suggested the presence of an unknown cell surface receptor that may be activated by a certain functional group, namely the imidazole of a histidine residue. The identity of the acid-activated receptor was later unmasked by Ludwig et al as a family of proton-sensing G protein-coupled receptors (GPCRs). This group identified human ovarian cancer GPCR 1 (OGR1) which upon activation will produce inositol phosphate and calcium efflux through the Gq pathway.19 These pH-sensing GPCR family members, including GPR4, GPR65 (TDAG8), and GPR68 (OGR1), will be discussed in this review (Figure 1). The proton-sensing GPCRs sense extracellular pH by protonation of several histidine residues on their extracellular domain. The activation of these proton-sensing GPCRs facilitates the downstream signaling through the Gq/11, Gs, and G12/13 pathways. Their expression varies in different cell types and play critical roles in sensing extracellular acidity and modulating cellular functions in several biological systems.
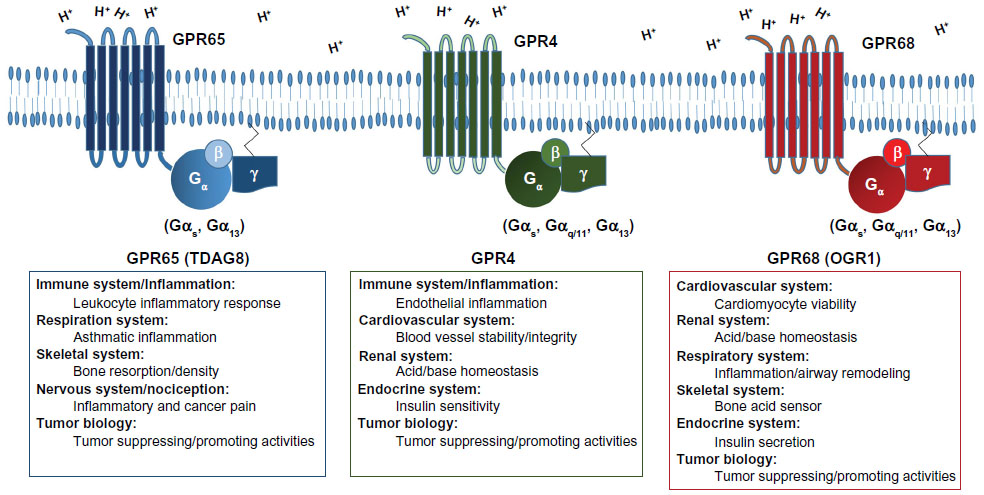 | Figure 1 Biological roles and G protein coupling of the pH-sensing GPCRs. |
Role for the pH-sensing GPCRs in the immune system and inflammation
Acidic pH is a main characteristic of the inflammatory loci.14,20,21 The acidic microenvironment in inflamed tissue is predominately due to the increased metabolic demand from infiltrating immune cells, such as the neutrophil. These immune cells increase oxygen consumption and glucose uptake for glycolysis and oxidative phosphorylation. When oxygen availability is limited, cells often undergo anaerobic glycolysis. This process generates increasing amounts of lactic acid, thereby creating a local acidic microenvironment within the inflammatory loci.22 This presents a role for the pH-sensing GPCR GPR65 (TDAG8) in inflammation and immune cell function.23 TDAG8 was originally identified by cloning as an orphan GPCR which was observed to be upregulated during thymocyte apoptosis.24,25 GPR65 (TDAG8) is predominately expressed in lymphoid tissues such as the spleen, lymph nodes, thymus, and leukocytes.24–26 It was demonstrated that GPR65 inhibited pro-inflammatory cytokine secretion, which includes IL-6 and TNF-α, in mouse peritoneal macrophages upon activation by extracellular acidification. This cytokine inhibition was shown to occur through the Gs-cAMP-protein kinase A (PKA) signaling pathway.23,27 Treatment with dexamethasone, a potent glucocorticoid, increased GPR65 expression in peritoneal macrophages. Following dexamethasone treatment, there was an inhibition of TNF-α secretion in a manner dependent on increased expression of GPR65.28 Another report provides an anti-inflammatory role for GPR65 in arthritis.29 Type II collagen-induced arthritis was increased in GPR65-null mice in comparison to wild-type mice. These studies taken together suggest GPR65 serves as a negative regulator in inflammation.30 However, one study provided a function for GPR65 as a positive modulator in inflammation.31 GPR65 was reported to increase eosinophil viability in the acidic microenvironment by reducing apoptosis through the cAMP pathway. As eosinophils are central in asthmatic inflammation and allergic airway disease, GPR65 may play a role in increasing asthmatic inflammation.31 On the other hand, GPR65 has shown little involvement in immune cell development. One report indicates that GPR65 knockout mice had normal immune development and function.26 Modulation of inflammation by GPR65 is complex and must be examined within each specific pathology.23
In addition to GPR65, GPR4 is also involved in the inflammatory response. Endothelial cells compose blood vessels that often penetrate acidic tissue microenvironments such as the inflammatory loci. Among the pH-sensing GPCR family, GPR4 has the highest expression in endothelial cells. Response to inflammation by vascular endothelial cells facilitates the induction of inflammatory cytokines that are involved in the recruitment of leukocytes for adherence and transmigration into inflamed tissues. Activation of GPR4 by acidosis in human umbilical vein endothelial cells, among other endothelial cell types, increased the expression of a broad range of pro-inflammatory genes including chemokines, cytokines, PTGS2, NF-κB pathway genes, and adhesion molecules.32 Moreover, human umbilical vein endothelial cells, when treated with acidic pH, increased GPR4-mediated endothelial adhesion to leukocytes.32,33 Altogether, GPR65 and GPR4 provide differential regulation of the inflammatory response through their acid sensing capabilities. GPR65 predominately demonstrates function in the inhibition of the inflammatory response whereas GPR4 activation exacerbates inflammation.
Role for the pH-sensing GPCRs in the cardiovascular system
The cardiovascular system is essential for the delivery of oxygen and nutrients while also removing metabolic waste from tissues. Vascular occlusion, however, results in tissue ischemia and acidification due to oxygen deprivation, anaerobic metabolism, and deficiency to remove acid byproducts. Within the cardiac system, acidosis disrupts the regulation of action potential duration associated with cardiac excitability.34 Most notably, this action potential disruption is caused by cardiac ischemia and acidosis, which impairs sodium channels and can lead to life-threatening events such as arrhythmia, myocardial infarction, and cardiac death.35–37
A decrease in interstitial tissue pH may alter angiogenesis.38 Angiogenesis involves the activation of endothelial cells, which line blood vessels, to switch from a quiescent state to a vascular-sprouting state, giving rise to a functional blood vessel network. This change in active growth can be regulated by signaling through hypoxia, acidosis, VEGF, nitric oxide, among others.39,40 A decrease in interstitial tissue pH, which creates an acidic microenvironment, has been attributed to abnormalities in VEGF and VEGF receptor expression and signaling. Furthermore, acidosis may inhibit angiogenesis depending on the extracellular proton concentration.38
Disturbances in acid-base balance within the cardiovascular system can also influence vascular tone by modulating endothelial and smooth muscle interactions and function. Intracellular acidosis is associated with vasodilation of large arteries; whereas intracellular acidosis initiates vasoconstriction within smaller arteries noted in the pulmonary vascular system.41,42 Additionally, a recent study showed that acidic pH increased relaxation of human placental arteries.43
The pH-sensing GPCRs have recently been studied as candidate acid sensors in the cardiovascular system. The GPR4 receptor is highly expressed in vascular endothelial cells.33,44 GPR4 is activated by extracellular acidic pH to initiate signaling cascades which regulate cellular function.19,33,45,46 It has been reported that GPR4 is a functional pH sensor in vascular endothelial cells and a regulator of blood vessel stability.47 Whereas adult GPR4-null mice in the C57BL6/129 mixed genetic background were fertile and phenotypically normal, there were smaller litter sizes and a higher perinatal mortality rate in partial correlation with spontaneous hemorrhaging and respiratory distress in a fraction of GPR4-null neonates.47 Histological analysis revealed GPR4-null neonates with hemorrhages exhibiting disorganized and tortuous blood vessels associated with reduced mural cell coverage.47 Studies on another GPR4 knockout mouse strain observed that the blood vessels of tumors formed in the GPR4-deficient mice were fragmented and fragile, also suggesting that GPR4 may regulate blood vessel integrity.44 Similar to the previous study,47 the phenotype of this GPR4 knockout mouse strain was also grossly normal.44 However no perinatal mortality was observed in this knockout strain, which might be related to differences in mouse strain genetic backgrounds, knockout constructs, and breeding conditions. Wyder et al also noted that the GPR4-deficient mice showed a reduced angiogenic response to VEGF, implicating the involvement of GPR4 in VEGF-driving angiogenesis.44 It has been shown that GPR4 activation by acidosis stimulated the expression of endoplasmic reticulum stress response genes such as ATF3 and CHOP (DDIT3) in vascular endothelial cells.32,48 Moreover, a recent study demonstrated that acidosis/GPR4-induced CHOP expression was involved in endothelial cell apoptosis in a renal ischemia/reperfusion mouse model.49 These studies provide evidence that implicate involvement of GPR4 in regulating the vascular system.
Another proton-sensing GPCR, GPR68 (OGR1), has also shown involvement in the response of the cardiovascular system to tissue acidosis. Russell et al identified 3,5-disubstituted isoxazoles (lsx), which are cardiomyogenic small molecules targeting Notch activated epicardium-derived cells, as agonists of the GPR68 receptor.50 GPR68-expressing cardiomyocytes formed a proton-sensing cellular buffer zone surrounding the infarcted myocardium. Activation of GPR68 by lsx induced the expression of cardiomyogenic and pro-survival genes.50 In human aortic smooth muscle cells, GPR68 is the major receptor responsible for extracellular acidic pH-induced production of inositol phosphate, PGI2, and cAMP.51 Additionally, acidic pH-induced vascular actions of aortic smooth muscle cells can be divided into GPR68-dependent effects such as COX-2 expression, PGI2 production, and MAPK phosphatase-1 expression and GPR68-independent effects such as PAI-1 expression and cell proliferation.52
Taken together, both GPR4 and GPR68 play roles in regulating the function of the cardiovascular system. GPR4 regulates blood vessel stability and endothelial cell function and GPR68 increases cardiomyogenic and pro-survival gene expression while also mediating aortic smooth muscle cell gene expression.
Role for the pH-sensing GPCRs in the renal system
The human body has several ways to buffer systemic pH to maintain normal physiological homeostasis. The physiological pH of arterial blood is around pH 7.4. Under pathological conditions the body’s blood pH can become acidic (metabolic or respiratory acidosis) or basic (metabolic or respiratory alkalosis). A physiological response to buffer blood pH can occur quickly (minutes) via respiratory activity but may also be modified slowly (days) by controlling the level of extra-cellular fluid electrolytes through the renal system. Respiratory activity can increase via hyperventilation in response to metabolic acidosis to excrete carbonic acid (H2CO3) in the form of a gas (CO2). When there are excess acids other than CO2 in the circulatory system the kidneys will excrete them in an effort to restore normal blood pH. Chronic kidney disease among other renal impairments can result in the damage of functioning glomeruli as well as nephrons, which may lead to severe metabolic acidosis. The loss of bases through intestinal bicarbonate malabsorption in the form of diarrhea may also lead to metabolic acidosis, which is one of the most common acid/base problems. Metabolic acidosis may lead to poor nutritional status, uremic bone disease, and may correlate with patient mortality.53–55 In addition, acidosis in the kidney has recently been found to exacerbate the progression of nephropathy.56,57 As some mechanisms of how circulatory pH may be buffered in response to metabolic acidosis have been discovered, exactly how kidney cells may sense acidosis remains to be fully elucidated. Two of the proton-sensing GPCRs, GPR4 and GPR68, have been studied as pH sensors in the renal system.58,59 GPR4 and GPR68 are expressed in the lung and kidney, which may both be necessary to successfully buffer the circulatory system and maintain pH homeostasis.
GPR4 is expressed in the kidney cortex, isolated kidney collecting ducts, inner and outer medulla, and in cultured inner and outer medullary collecting duct cells.59 In mice deficient for GPR4, renal acid excretion and the ability to respond to metabolic acidosis was reduced.59 In response to acidosis, inner and outer medullary collecting duct cells produced cAMP, a second messenger for the Gs G-protein pathway, through the GPR4 receptor.59 In renal HEK293 epithelial cells GPR4 overexpression was found to increase the activity of PKA.60 In addition, the protein expression of H+-K+-ATPase α-subunit (HKα2) was increased following GPR4 overexpression dependent on increased PKA activity.60
GPR68 has also been reported to alter proton export of HEK293 cells by stimulating the Na+/H+ exchanger and H+-ATPase.58 The activation of GPR68 by acidosis was found to stimulate this effect through a cluster of extracellular histidine residues and the Gq/PKC signaling pathway.58 In GPR68-null mice the expression of the pH-sensitive kinase Pyk2 in the kidney proximal tubules was upregulated which might compensate for GPR68 deficiency.58 Taken together, GPR4 and GPR68 may both be necessary for successful systemic pH buffering by controlling renal acid excretion.
Role for the pH-sensing GPCRs in the respiratory system
The primary function of the respiratory system is to provide an adequate oxygen supply to tissues while subsequently removing carbon dioxide by exhalation. Enhancement of gas transport occurs by hemoglobin binding of oxygen and bicarbonate transport of carbon dioxide. This mechanism maintains an acid/base homeostasis that serves as a required pH buffering system. Respiratory acidosis results from an alteration in the partial pressures of carbon dioxide, a product of cellular metabolism. An increase in carbon dioxide in the blood circulation will facilitate proton accumulation by means of carbonic acid conversion by carbonic anhydrase. Respiratory acidosis has been observed in several respiratory diseases, such as chronic obstructive pulmonary disease (COPD) and asthma.12,61–63
Asthma is a chronic inflammatory disease, which is associated with bronchial hyper-responsiveness, airway inflammation, remodeling, and acidic features. Ichimonji et al demonstrated in human airway smooth muscle cells that GPR68 mediated extracellular acidification-induced production of pro-inflammatory cytokine IL-6 and increased intracellular Ca2+ concentration.64 The same group also found that extracellular acidification of pH 6.3 induced the expression of CTGF, which was involved in the formation of extracellular matrix proteins and associated with airway remodeling, through the GPR68/Gq/11/IP3/Ca2+ pathway.65 Furthermore, Saxena et al demonstrated that GPR68 receptor mediated airway smooth muscle cell contraction in an acidic extracellular environment.66 Inflammatory airway disease is also known to have excess mucus excretion, which can be exacerbated by exposure to an acidic microenvironment. Liu et al showed that airway acidification could induce the hyper-secretion of MUC5AC, a protein associated with mucus production within the pulmonary tracts, in human airway epithelial cells.67
Aoki et al demonstrated that GPR68-deficient mice were resistant to asthma along with inhibiting Th2 cytokine and immunoglobulin E production.68 This study concludes that GPR68 in dendritic cells is crucial for the onset of asthmatic responses.68 Moreover, GPR65 has been implicated as having a role in respiratory disorders as it is highly expressed in eosinophils, hallmark cells for asthmatic inflammation.69 Kottyan et al showed that GPR65 increased the viability of eosinophils within an acidic environment through the cAMP pathway in murine asthma models.31 In summary, GPR68 and GPR65 play important roles in the respiratory system and asthma. GPR68 regulates gene expression in airway epithelial, smooth muscle and immune cells while GPR65 enhances the survival of airway eosinophils in response to acidosis.
Role for the pH-sensing GPCRs in the skeletal system
Recently, pH-sensing GPCRs have been found to be important for response to metabolic acidosis in the skeletal system. During metabolic acidosis there are several mechanisms to restore normal physiological pH as mentioned previously. In addition to respiratory CO2 excretion and renal acid excretion, the skeletal system also carefully balances systemic pH. Acidosis has been reported to directly inhibit osteoblast activity, resulting in a reduction in the bone resorbing of minerals and Ca2+.3,70,71 Acidosis may also activate osteoclast activity to increase Ca2+ release in vitro.71 Ca2+ release and mineral dissolution from bone may help systemically buffer protons and restore normal physiological pH.72,73 Chronic metabolic acidosis can result in a reduction of total volumetric bone density and over time can result in osteoporosis.72 The reduction in osteoblast activity may also reduce the gene expression of several matrix proteins such as COL1A, OPN, and MGP, which may contribute to increased osteoarthritis development.71,74–76 Two of the pH-sensing G protein-coupled receptors, GPR68 and GPR65, have been reported to sense acidosis in bone cells.
Among other cell types, there is high expression of GPR68 in osteoblasts and osteoclasts as well as chondrocytes.19 As osteoclast and osteoblast homeostasis may regulate systemic pH by controlling the release of minerals from bone, GPR68 has been speculated as a possible bone acidosis sensor.19,76 In addition, GPR68 is expressed during osteoclastogenesis and may be involved in osteoclast differentiation.77 Acidosis has been determined to increase the accumulation of NFATc1 protein through NF-kappa B ligand (RANKL) in the nuclei of rat and rabbit osteoclasts.78 This was proposed to occur through prolonged Ca2+ release and the sequential activation of the calcineurin/NFAT pathway in response to GPR68 activation by acidosis.78 In normal human osteoblast (NHOst) cells acidosis has been found to stimulate the expression of COX2 and PGE2 through the GPR68-mediated Gq/PLC/Ca2+ signaling pathway.79 GPR68 is also expressed in cultured neonatal calvariae osteoblasts and in response to acidosis may increase intracellular Ca2+ release.76 Furthermore, following the transfection of a heterologous GPR68 construct into Chinese hamster ovary cells, the response to acidosis stimulated the intracellular release of Ca2+ as well.76 GPR68 expression in rat endplate chondrocytes was found to induce apoptosis in response to acidosis,80 which could reduce collagen production and lead to intervertebral disk degeneration.
GPR65 has also been reported as a pH sensor in bone. GPR65 is expressed in osteoclasts and its activity may inhibit Ca2+ resorption.81 Disruption of GPR65 gene exacerbated osteoclastic bone resorption in ovariectomized mice.81 The relative bone density of GPR65-null mice was less than control mice.81 In cultured osteoclast cells from mice deficient for GPR65, the normal inhibition of osteoclast formation in response to acidosis was abrogated.81 Taken together, this data suggest that the activation of GPR65 may enhance bone density, thus the GPR65 signaling may be important for disease processes such as osteoporosis and other bone density disorders.
Role for the pH-sensing GPCRs in the endocrine system
The effects of metabolic acidosis have been studied in the endocrine system. Metabolic acidosis can alter insulin secretion as well as pancreatic function,82 which is relevant in disease states such as diabetes. Recently, the pH-sensing GPCRs, GPR4 and GPR68, have been found to be involved in insulin secretion and tissue sensitivity to insulin.83,84 In GPR4 knockout mice glucose tolerance was augmented by increasing the sensitivity to insulin.83 The balance of pro-inflammatory and anti-inflammatory molecules are crucial for the maintenance of insulin sensitivity as pro-inflammatory cytokines can further promote insulin resistance and type II diabetes.85 The absence of GPR4 may reduce the expression of several inflammatory modulators in white adipose tissue such as IL-6 and PPARα and an increase in TNF-α and TGF1-β.83 GPR4 is also highly expressed in endothelial cells, which when activated has been reported to stimulate the expression of inflammatory genes.32,33
GPR68 has also been found to modify insulin production and secretion. In GPR68 knockout mice insulin secretion in response to glucose administration was reduced when compared to wild-type mice although blood glucose was not significantly altered.84 GPR68 deficiency in this respect may reduce insulin secretion but at the same time increase insulin sensitivity. In addition, stimulation of GPR68 in islet cells by acidosis increased the secretion of insulin through the Gq/11 G-protein signaling.84
Role for the pH-sensing GPCRs in the nervous system and nociception
Acidosis causes pain by exciting nociceptors located in sensory neurons. Several types of ion channels and receptors, such as ASICs, TRPV1, and proton-sensing GPCRs, have been identified as nociceptors in response to acidosis. ASICs and TRPV act as proton-gated membrane-bound channels, which are activated by acidic pH and mediate multimodal sensory perception including nociception.86–88 It has also been reported that acidosis causes excitation or modulation of nociceptive sensory neurons through proton-sensing GPCRs.89 All proton-sensing GPCRs are localized in the dorsal root ganglia that are the pain-relevant loci.89 Chen et al demonstrated the involvement of GPR65 in inflammatory pain by using mouse models with injection of capsaicin, carrageenan, and complete Freund’s adjuvant.89 Notably, GPR65 activation sensitized the response of TRPV1 to capsaicin. The results suggest high accumulation of protons post inflammation may not only stimulate nociceptive ion channels such as TRPV1 to trigger pain, but also activate proton-sensing GPCRs to regulate heightened sensitivity to pain.89 Furthermore, Hang et al demonstrated GPR65 activation elicited cancer-related bone pain through the PKA and phosphorylated CREB (pCREB) signaling pathway in the rat model.90 Collectively, GPR4, GPR65, and GPR68 are all expressed in the dorsal root ganglia; GPR65 is a functional receptor involved in nociception and the nervous system by sensitizing inflammatory pain and the evocation of cancer-related bone pain.
Role for the pH-sensing GPCRs in tumor biology
The tumor microenvironment is highly heterogeneous. Hypoxia, acidosis, inflammation, defective vasculature, poor blood perfusion, and deregulated cancer cell metabolism are hallmarks of the tumor microenvironment.91–93 The acidity in the tumor microenvironment is owing to the altered cancer cell metabolism termed the “Warburg Effect”. This metabolic phenotype allows the cancer cells to preferentially utilize glycolysis over oxidative phosphorylation as a primary means of energy production.94 This process occurs even in normoxic tissue environments where sufficient oxygen is available. Due to this phenomenon, the Warburg Effect is often termed “aerobic glycolysis”. This unique metabolic phenotype produces vast quantities of lactic acid, which serve as a proton source for acidification. Upon disassociation of lactic acid to one lactate molecule and one proton, the monocarboxylate transporter and proton transporters export lactate and protons into the extracellular tumor microenvironment.95 The proton-sensing GPCRs are activated by acidic pH and facilitate tumor cell modulation in response to extracellular acidification. GPR4, GPR65, and GPR68 play roles in tumor cell apoptosis, proliferation, metastasis, angiogenesis, and immune cell function.19,27,32,33,44,45,96,97
GPR4 has had conflicting reports in terms of tumor suppressing or promoting activities. One study demonstrated that GPR4 could act as a tumor metastasis suppressor, when overexpressed and activated by acidic pH in B16F10 melanoma cells, by impeding migration and invasion of tumor cells.45 GPR4 overexpression also significantly inhibited the lung metastasis of B16F10 melanoma cells in mice.45 Another study utilizing the B16F10 melanoma cell line which overexpressed GPR4 showed an increase in mitochondrial surface area and a significant reduction in membrane protrusions by quantification of 3D morphology.98 These data point to a decrease in cancer cell migration when GPR4 is overexpressed and provides another example of GPR4 as exhibiting tumor metastasis suppressor function.98 However, in another report GPR4 malignantly transformed immortalized NIH3T3 fibroblasts.99 This presents GPR4 with tumor-promoting capabilities. The conflicting reports seem to indicate the functional ability of GPR4 to act as a tumor promoter and a tumor suppressor depending on the context of certain cell types and biological systems.
Reports with GPR65 involvement in cancer cells provide evidence in favor for cancer cell survival; however, opposing evidences suggest GPR65 functions as a tumor suppressor. In the same report suggesting GPR4 is oncogenic due to GPR4 transforming immortalized NIH3T3 fibroblasts, GPR65 overexpression was able to transform the mouse NMuMG mammary epithelial cell line.99 Another group demonstrated in NCI-H460 human non-small cell lung cancer cells that GPR65 promotes cancer cell survival in an acidic microenvironment.100 Conversely, a recent study showed that GPR65 inhibited c-Myc oncogene expression in human lymphoma cells.101 Furthermore, GPR65 messenger ribonucleic acid expression was reduced by more than 50% in a variety of human lymphoma samples when compared to normal lymphoid tissues, therefore implying GPR65 has a tumor suppressor function in lymphoma.101 GPR65 has also been shown to increase glucocorticoid-induced apoptosis in murine lymphoma cells.102 These reports highlight cell type dependency and biological context for GPR65 activity as a tumor suppressor or promoter.
GPR68 also has roles in tumor biology as a potential tumor suppressor or a tumor promoter. Reports have shown that GPR68 can inhibit cancer metastasis, reduce cancer cell proliferation, and inhibit migration. One study showed that when GPR68 was overexpressed in prostate cancer cells, metastasis to the lungs, diaphragm, and spleen was inhibited.97 When GPR68 was overexpressed in ovarian cancer (HEY) cells, cellular proliferation and migration were significantly reduced, and cell adhesion to the extracellular matrix was increased.96 Another study reported GPR68 expression was critical for the tumor cell induced immunosuppression in myeloid-derived cells. This study proposed that GPR68 promotes M2 macrophage development and inhibits T-cell infiltration, and thereby facilitates tumor development.103 In summary, the biological roles of GPR4, GPR65, and GPR68 in tumor biology are complex and both tumor-suppressing and tumor-promoting functions have been reported, primarily dependent on cell type and biological milieu.
Development of small molecule modulators of the pH-sensing GPCRs
GPCRs are critical receptors for the regulation of many physiological operations. It is of little surprise that GPCRs have become a central focus of pharmaceutical development. In fact, 30%–50% of therapeutics focuses on modulating GPCR activity.104,105 In view of the diverse roles of the pH-sensing GPCRs in the context of multiple biological systems, targeting these receptors with small molecules and other modulators could serve as potential therapeutics for diseases associated with deregulated pH homeostasis. There have been recent developments in the characterization of GPR4 antagonists along with agonists for GPR65 and GPR68.29,32,50,106 The GPR4 antagonist demonstrated effectiveness in vitro to reduce the GPR4-mediated inflammatory response to acidosis in endothelial cells.32 The GPR65 agonist, BTB09089, showed in vitro effects in GPR65 activation of immune cells to inhibit inflammatory response; however, the activity of BTB09089 was not strong enough for the use in animal models in vivo.29 The GPR68 agonist, lsx, exhibited pro-neurogenic activity and induced hippocampal neurogenesis in young mice.107 It was also demonstrated that lsx suppressed the proliferation of malignant astrocytes.108 To date, however, much advancement needs to be done in development of efficacious agonists and antagonists of the pH-sensing GPCRs coupled with a capacity to target specific tissue dysfunction in the midst of systemic drug administration to optimize therapeutic effects and minimize potential adverse effects.
Concluding remarks
Cells encounter acidotic stress in many pathophysiologic conditions such as inflammation, cancer, and ischemia. Intricate molecular mechanisms, including a large array of acid/base transporters and acid sensors, have evolved for cells to sense and respond to acidotic stress. Emerging evidence has demonstrated that a family of the pH-sensing GPCRs can be activated by extracellular acidotic stress and regulate the function of multiple physiological systems (Table 1). The pH-sensing GPCRs also play important roles in various pathological disorders. Agonists, antagonists and other modulators of the pH-sensing GPCRs are being actively developed and evaluated as potential novel treatment for acidosis-related diseases.
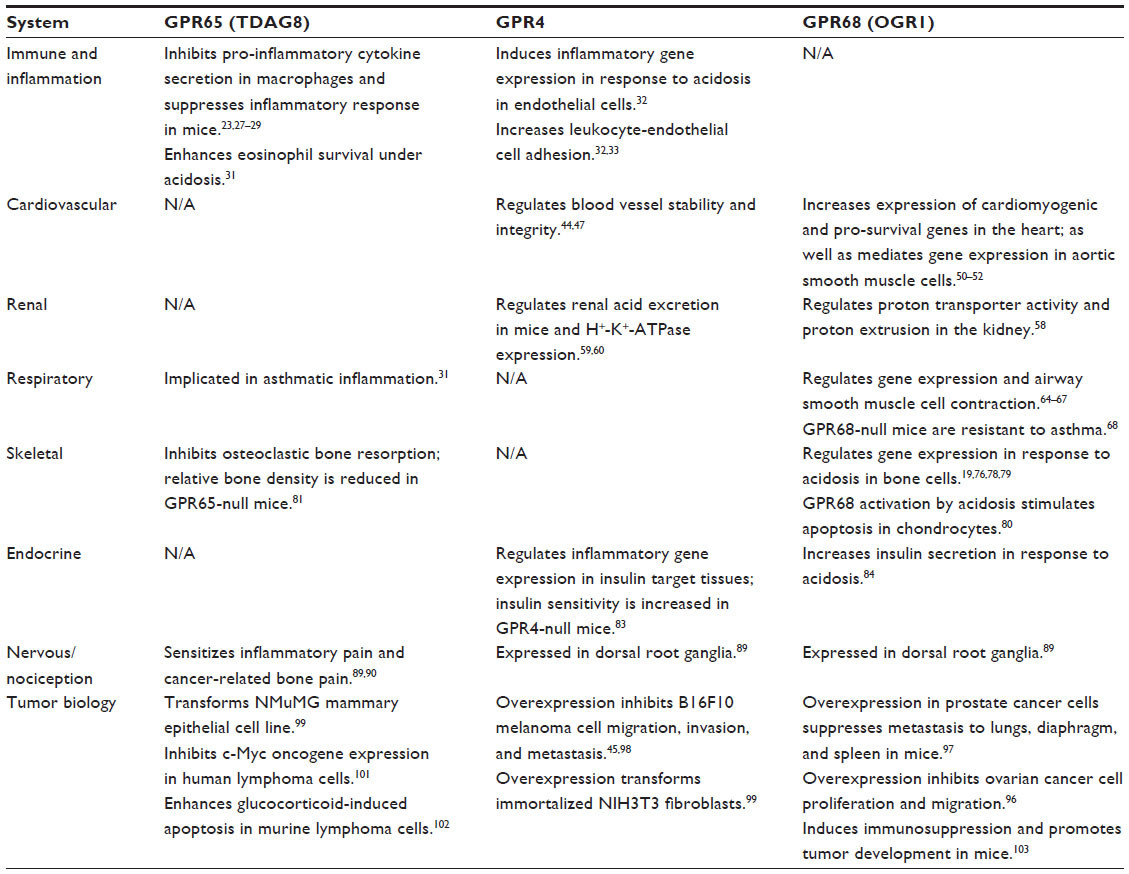 | Table 1 The main biological functions of the pH-sensing GPCRs |
Acknowledgments
The research in the authors’ laboratory was supported in part by grants from the American Heart Association National Center, Brody Brothers Endowment Fund, Brody School of Medicine Seed Grant, Golfers against Cancer Foundation, North Carolina Biotechnology Center, and Vidant Medical Center Cancer Research and Education Fund (to LVY).
Disclosure
Dr Li Yang is the inventor of a US patent (US 8207139 B2) “Function of GPR4 in vascular inflammatory response to acidosis and related methods”, but there is no current financial interest. The authors report no conflicts of interest in this work.
References
Koul PB. Diabetic ketoacidosis: a current appraisal of pathophysiology and management. Clin Pediatr (Phila). 2009;48(2):135–144. | |
Kraut JA, Madias NE. Approach to patients with acid-base disorders. Respir Care. 2001;46(4):392–403. | |
Krieger NS, Frick KK, Bushinsky DA. Mechanism of acid-induced bone resorption. Curr Opin Nephrol Hypertens. 2004;13(4):423–436. | |
Lemann J Jr, Bushinsky DA, Hamm LL. Bone buffering of acid and base in humans. Am J Physiol Renal Physiol. 2003;285(5):F811–F832. | |
Aalkjaer C, Peng HL. pH and smooth muscle. Acta Physiol Scand. 1997;161(4):557–566. | |
De Vito P. The sodium/hydrogen exchanger: a possible mediator of immunity. Cell Immunol. 2006;240(2):69–85. | |
Fang J, Quinones QJ, Holman TL, et al. The H+-linked monocarboxylate transporter (MCT1/SLC16A1):a potential therapeutic target for high-risk neuroblastoma. Mol Pharmacol. 2006;70(6):2108–2115. | |
Izumi H, Torigoe T, Ishiguchi H, et al. Cellular pH regulators: potentially promising molecular targets for cancer chemotherapy. Cancer Treat Rev. 2003;29(6):541–549. | |
Curley G, Contreras MM, Nichol AD, Higgins BD, Laffey JG. Hypercapnia and acidosis in sepsis: a double-edged sword? Anesthesiology. 2010;112(2):462–472. | |
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–899. | |
Huang WC, Swietach P, Vaughan-Jones RD, Ansorge O, Glitsch MD. Extracellular acidification elicits spatially and temporally distinct Ca2+ signals. Curr Biol. 2008;18(10):781–785. | |
Hunt JF, Fang K, Malik R, et al. Endogenous airway acidification. Implications for asthma pathophysiology. Am J Respir Crit Care Med. 2000;161(3 Pt 1):694–699. | |
Kellum JA. Determinants of blood pH in health and disease. Crit Care. 2000;4(1):6–14. | |
Lardner A. The effects of extracellular pH on immune function. J Leukoc Biol. 2001;69(4):522–530. | |
Nedergaard M, Kraig RP, Tanabe J, Pulsinelli WA. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am J Physiol. 1991;260(3 Pt 2):R581–R588. | |
Holzer P. Acid-sensitive ion channels and receptors. Handb Exp Pharmacol. 2009;(194):283–332. | |
Smith JB, Dwyer SD, Smith L. Lowering Extracellular Ph Evokes Inositol Polyphosphate Formation and Calcium Mobilization. J Biol Chem. 1989;264(15):8723–8728. | |
Dwyer SD, Zhuang Y, Smith JB. Calcium mobilization by cadmium or decreasing extracellular Na+ or pH in coronary endothelial cells. Exp Cell Res. 1991;192(1):22–31. | |
Ludwig MG, Vanek M, Guerini D, et al. Proton-sensing G-protein-coupled receptors. Nature. 2003;425(6953):93–98. | |
Park SY, Bae DJ, Kim MJ, Piao ML, Kim IS. Extracellular low pH modulates phosphatidylserine-dependent phagocytosis in macrophages by increasing stabilin-1 expression. J Biol Chem. 2012;287(14):11261–11271. | |
Simmen HP, Battaglia H, Giovanoli P, Blaser J. Analysis of pH, pO2 and pCO2 in drainage fluid allows for rapid detection of infectious complications during the follow-up period after abdominal surgery. Infection. 1994;22(6):386–389. | |
Grinstein S, Swallow CJ, Rotstein OD. Regulation of cytoplasmic pH in phagocytic cell function and dysfunction. Clin Biochem. 1991;24(3):241–247. | |
Okajima F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal. 2013;25(11):2263–2271. | |
Choi JW, Lee SY, Choi Y. Identification of a putative G protein-coupled receptor induced during activation-induced apoptosis of T cells. Cell Immunol. 1996;168(1):78–84. | |
Kyaw H, Zeng Z, Su K, et al. Cloning, characterization, and mapping of human homolog of mouse T-cell death-associated gene. DNA Cell Biol. 1998;17(6):493–500. | |
Radu CG, Cheng D, Nijagal A, et al. Normal immune development and glucocorticoid-induced thymocyte apoptosis in mice deficient for the T-cell death-associated gene 8 receptor. Mol Cell Biol. 2006;26(2):668–677. | |
Mogi C, Tobo M, Tomura H, et al. Involvement of proton-sensing TDAG8 in extracellular acidification-induced inhibition of proinflammatory cytokine production in peritoneal macrophages. J Immunol. 2009;182(5):3243–3251. | |
He XD, Tobo M, Mogi C, et al. Involvement of proton-sensing receptor TDAG8 in the anti-inflammatory actions of dexamethasone in peritoneal macrophages. Biochem Biophys Res Commun. 2011;415(4):627–631. | |
Onozawa Y, Fujita Y, Kuwabara H, Nagasaki M, Komai T, Oda T. Activation of T cell death-associated gene 8 regulates the cytokine production of T cells and macrophages in vitro. Eur J Pharmacol. 2012; 683(1–3):325–331. | |
Onozawa Y, Komai T, Oda T. Activation of T cell death-associated gene 8 attenuates inflammation by negatively regulating the function of inflammatory cells. Eur J Pharmacol. 2011;654(3):315–319. | |
Kottyan LC, Collier AR, Cao KH, et al. Eosinophil viability is increased by acidic pH in a cAMP- and GPR65-dependent manner. Blood. 2009; 114(13):2774–2782. | |
Dong L, Li Z, Leffler NR, Asch AS, Chi JT, Yang LV. Acidosis Activation of the Proton-Sensing GPR4 Receptor Stimulates Vascular Endothelial Cell Inflammatory Responses Revealed by Transcriptome Analysis. PLoS One. 2013;8(4):e61991. | |
Chen A, Dong L, Leffler NR, Asch AS, Witte ON, Yang LV. Activation of GPR4 by acidosis increases endothelial cell adhesion through the cAMP/Epac pathway. PLoS One. 2011;6(11):e27586. | |
Jones DK, Ruben PC. Proton modulation of cardiac I Na: a potential arrhythmogenic trigger. Handb Exp Pharmacol. 2014;221:169–181. | |
Antzelevitch C, Belardinelli L. The role of sodium channel current in modulating transmural dispersion of repolarization and arrhythmogenesis. J Cardiovasc Electrophysiol. 2006;17 Suppl 1: S79–S85. | |
Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497(Pt 2):337–347. | |
Yatani A, Brown AM, Akaike N. Effect of extracellular pH on sodium current in isolated, single rat ventricular cells. J Membr Biol. 1984;78(2):163–168. | |
Burbridge MF, West DC, Atassi G, Tucker GC. The effect of extracellular pH on angiogenesis in vitro. Angiogenesis. 1999;3(3):281–288. | |
De Bock K, Georgiadou M, Schoors S, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154(3):651–663. | |
Hellstrom M, Phng LK, Gerhardt H. VEGF and Notch signaling: the yin and yang of angiogenic sprouting. Cell Adh Migr. 2007;1(3):133–136. | |
Aalkjaer C. Regulation of intracellular pH and its role in vascular smooth muscle function. J Hypertens. 1990;8(3):197–206. | |
Crimi E, Taccone FS, Infante T, Scolletta S, Crudele V, Napoli C. Effects of intracellular acidosis on endothelial function: an overview. J Crit Care. 2012;27(2):108–118. | |
Ali TY, Broughton Pipkin F, Khan RN. The Effect of pH and Ion Channel Modulators on Human Placental Arteries. PLoS One. 2014; 9(12):e114405. | |
Wyder L, Suply T, Ricoux B, et al. Reduced pathological angiogenesis and tumor growth in mice lacking GPR4, a proton sensing receptor. Angiogenesis. 2011;14(4):533–544. | |
Castellone RD, Leffler NR, Dong L, Yang LV. Inhibition of tumor cell migration and metastasis by the proton-sensing GPR4 receptor. Cancer Lett. 2011;312(2):197–208. | |
Tobo M, Tomura H, Mogi C, et al. Previously postulated “ligand-independent” signaling of GPR4 is mediated through proton-sensing mechanisms. Cell Signal. 2007;19(8):1745–1753. | |
Yang LV, Radu CG, Roy M, et al. Vascular abnormalities in mice deficient for the G protein-coupled receptor GPR4 that functions as a pH sensor. Mol Cell Biol. 2007;27(4):1334–1347. | |
Dong L, Krewson E, Bliss D, Tulis DA, Yang LV. Abstract 11587: Acidosis/GPR4 signaling regulates inflammatory and endoplasmic reticulum stress responses in vascular endothelial cells. Circulation. 2013;128(22 Suppl):A11587. | |
Dong B, Zhou H, Han C, et al. Ischemia/Reperfusion-Induced CHOP Expression Promotes Apoptosis and Impairs Renal Function Recovery: The Role of Acidosis and GPR4. PLoS One. 2014;9(10):e110944. | |
Russell JL, Goetsch SC, Aguilar HR, et al. Regulated expression of pH sensing G Protein-coupled receptor-68 identified through chemical biology defines a new drug target for ischemic heart disease. ACS Chem Biol. 2012;7(6):1077–1083. | |
Tomura H, Wang JQ, Komachi M, et al. Prostaglandin I(2) production and cAMP accumulation in response to acidic extracellular pH through OGR1 in human aortic smooth muscle cells. J Biol Chem. 2005;280(41):34458–34464. | |
Liu JP, Komachi M, Tomura H, et al. Ovarian cancer G protein-coupled receptor 1-dependent and -independent vascular actions to acidic pH in human aortic smooth muscle cells. Am J Physiol Heart Circ Physiol. 2010;299(3):H731–H742. | |
Bommer J, Locatelli F, Satayathum S, et al. Association of predialysis serum bicarbonate levels with risk of mortality and hospitalization in the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2004;44(4):661–671. | |
Bushinsky DA, Ori Y. Effects of metabolic and respiratory acidosis on bone. Curr Opin Nephrol Hypertens. 1993;2(4):588–596. | |
Pickering WP, Price SR, Bircher G, Marinovic AC, Mitch WE, Walls J. Nutrition in CAPD: serum bicarbonate and the ubiquitin-proteasome system in muscle. Kidney Int. 2002;61(4):1286–1292. | |
Goraya N, Simoni J, Jo CH, Wesson DE. Treatment of metabolic acidosis in patients with stage 3 chronic kidney disease with fruits and vegetables or oral bicarbonate reduces urine angiotensinogen and preserves glomerular filtration rate. Kidney Int. 2014;86(5):1031–1038. | |
de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol. 2009;20(9):2075–2084. | |
Mohebbi N, Benabbas C, Vidal S, et al. The proton-activated G protein coupled receptor OGR1 acutely regulates the activity of epithelial proton transport proteins. Cell Physiol Biochem. 2012;29(3–4):313–324. | |
Sun X, Yang LV, Tiegs BC, et al. Deletion of the pH sensor GPR4 decreases renal acid excretion. J Am Soc Nephrol. 2010;21(10):1745–1755. | |
Codina J, Opyd TS, Powell ZB, et al. pH-dependent regulation of the alpha-subunit of H+-K+-ATPase (HKalpha2). Am J Physiol Renal Physiol. 2011;301(3):F536–F543. | |
Epstein SK, Singh N. Respiratory acidosis. Respir Care. 2001;46(4):366–383. | |
Kodric M, Shah AN, Fabbri LM, Confalonieri M. An investigation of airway acidification in asthma using induced sputum: a study of feasibility and correlation. Am J Respir Crit Care Med. 2007;175(9):905–910. | |
Ricciardolo FL, Gaston B, Hunt J. Acid stress in the pathology of asthma. J Allergy Clin Immunol. 2004;113(4):610–619. | |
Ichimonji I, Tomura H, Mogi C, et al. Extracellular acidification stimulates IL-6 production and Ca(2+) mobilization through proton-sensing OGR1 receptors in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2010;299(4):L567–L577. | |
Matsuzaki S, Ishizuka T, Yamada H, et al. Extracellular acidification induces connective tissue growth factor production through proton-sensing receptor OGR1 in human airway smooth muscle cells. Biochem Biophys Res Commun. 2011;413(4):499–503. | |
Saxena H, Deshpande DA, Tiegs BC, et al. The GPCR OGR1 (GPR68) mediates diverse signalling and contraction of airway smooth muscle in response to small reductions in extracellular pH. Br J Pharmacol. 2012;166(3):981–990. | |
Liu C, Li Q, Zhou X, Kolosov VP, Perelman JM. Regulator of G-protein signaling 2 inhibits acid-induced mucin5AC hypersecretion in human airway epithelial cells. Respir Physiol Neurobiol. 2013;185(2):265–271. | |
Aoki H, Mogi C, Hisada T, et al. Proton-sensing ovarian cancer G protein-coupled receptor 1 on dendritic cells is required for airway responses in a murine asthma model. PLoS One. 2013;8(11):e79985. | |
Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol. 2006;24:147–174. | |
Brandao-Burch A, Utting JC, Orriss IR, Arnett TR. Acidosis inhibits bone formation by osteoblasts in vitro by preventing mineralization. Calcif Tissue Int. 2005;77(3):167–174. | |
Krieger NS, Sessler NE, Bushinsky DA. Acidosis inhibits osteoblastic and stimulates osteoclastic activity in vitro. Am J Physiol. 1992;262(3 Pt 2):F442–F448. | |
Barzel US. The skeleton as an ion exchange system: implications for the role of acid-base imbalance in the genesis of osteoporosis. J Bone Miner Res. 1995;10(10):1431–1436. | |
Bushinsky DA, Goldring JM, Coe FL. Cellular contribution to pH-mediated calcium flux in neonatal mouse calvariae. Am J Physiol. 1985;248(6 Pt 2):F785–F789. | |
Frick KK, Bushinsky DA. Effect of metabolic and respiratory acidosis on intracellular calcium in osteoblasts. Am J Physiol Renal Physiol. 2010;299(2):F418–F425. | |
Frick KK, Jiang L, Bushinsky DA. Acute metabolic acidosis inhibits the induction of osteoblastic egr-1 and type 1 collagen. Am J Physiol. 1997;272(5 Pt 1):C1450–C1456. | |
Frick KK, Krieger NS, Nehrke K, Bushinsky DA. Metabolic acidosis increases intracellular calcium in bone cells through activation of the proton receptor OGR1. J Bone Miner Res. 2009;24(2):305–313. | |
Yang M, Mailhot G, Birnbaum MJ, MacKay CA, Mason-Savas A, Odgren PR. Expression of and role for ovarian cancer G-protein-coupled receptor 1 (OGR1) during osteoclastogenesis. J Biol Chem. 2006;281(33):23598–23605. | |
Komarova SV, Pereverzev A, Shum JW, Sims SM, Dixon SJ. Convergent signaling by acidosis and receptor activator of NF-kappaB ligand (RANKL) on the calcium/calcineurin/NFAT pathway in osteoclasts. Proc Natl Acad Sci U S A. 2005;102(7):2643–2648. | |
Tomura H, Wang JQ, Liu JP, et al. Cyclooxygenase-2 expression and prostaglandin E2 production in response to acidic pH through OGR1 in a human osteoblastic cell line. J Bone Miner Res. 2008;23(7):1129–1139. | |
Yuan FL, Wang HR, Zhao MD, et al. Ovarian cancer G protein-coupled receptor 1 is involved in acid-induced apoptosis of endplate chondrocytes in intervertebral discs. J Bone Miner Res. 2014;29(1):67–77. | |
Hikiji H, Endo D, Horie K, et al. TDAG8 activation inhibits osteoclastic bone resorption. FASEB J. 2014;28(2):871–879. | |
Mak RH. Effect of metabolic acidosis on insulin action and secretion in uremia. Kidney Int. 1998;54(2):603–607. | |
Giudici L, Velic A, Daryadel A, et al. The proton-activated receptor GPR4 modulates glucose homeostasis by increasing insulin sensitivity. Cell Physiol Biochem. 2013;32(5):1403–1416. | |
Nakakura T, Mogi C, Tobo M, et al. Deficiency of proton-sensing ovarian cancer G protein-coupled receptor 1 attenuates glucose-stimulated insulin secretion. Endocrinology. 2012;153(9):4171–4180. | |
Tateya S, Kim F, Tamori Y. Recent advances in obesity-induced inflammation and insulin resistance. Front Endocrinol (Lausanne). 2013;4:93. | |
Jones NG, Slater R, Cadiou H, McNaughton P, McMahon SB. Acid-induced pain and its modulation in humans. J Neurosci. 2004; 24(48):10974–10979. | |
Krishtal O. The ASICs: signaling molecules? Modulators? Trends Neurosci. 2003;26(9):477–483. | |
Wemmie JA, Price MP, Welsh MJ. Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci. 2006;29(10):578–586. | |
Chen YJ, Huang CW, Lin CS, Chang WH, Sun WH. Expression and function of proton-sensing G-protein-coupled receptors in inflammatory pain. Mol Pain. 2009;5:39. | |
Hang LH, Yang JP, Yin W, et al. Activation of spinal TDAG8 and its downstream PKA signaling pathway contribute to bone cancer pain in rats. Eur J Neurosci. 2012;36(1):2107–2117. | |
Cairns R, Papandreou I, Denko N. Overcoming physiologic barriers to cancer treatment by molecularly targeting the tumor microenvironment. Mol Cancer Res. 2006;4(2):61–70. | |
Justus CR, Dong L, Yang LV. Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front Physiol. 2013;4:354. | |
Yang LV, Castellone RD, Dong L. Targeting Tumor Microenvironments for Cancer Prevention and Therapy In: Georgakilas AG, editor. Cancer Prevention – From Mechanisms to Translational Benefits. InTech; 2012:3–40. | |
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. | |
Izumi H, Takahashi M, Uramoto H, et al. Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci. 2011;102(5):1007–1013. | |
Ren J, Zhang L. Effects of ovarian cancer G protein coupled receptor 1 on the proliferation, migration, and adhesion of human ovarian cancer cells. Chin Med J (Engl). 2011;124(9):1327–1332. | |
Singh LS, Berk M, Oates R, et al. Ovarian cancer G protein-coupled receptor 1, a new metastasis suppressor gene in prostate cancer. J Natl Cancer Inst. 2007;99(17):1313–1327. | |
Zhang Y, Feng Y, Justus CR, et al. Comparative study of 3D morphology and functions on genetically engineered mouse melanoma cells. Integr Biol (Camb). 2012;4(11):1428–1436. | |
Sin WC, Zhang Y, Zhong W, et al. G protein-coupled receptors GPR4 and TDAG8 are oncogenic and overexpressed in human cancers. Oncogene. 2004;23(37):6299–6303. | |
Ihara Y, Kihara Y, Hamano F, et al. The G protein-coupled receptor T-cell death-associated gene 8 (TDAG8) facilitates tumor development by serving as an extracellular pH sensor. Proc Natl Acad Sci U S A. 2010;107(40):17309–17314. | |
Li Z, Dong L, Dean E, Yang LV. Acidosis decreases c-Myc oncogene expression in human lymphoma cells: a role for the proton-sensing G protein-coupled receptor TDAG8. Int J Mol Sci. 2013;14(10):20236–20255. | |
Malone MH, Wang Z, Distelhorst CW. The glucocorticoid-induced gene tdag8 encodes a pro-apoptotic G protein-coupled receptor whose activation promotes glucocorticoid-induced apoptosis. J Biol Chem. 2004;279(51):52850–52859. | |
Yan L, Singh LS, Zhang L, Xu Y. Role of OGR1 in myeloid-derived cells in prostate cancer. Oncogene. 2014;33(2):157–164. | |
Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. The 7 TM G-protein-coupled receptor target family. ChemMedChem. 2006;1(8):761–782. | |
Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10(1):47–60. | |
Taracido IC, Harrington EM, Hersperger R, Lattmann R, Miltz W, Weigand K, inventors. Imidazo pyridine derivatives. United States patent application 12/468706. November 26, 2009. | |
Schneider JW, Goetsch SC, Leng X, et al. Coupling Hippocampal Neurogenesis to Brain pH through Proneurogenic Small Molecules That Regulate Proton Sensing G Protein-Coupled Receptors. ACS Chem Neurosci. 2012;3(7):557–568. | |
Zhang L, Li P, Hsu T, et al. Small-molecule blocks malignant astrocyte proliferation and induces neuronal gene expression. Differentiation. 2011;81(4):233–242. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.