")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease
Authors Bennett L, Turcotte K
Received 26 June 2015
Accepted for publication 10 July 2015
Published 18 August 2015 Volume 2015:9 Pages 4639—4647
DOI https://doi.org/10.2147/DDDT.S77760
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Lunawati L Bennett, Kelsey Turcotte
School of Pharmacy, Union University, Jackson, TN, USA
Abstract: The purpose of this article is to review eliglustat tartrate, a substrate reduction therapy, for the treatment of Gaucher disease type 1 (GD1). GD is an rare inborn error of metabolism caused by accumulation of lipid substrates such as glucosylceramide within the monocyte-macrophage system that affects the body by causing enlargement of the spleen and liver, destruction of bone, and abnormalities of the lungs and blood, such as anemia, thrombocytopenia, and leukopenia. GD is classified into three types: GD1, a chronic and non-neuronopathic disease accounting for 95% of GD cases; and types 2 and 3 (GD2 GD3) which are more progressive diseases with no approved drugs available at this time. Treatment options for GD1 include enzyme replacement therapy and substrate reduction therapy. Eliglustat works by inhibiting UDP-glucosylceramide synthase, the first enzyme that catalyzes the biosynthesis of glycosphingolipids, thus reducing the load of glucosylceramide influx into the lysosome. Eliglustat was approved by the US Food and Drug Administration after three Phase I, two Phase II, and two Phase III clinical trials. The dose of eliglustat is 84 mg twice a day or once daily depending on the cytochrome P450 2D6 genotype of the patient.
Keywords: Gaucher disease, glucocerebrosidase, glucosylceramide synthase, eliglustat tartrate, substrate reduction therapy
Corrigendum for this paper has been published
Introduction
Gaucher disease (GD), an extremely rare inherited autosomal recessive lipid storage disease (LSD), was first described by Philippe Gaucher in 1882.1 This was the first identified LSD caused by deficiency or absence of activity of the enzyme acid β-glucosidase, also known as β-glucocerebrosidase or glucosylceramidase E.C.3.2.1.45 (GBA1), leading to accumulation of glucocerebroside, also known as glucosylceramide (GLC) in tissue monocyte macrophages.2
Treatment options for GD include enzyme replacement therapy (ERT) and substrate reduction therapy (SRT). To date, there are three ERTs available: imiglucerase, velaglucerase alfa, and taliglucerase alfa, and two SRTs available, miglustat and the newest drug, eliglustat tartrate. These have been approved as orphan drugs for the treatment of GD. This review summarizes the epidemiology of the disease, its pathophysiology, diagnosis, and useful biomarkers, and the treatment options available for GD1 with a focus on eliglustat. Supportive care and cost of therapies are also briefly discussed.
Epidemiology and pathophysiology of GD
GD is a systemic metabolic disorder caused by accumulation of the lipid substrate GLC within the monocyte-macrophage system, resulting in formation of Gaucher cells. These cells are the hallmark of the disease and are found in many organs, mostly in bone, bone marrow, liver, spleen, and lymph node parenchyma.3–8 Accumulation of Gaucher cells can also enhance production of inflammatory cytokines, which cause enlargement of the spleen and liver, destruction of bone, abnormalities of the lungs, and anemia, thrombocytopenia, and leukopenia.3–8 Gaucher cells are about 20–100 μm in diameter, have small eccentric nuclei, and cytoplasm with crinkles and striation.7
GD affects males and females equally. GD is classified into GD1 (non-neuronopathic), GD2 (acute neuronopathic), and GD3 (chronic neuronopathic) according to the presence of neurological deterioration, age at identification, and rate of disease progression.9,10 Patients with GD2 or GD3 manifest more severe complications than those with GD1, and many of these patients died at a very young age or in early adulthood.5,9,10 GD1 occurs mainly in adults, accounting for 95% of cases. If the onset of GD occurs prior to adulthood, more rapidly progressive disease is likely.11 The National Gaucher Foundation estimated the incidence of GD1 in the USA to be about one in 20,000 live births, or a prevalence of one in 40,000.12 The incidence of GD may be as high as one in 450 births among individuals with Ashkenazi Jewish ancestry and 1:20,000 to 1:200,000 in the general population.13–16
The initial manifestations of GD normally start with splenomegaly, hepatomegaly, anemia, leukopenia, and thrombocytopenia.17 Further progression involves gastrointestinal complications, such as portal hypertension, cirrhosis, ascites, esophageal hemorrhage, and bone lesions manifested as chronic bone pain, skeletal deformities, osteonecrosis, osteopenia, and osteoarticular infections.17–22 Increased risk of cholelithiasis is present in women older than 40 years.17 Interstitial lung disease, pulmonary hypertension, polyclonal gammopathy, and peripheral neuropathy have also been observed in GD1 patients.17,21–24
The pathophysiology of GD possibly includes enzyme, gene, and/or lysosome packing defects.7,25–29 Defects in the function of lysosomes result in missorting or loss of function of lysosomal proteins.26 Normal lysosomal proteins are usually tagged with a carbohydrate that allows their recognition and transport via the mannose-6-phosphate receptor.25,27 However, mutation in mannose-6-phosphate has been identified in GD patients.25,27,28 Additionally, one of the lysosomal hydrolases, GBA1, which is important for degradation of GLC into glucose and ceramide, was found to be defective/deficient and this enzyme is improperly packed in GD patients, leading to accumulation of GLC in the monocyte-macrophage system.7,27,30
Diagnosis and biomarkers
GD is normally diagnosed during initial clinical examination by the presence of unexpected anemia, thrombocytopenia, and organomegaly.9 Clinical diagnoses are confirmed by biochemical diagnosis.7 Detection of low enzymatic activity of GBA1 in peripheral blood compared with normal controls is still the “gold standard” for diagnosing GD. Despite the availability of this testing for nearly four decades, many patients are still incorrectly diagnosed.9,19,31 The assay is performed in 10 cc of blood leukocytes using a fluorescent substrate, 4-methyumbelliferone β-glucosidase. The sample can be shipped at ambient temperature overnight to diagnostic laboratories.19,32–34
Biomarkers can add quality assurance to biochemical diagnosis. Ongoing studies are being conducted to detect useful protein biomarkers for GD through a survey of the protein composition of body fluids, cells, and tissue specimens from symptomatic Gaucher patients. Non-specific biomarkers such as tartrate-resistant acid phosphatase, angiotensin-converting enzyme, hexosaminidase, and cathepsin K have been used for routine monitoring; however, these biomarkers are also observed in healthy individuals.35–37 Increases in interleukin (IL)-1β, IL-6, IL-10, tumor necrosis factor alpha, macrophage inflammatory protein (MIP)-1α, MIP-1β, and soluble CD 163 have also been used as biomarkers for GD; however, corrections in plasma MIP-1α and MIP-1β after treatment are not proportional to those found with true Gaucher cell biomarkers.7,38–40
Activated macrophages also cause secretion of the enzyme chitotriosidase (CT), which plays a role during the remodeling phase of tissue healing and in immune chemotaxis.41,42 CT, a macrophage-derived chitin-fragmenting hydrolase, is abundantly expressed in lipid macrophages. Common tissue macrophages do not produce CT.8,25 Patients with diseases such as LSD, sarcoidosis, thalassemia, visceral leishmaniasis, and leprosy have been identified to have elevated CT levels.25,41,42 Recently, plasma CT has been used as the first screening test in diagnosing GD. In patients with high clinical severity scores, CT levels were usually greater than 20,000 nmol/mL/hour.43,44 After treatment, the CT value is expected to decrease. However, even after treatment, more severely affected patients will have less reduction in their plasma CT activity. A smaller than expected reduction in plasma CT activity after the initial treatment can also be used as a clinical parameter to increase the dose of drugs being used, such as ERTs or SRTs.43
Massive overproduction and secretion of pulmonary and activation-regulated chemokines (PARC/CCL18), which are elevated by 10- to 40-fold in symptomatic patients with GD, can also be used as a biomarker.45,46 Because PARC/CCL18 is a small molecule, its level in urine is proportional to the level in the circulation.47 Measurement of plasma PARC/CCL18 has been a useful additional tool to monitor changes in Gaucher cells and as a useful tool for evaluating GD patients who are CT-deficient.48 Therefore, regular monitoring of CT or PARC/CCL18 in CT-deficient patients, along with radiological monitoring of the bone marrow and skeleton, and other sensitive assays are needed to confirm the diagnostic of GD and to monitor the effectiveness of treatment.
As part of their search for a better biomarker for GD, Rolfs et al have patented glucosylsphingosine as being more specific and sensitive than CT and PARC/CCL18 as a biomarker in normal subjects, GD patients, GD carriers, and patients with other LSDs.49 The glucosylsphingosine from plasma is labeled with fluorescent dye, and total and free glucosylsphingosine is then detected by reverse phase high performance liquid chromatography and a fluorescence detector. GD patients show elevated glucosylsphingosine above 12 ng/mL as compare to non-GD patients. Glucosylsphingosine can measure 100% sensitivity compare to CT or PAR/CCL18 at 91.7% or 76.2%, respectively; and 100% specificity as compared with CT or PAR/CCL18 at 86.1% or 79.4%, respectively.49
The severity of GD1 is usually classified using the Gaucher disease severity score index-type I (GauSSI-I).44 This scoring index was developed to provide a thorough and reliable method for correlating differences in genotypes and phenotypes of patients, to correlate patients’ response to biological markers, and to account for variability in clinical response and severity of disease. There are six domains, ie, skeletal, hematological, biomarker, visceral, lung, and neurological, giving a total score of 42 points. A higher score indicates more severe GD.44 For detailed information on classification of GauSSI-I, please refer to DiRocco et al.44
Therapeutic options
Five treatments are available for GD, comprising three ERTs and two SRTs. The available ERTs are: imiglucerase (Cerezyme®), velaglucerase alfa (VPRIV®), and taliglucerase alfa (Elelyso®). ERTs work by supplementing defective GBA1 with the active enzyme, which catalyzes the hydrolysis of GLC into glucose and ceramide, thus reducing the amount of GLC accumulated in the liver, spleen, bone marrow, and other organs.50,51 All ERTs are given as an intravenous infusion, which makes this therapeutic option costly and inconvenient.
The two small molecule SRTs available, ie, miglustat and the newer eliglustat tartrate, are taken as oral drugs. In contrast with ERTs, which aim to replace the defective enzyme with active enzyme, SRTs work by inhibiting UDP-GLC synthase, the first enzyme catalyzing the biosynthesis of glycosphingolipids from UDP-glucose and ceramide, thus reducing the influx of GLC into the lysosome. Figure 1 shows the pathway for synthesis of glycosphingolipids and where eliglustat works.52,53
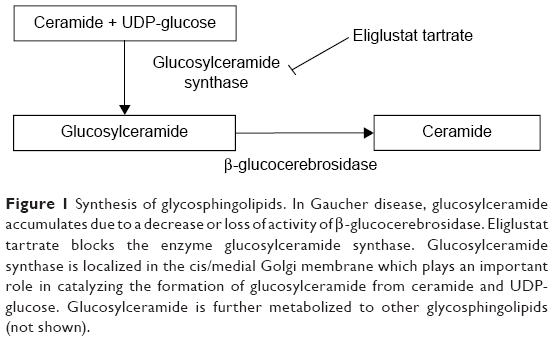 | Figure 1 Synthesis of glycosphingolipids. Iin Gaucher disease, glucosylceramide accumulates due to a decrease or loss of activity of β-glucocerebrosidase. Eliglustat tartrate blocks the enzyme glucosylceramide synthase. Glucosylceramide synthase is localized in the cis/medial Golgi membrane which plays an important role in catalyzing the formation of glucosylceramide from ceramide and UDP-glucose. Glucosylceramide is further metabolized to other glycosphingolipids (not shown). |
Miglustat (Zavesca®, Actelion Pharmaceutical Limited, Allschwil, Switzerland), the first SRT, is a synthetic d-glucose analog.54,55 The drug was approved in Europe, Australia, and the USA for treatment of mild to moderate GD3. However, miglustat also inhibits other glucosides, so comes with a higher incidence of adverse reactions, such as tremor (30%), diarrhea (85%), weight loss (65%), reduced platelet counts, numbness, and a feeling of burning in the hands and feet.54–57 Because of the essential roles of glycosphingolipids, long-term reduction in levels of these lipids affects a variety of normal cell functions.58 Miglustat is now only approved in Europe, Israel, Australia, and the USA for patients who cannot take ERTs due to anaphylactic reactions.7 This led to the formulation of eliglustat tartrate (GENZ 112638) by the Michigan Group and licensing of this compound for clinical development by Genzyme Corporation.52,53
Eliglustat tartrate
Eliglustat tartrate is a potent ceramide analog and has a selective inhibitory action on GLC synthase. This compound was found to be effective in reducing GLC in a murine model of GD (GBA D409/null).52,53,59–62 Eliglustat caused decrease GLC and infiltration of abnormal macrophages in the tissues in concentration dependent manner.62 The structure of eliglustat is similar to that of D-threo-1-phenyl-2-decanoylamino 3-morpholino-propanol.52,58 Eliglustat tartrate (Cardelga®) is manufactured by Genzyme Corporation (a subsidiary of Sanofi), and received approval in the USA on August 19, 2014 for treatment-naïve and treatment-experienced adult patients with GD1.63–67 Table 1 lists the features of eliglustat and Table 2 compares the characteristics of miglustat with those of eliglustat.63–67
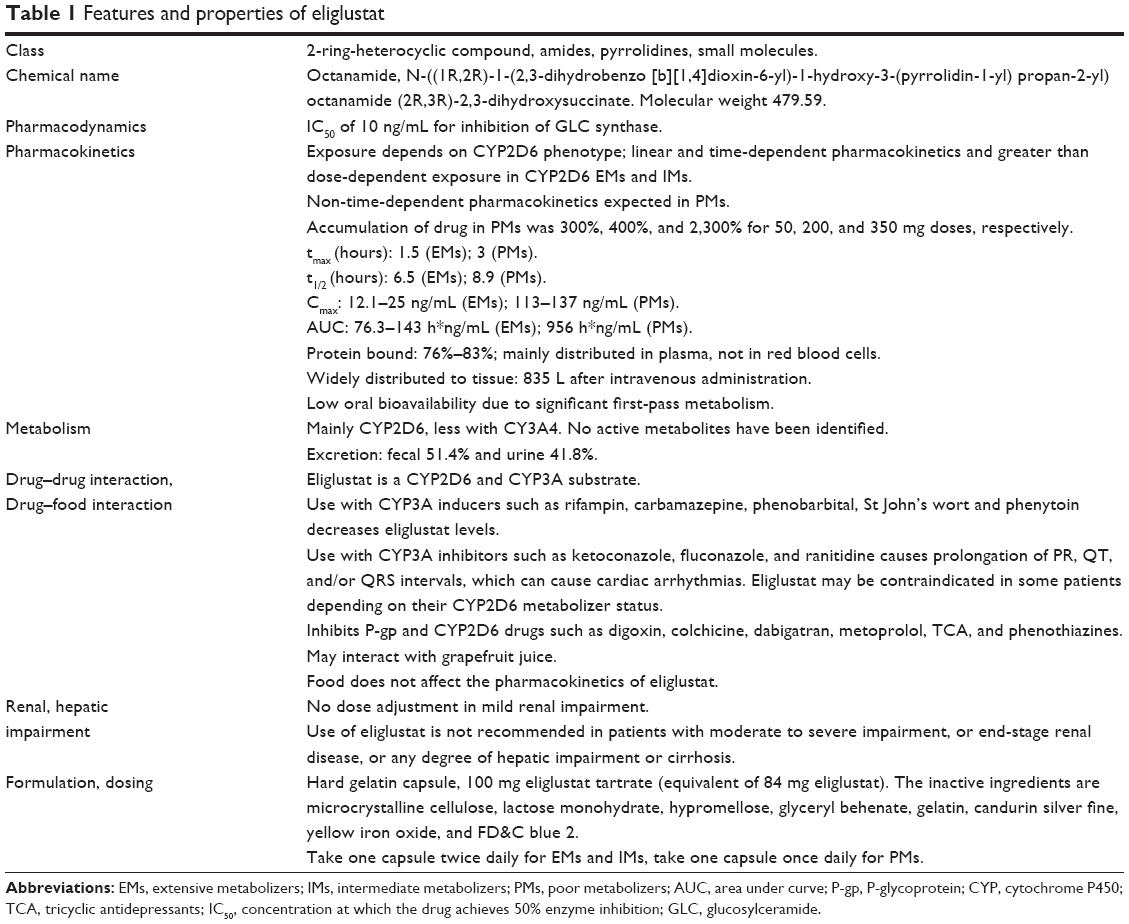 | Table 1 Features and properties of eliglustat |
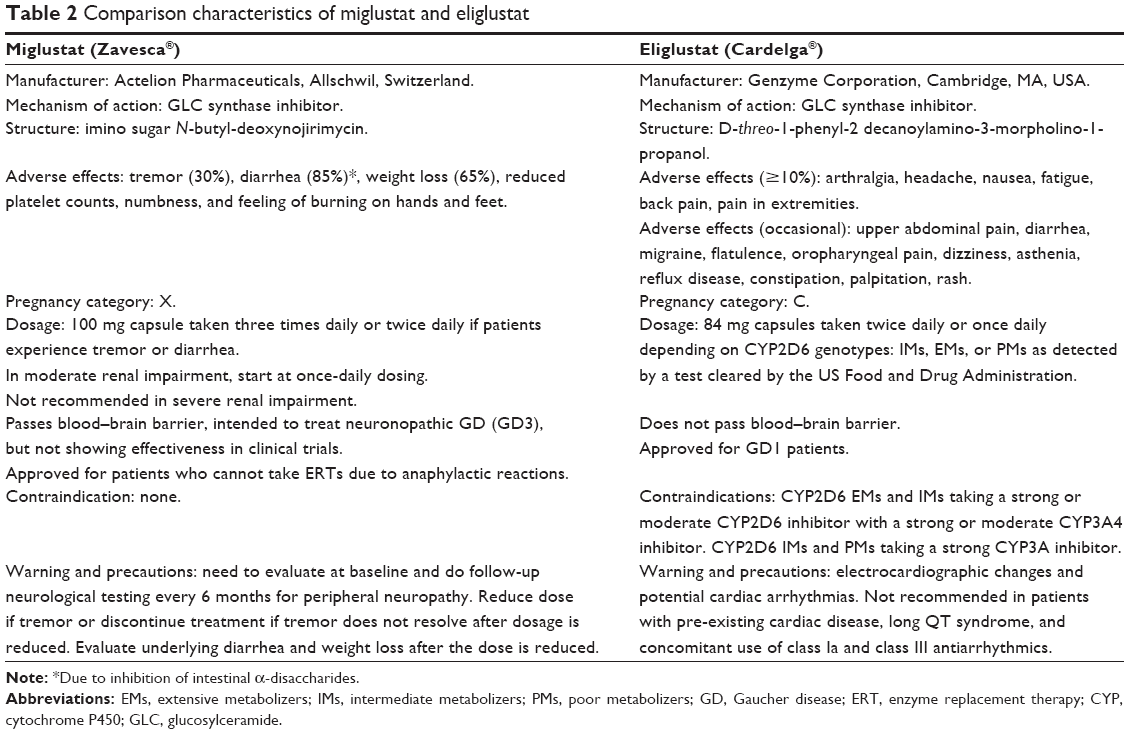 | Table 2 Comparison characteristics of miglustat and eliglustat |
Clinical trials of eliglustat
Eliglustat tartrate has been studied in three Phase I, two Phase II, and three Phase III trials, with one study ongoing at the time this paper was written.53 The three Phase I studies were performed to determine the safety, tolerability, and pharmacokinetics of eliglustat in healthy volunteers.52 These studies examined the safety and tolerability of eliglustat in single dose and in multiple dose, and the effect of food on eliglustat.
The first Phase I trial used single-dose eliglustat, recruited 99 healthy men aged 18–44 years, and was a randomized, placebo-controlled, double-blind, parallel-group study. There were 74 participants in the eliglustat group and 24 in the placebo group. The design of the study consisted a 21-day screening period, a 72-hour evaluation period, and a safety follow-up at 9–11 days later. Eliglustat was dosed at 0.01 mg/kg to 30 mg/kg based on allometric scaling data from previous animal studies.67 The second Phase I randomized, placebo-controlled, double-blind, parallel-group study used multiple doses of eliglustat and recruited 18 men and 18 women aged 18–44 years. The design of the study consisted of a 21-day screening period, a 13-day evaluation period, and a safety follow-up at 19–21 days later. Three eliglustat doses (50 mg, 200 mg, or 350 mg) were given to eight participants in the eliglustat group and to four in the placebo group. Each participant received 21 doses of eliglustat or placebo once daily on days 1, 2, and 12, and twice daily on days 3–11 under fasting conditions.52,67 The next cohort was enrolled after the previous dose of eliglustat was determined to be safe and tolerable. The third Phase I trial recruited 24 men aged 19–43 years and was designed to study the effect of food on eliglustat. It was a randomized, unblended, two-period, crossover study that also utilized screening, evaluation, washout, evaluation, and safety follow-up periods.67 Twelve participants received eliglustat 300 mg under fasting conditions followed by a 6-day washout and a second 300 mg dose on day 7 immediately after a meal. The other 12 participants were randomized to receive the first dose of the drug in the fed state and the second dose in the fasted state.
In all three Phase I trials, blood samples were analyzed at pre-dose and post-dose intervals. Safety and tolerability were analyzed by vital signs, hematology, urinalysis, 12-lead electrocardiography, and continuous electrocardiographic monitoring.67 No serious adverse effects were reported. Dose-limiting toxicities included nausea, vomiting, dizziness, and mild prolongation of the PR, QRS, and QTc intervals observed in the single-dose group taking eliglustat 10–30 mg/kg. Although no clinically significant cardiac problems were observed at the therapeutic dose of 200 mg or the supratherapeutic dose of 800 mg, patients with a predisposition to arrhythmia should not take eliglustat and patients with cardiovascular disease should take this drug cautiously due to possible mild prolongation of PR, QRS, and QTc intervals. The adverse effects observed due to the increased dosage of eliglustat were transient and resolved without further problems.67 In the trial investigating the effects of food, eliglustat caused diarrhea (8.3%), flatulence (8.3%), and nausea (4.2%).67 Common adverse effects reported from the Phase I trials were nausea (45.8%), headache (25.0%), dizziness (20.8%), constipation (16%), vomiting (16%), and decreased appetite (16%).67 Based on the favorable Phase I results, Phase II trials were initiated in GD1 patients using a dose of 50 or 100 mg twice daily.
A Phase II, multinational, open-label, single-arm clinical trial was conducted in 28 GD1 patients aged 18–65 years to evaluate the efficacy, safety, and pharmacokinetics of eliglustat administered twice daily orally at 50 or 100 mg for 52 weeks.3,68 Inclusion criteria for this study were GBA1 enzyme deficiency, a spleen volume ten times that of normal, thrombocytopenia, and/or anemia. Statistically significant improvements in hemoglobin level (1.62 g/dL; 95% confidence interval [CI] 1.05–2.18), platelet count (40.3%; 95% CI 23.7–57), spleen volume (−38.5%; 95% CI −43.5, −33.5), liver volume (−17%; 95% CI −21.6, −12.3), and bone mineral density at the lumbar spine (Z score 0.31; 95% CI 0.09–0.53) were observed. A decrease in biomarkers (CT, angiotensin-converting enzyme, PARC/CCL18, tartrate-resistant acid phosphatase) of 35%–50% was also achieved. During this trial, four patients withdrawn from the study due to asymptomatic non-sustained ventricular tachycardia detected on day 1 (n=2) and pregnancy during weeks 17 and 26 (n=2).68 A Phase II, multisite, open-label, single-arm clinical study involving the same patients (total 20) and the same group of researchers was conducted for one further year. Statistically significant (P<0.001) improvements were seen in the primary and secondary end points, and in platelet count, hemoglobin level, and spleen and liver volumes.69 Significant increases in bone mineral density at the lumbar spine and T score and a decrease in bone marrow infiltration by Gaucher cells were also recorded.69 Nineteen patients continued on the study for 2 further years (a total of 4 years). Fifteen patients received eliglustat 100 mg twice daily, three received 50 mg twice daily, and one received 50 mg twice daily for 3 years then increased to 100 mg twice daily in year 4.70,71 Improvements observed in spleen and liver volume, hemoglobin level, and platelet counts during the 1st and 2nd years of treatment were maintained and extended through the 4 years, demonstrating the long-term efficacy of eliglustat.70,71
The three Phase III clinical trials are known as ENGAGE, ENCORE, and EDGE. Table 3 provides more detailed information on these trials. ENGAGE was a randomized, double-blind, multicenter, placebo-controlled trial that recruited 40 patients to evaluate the manifestations of 9 months of treatment with eliglustat in patients with untreated GD1.72 Patients were stratified to receive eliglustat 50 mg or 100 mg twice daily based on their spleen volume (n=20) or placebo (n=20). The primary endpoint was the percent change in spleen volume, and secondary endpoints were change in hemoglobin level and percent changes in liver volume and platelet count from baseline.
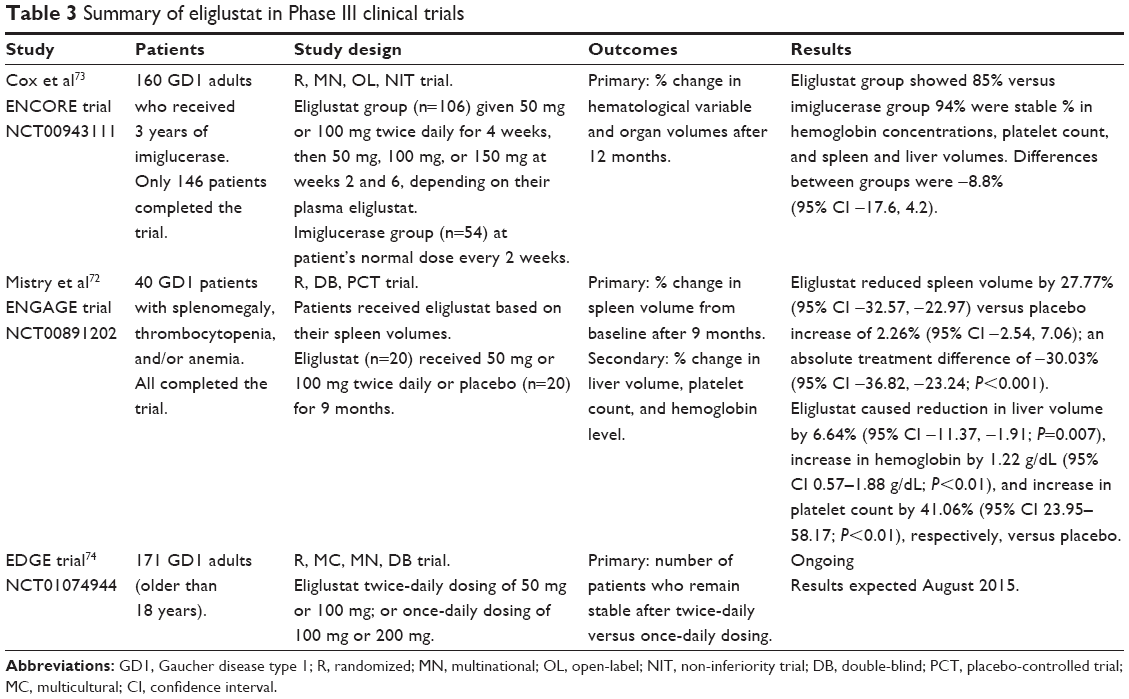 | Table 3 Summary of eliglustat in Phase III clinical trials |
ENCORE was a randomized, multinational, open-label, Phase III non-inferiority trial that examined the stability of patients who had been on the ERTs imiglucerase or velaglucerase for over 3 years when switched to oral eliglustat. The patients were randomized to receive an infusion of imiglucerase every other week at their usual doses (n=54) or to receive eliglustat 50 mg, 100 mg, or 150 mg twice daily (n=106). The primary endpoint was change in hematological or organ volumes after 12 months of treatment. Eighty-five percent of the patients treated with eliglustat and 94% of those treated with imiglucerase met the primary efficacy endpoint of the study (−8.8%, 95% CI −17.6, 4.2).73 Using the preliminary screening questionnaire, 94% of patients preferred taking oral therapy over intravenous infusion.8 Serious adverse effects reported were hepatocellular carcinoma in 10% of the eliglustat group, cholecystitis, and joint dislocation possibly related to underlying GD. Three patients withdrew from the study due to psychotic disorders or palpitations without clinically relevant electrocardiographic findings. Common adverse effects associated with switching from imiglucerase to eliglustat included diarrhea (5%), arthralgia (4%), fatigue (4%), and headache (4%). Overall, oral eliglustat therapy was found to be non-inferior to imiglucerase.73 Further details of this study are reported in Table 3.
The EDGE trial is currently ongoing and has not reported any data to date. It is a randomized, multicenter, multinational, double-blind trial investigating the efficacy, safety, and pharmacokinetics of once-daily versus twice-daily dosing of eliglustat in patients with GD1 who have demonstrated clinical stability on eliglustat twice daily.74
Dosing and administration
Eliglustat is dosed according to cytochrome P450 (CYP)2D6 genotype, whereby patients are classified as extensive metabolizers (EMs), intermediate metabolizers (IMs), or poor metabolizers (PMs). It is recommended that patients receive a test approved by the US Food and Drug Administration for determining CYP2D6 genotype prior to taking eliglustat. CYP2D6 ultra-rapid metabolizers may not reach therapeutic efficacy of the drug and a specific dosage cannot be recommended for patients without confirmed CYP2D6 genotype assessment.
The recommended dosing of eliglustat is 84 mg twice a day in CYP2D6 EMs and IMs, and 84 mg once daily in CYP2D6 PMs. Caution is required when patients are taking other CYP2D6 and CYP3A4 inhibitors because this may increase the availability of eliglustat and worsen adverse effects. CYP2D6 EMs and IMs taking strong to moderate CYP2D6 inhibitors need to reduce the dose of eliglustat to 84 mg once daily, and CYP2D6 EMs taking strong to moderate CYP3A4 inhibitors need to reduce the dose to 84 mg once daily. Eliglustat has not been adequately studied in geriatric or pediatric patients. Eliglustat capsules should be swallowed whole, preferably with water, and not crushed, dissolved, or opened. Food does not affect the availability of this drug, but patients should avoid grapefruit juice because it is a strong CYP3A4 inhibitor. Missed doses should not be replaced, but the patient should take the next dose. When switching therapies, eliglustat can be given 24 hours after the last dose of ERT. For additional information on eliglustat, refer to Table 1.
Adverse effects
Common adverse effects (≥10%) of eliglustat included fatigue, headache, nausea, diarrhea, back pain, pain in the extremities, and upper abdominal pain. Eliglustat is not recommended for patients with pre-existing cardiac disease or long QT syndrome, or for patients taking class IA or class IIIA antiarrhythmics.
Supportive care for GD
In addition to taking an ERT or SRT, GD patients usually need to take other drugs to alleviate specific disease symptoms such as bone disease, hepatosplenomegaly, bleeding, pulmonary hypertension, seizures, and Parkinsonism. Bone disease usually indicates advanced GD, but susceptibility to fractures, osteopenia, and osteonecrosis can also be a sign of GD in asymptomatic patients.26 Treatment of bone disease with oral bisphosphonates such as alendronate disodium 40 mg/day, calcium 1,500 mg/day, and vitamin D 400 IU/day for 24 months significantly improved bone mineral density and content, and decreased the fracture risk in comparison to patients who took only calcium and vitamin D at the same dosage without oral biphosphonates.75,76 However, alendronate did not improve focal bone lesions (deformity in distal femur, vertebral, pelvic bones), suggesting a more complex osteoclast-mediated mechanism in GD that needs further study. Alendronate at a high dose of 40 mg/day may provide benefit and could be an effective and safe strategy to increase bone mineral density and bone mineral content.75,76 Defects in the platelet, coagulation, and non-corrected and thrombocytopenia coagulation pathways may increase the bleeding risk in GD patients and require constant monitoring.26
Although the majority of GD patients never develop clinical signs of Parkinsonism, those who manifest such symptoms may experience improvement or worsening of their symptoms despite optimal treatment of GD. Due to the severity and complexity of GD, providers need to individualize treatment options for complicated GD, given that few guidelines are available.
Therapeutic costs
The cost of ERTs, not including nurses/doctors who administered the intravenous or other hospital expenses, for imiglucerase is US $1,729 per vial of 400 units, or velaglucerase alfa is US $1,501 per vial of 400 units, or taliglucerase alfa is $700 per vial of 200 units, respectively.77 Patients are generally given 60 units/kg every 2 weeks, so the monthly cost for a 70 kg patient is approximately $36,307 for imiglucerase, $31,521 for velaglucerase alfa, and $29,200 for taliglucerase. The additional cost of administering these drugs by intravenous infusion in hospital should also be considered. The monthly cost of 90 capsules of miglustat is $11,255, while the monthly cost of 56 eliglustat capsules is $24,284. If a patient can take miglustat, the cost of the drug is the cheaper than all the other drugs. Patients taking eliglustat can save from $4,900 to $12,000 monthly, when switching from any of the ERTs. Insurance is not factored into these values.
Conclusion
Eliglustat tartrate is a the newest SRT for use in the treatment of GD1, an autosomal recessive LSD affecting the bone marrow, bone structure, spleen, liver, and lymph node parenchyma. This drug is a potent ceramide analog that works by selectively inhibiting the action of enzyme GLC synthase, causing reduction of endogenous GLC and thus preventing its influx in the lysosome. GLC causes accumulation of Gaucher cells responsible for destruction of bone, enlargement of the spleen and liver, abnormalities in the lung, and anemia, thrombocytopenia and leukopenia. Common adverse effects (≥10%) include fatigue, headache, nausea, diarrhea, back pain, pain in the extremities, and upper abdominal pain. The recommended dosing of eliglustat is 84 mg twice a day in CYP2D6 EMs and IMs, and 84 mg once daily in CYP2D6 PMs. Eliglustat is not recommended for patients with pre-existing cardiac disease or long QT syndrome, or patients taking class IA or class IIIA antiarrhythmics. Caution is needed in patients taking strong CYP2D6 and CYP3A4 inhibitors. Based on its safety, efficacy, convenience, and cost-effectiveness, eliglustat tartrate provides a better option than all other available drugs for patients with GD1, who usually require lifetime treatment.
Disclosure
The authors of this paper have nothing to disclose concerning possible financial or personal relationships with commercial entities that may have a direct or indirect interest in the subject matter of this paper.
References
Mehta A. Epidemiology and natural history of Gaucher’s disease. Eur J Intern Med. 2006;17:S2–S5. | ||
Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid. 2012;7:15–20. | ||
Goker-Alpan O. Therapeutic approaches to bone pathology in Gaucher disease: past, present and future. Mol Genet Metab. 2011;104:438–447. | ||
Conradi NG, Kalimo H, Sourander P. Reactions of vessel walls and brain parenchyma to the accumulation of Gaucher cells in the Norrbottnian type (type III) of Gaucher disease. Acta Neuropathol. 1988;75:385–390. | ||
Grabowski GA. Gaucher disease: lessons from a decade of therapy. J Pediatr. 2004;144:S15–S19. | ||
Barneveld RA, Keijzer W, Tegelaers FP, et al. Assignment of the gene coding for human beta-glucocerebrosidase to the region q21-q31 of chromosome 1 using monoclonal antibodies. Hum Genet. 1983;64: 227–231. | ||
Aerts JM, Hollak C, Boot R, Groener A. Biochemistry of glycosphingolipid storage disorders: implications for therapeutic intervention. Philos Tran R Soc Lond B Biol Sci. 2003;358:905–914. | ||
Pastores GM, Hughes DA. Gaucher disease. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1269/?report. Accessed June 8, 2015. | ||
Zimran A, Elstein D. Lipid storage diseases. In: Lichtman MA, Kipps T, Seligsohn U, Kaushansky K, Prchal JT, editors. Williams Hematology. 8th ed. New York, NY, USA: McGraw-Hill; 2010. | ||
Lee JY, Lee BH, Kim GH, et al. Clinical and genetic characteristics of Gaucher disease according to phenotypic subgroups. Korean J Pediatr. 2012;55:48–53. | ||
Martins AM, Valadares ER, Porta G, et al. Recommendations on diagnosis, treatment, and monitoring for Gaucher disease. J Pediatr. 2009;155:S10–S18. | ||
National Gaucher Foundation Inc. Available from: http://www.gaucherdisease.org/. Accessed June 1, 2015. | ||
National Organization of Rare Disorders. Gaucher disease. Available from: http://www.rarediseases.org/rare-disease-information/rare-diseases/byID/12/viewFullReport. Accessed June 1, 2015. | ||
Altarescu G, Schiffmann R, Parker CC, et al. Comparative efficacy of dose regimens in enzyme replacement therapy of type I Gaucher disease. Blood Cells Mol Dis. 2000;26:285–290. | ||
[No authors listed]. Gaucher disease. Current issues in diagnosis and treatment. NIH Technology Assessment Panel on Gaucher Disease. JAMA. 1996;275:548–553. | ||
Zimran A, Gelbart T, Westwood B, Grabowski GA, Beutler E. High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews. Am J Hum Genet. 1991;49:855–889. | ||
Cox TM. Gaucher disease: clinical profile and therapeutic developments. Biologics. 2010;4:299–213. | ||
Harmanci O, Bayraktar Y. Gaucher disease: new developments in treatment and etiology. World J Gastroenterol. 2008;14:3968–3973. | ||
Mistry PK, Cappellini MD, Lukina E, et al. A reappraisal of Gaucher disease – diagnosis and disease management algorithms. Am J Hematol. 2011;86:110–115. | ||
Beighton P, Goldblatt J, Sacks S. Bone involvement in Gaucher disease. In: Desnick R, Gatt S, Grabowski G, editors. Gaucher Disease: A Century of Delineation. New York, NY, USA: Alan R Liss; 1982. | ||
Balwani M, Fuerstman L, Kornreich R, Edelmann L, Desnick RJ. Type 1 Gaucher disease: significant disease manifestations in “asymptomatic” homozygotes. Arch Intern Med. 2010;170:1463–1469. | ||
Mikosch P. Gaucher disease and bone: best practice and clinical research. Rheumatology. 2011;21:665–681. | ||
Wine E, Yaniv I, Cohen IJ. Hyper-immunoglobulinemia in pediatric onset type 1 Gaucher disease and effects of enzyme replacement. J Pediatr Hematol Oncol. 2007;29:451–457. | ||
Capablo JL, de Cabezón AS, Fraile J, Alfonso P, Pocovi M, Giraldo P. Neurological evaluation of patients with Gaucher disease diagnosed as type 1. J Neurol Neurosurg Psychiatry. 2008;79:219–222. | ||
Griffiths GM. Gaucher disease: forging a new path to the lysosomes. Cell. 2007;131:647–649. | ||
Dvir H, Harel M, McCarthy AA, et al. X-ray structure of human acid-beta glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003;4:704–709. | ||
Kornfeld S. Structure and function of the mannose 6-phosphate/insulin like growth factor II receptors. Annu Rev Biochem. 1992;61:307–330. | ||
Reczek D, Schwake M, Schröder J, et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta glucocerebrosidase. Cell. 2007;131:770–783. | ||
Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Hematol. 2005;129:178–188. | ||
Aerts JM, Kallemeijn WW, Wegdam W, et al. Biomarkers in the diagnosis of lysosomal storage disorders: proteins, lipids, and inhibodies. J Inherit Metab Dis. 2011;34:605–619. | ||
Beutler E, Kuhl W. The diagnosis of the adult type of Gaucher’s disease and its carrier state by demonstration of deficiency of beta-glucosidase activity in peripheral blood leukocytes. J Lab Clin Med. 1970;76:747–755. | ||
Zimran A. How I treat Gaucher disease. Blood. 2011;118:1463–1471. | ||
Aerts JM, Donker-Koopman WE, van der Vliet MK, et al. The occurrence of two immunologically distinguishable beta glucocerebrosidases in human spleen. Eur J Biochem. 1985;150:565–574. | ||
Aerts JM, Donker-Koopman WE, Koot M, Barranger JA, Tager JM, Schram AW. Deficient activity of glucocerebrosidase in urine from patients with type 1 Gaucher disease. Clin Chim Acta. 1986;158:155–163. | ||
Aerts JM, Hollak CE. Plasma and metabolic abnormalities in Gaucher’s disease. Baillieres Clin Haematol. 1997;10:691–709. | ||
Moran MT, Schofield JP, Hayman AR, Shi GP, Young E, Cox TM. Pathologic gene expression in Gaucher disease: upregulation of cysteine proteinases including osteoclastic cathepsin K. Blood. 2000;96:1969–1978. | ||
van Breemen MJ, Aerts JM, Sprenger RR, Speijer D. Potential artifacts in proteome analysis of plasma of Gaucher patients due to protease abnormalities. Clin Chim Acta. 2008;396:26–32. | ||
Barak V, Acker M, Nisman B, et al. Cytokines in Gaucher’s disease. Eur Cytokine Netw. 1999;10:205–210. | ||
van Breemen MJ, de Fost M, Voerman JS, et al. Increased plasma macrophage inflammatory protein (MIP)-1 alpha and MIP-1 beta levels in type 1 Gaucher disease. Biochim Biophys Acta. 2007;1772:788–796. | ||
Moller HJ, de Frost M, Aerts H, Hollak C, Noetrup SK. Plasma level of macrophage-derived soluble CD163 is increased and positively correlates with severity in Gaucher’s disease. Eur J Haematol. 2004;72:135–139. | ||
Hakala BE, White C, Recklies AD. Human cartilage gp-39, a major secretory product of articular chondrocytes and synovial cells, is a mammalian member of a chitinase protein family. J Biol Chem. 1993;268:25803–25810. | ||
Owhashi M, Arita H, Hayai N. Identification of a novel eosinophil chemotactic cytokine (ECF-L) as a chitinase family protein. J Biol Chem. 2000;275:1279–1286. | ||
Hollak CE, Maas M, Aerts JM .Clinically relevant therapeutic endpoints in type 1 Gaucher disease. J Inherit Metab Dis. 2001;24 Suppl 2:97–105. | ||
DiRocco M, Giona F, Carubbi F, et al. A new severity score index for phenotypic classification and evaluation of responses to treatment in type I Gaucher disease. Haematologica. 2008;93:1211–1218. | ||
Boot RG, Verhoek M, de Fost M, et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic interventions. Blood. 2004;103:33–39. | ||
Deegan B, Moran MT, McFarlane I, et al. Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cells Mol Dis. 2005;35:259–267. | ||
Boot RG, Verhoek M, Langeveld M, et al. CCL18: a urinary marker of Gaucher cell burden in Gaucher patients. J Inherit Metab Dis. 2006;29:564–571. | ||
Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuropathic Gaucher disease with special reference to pregnancy, splenectomy, biphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis. 2008;31:319–336. | ||
Rolfs A, Giese AK, Grittner U, et al. Glucosylsphingosine is highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS One. 2013;8:e79732. | ||
http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=ca02f7a4-ae4f-43c1-a06a-259fe4fcf9cf. Search word: velaglucerase alfa. Accessed July 24, 2015. | ||
http://dailymed.nlm.gov/dailymed/lookup.cfm? Search word: taliglucerase alfa. Accessed December 28, 2012. | ||
Shayman JA. Eliglustat tartrate: glucosylceramide synthase inhibitor treatment of type 1 Gaucher disease. Drugs Future. 2010;35:613–620. | ||
Shayman JA. The design and clinical development of inhibitors of glycosphingolipid synthesis: will invention be the mother of necessity? Trans Am Clin Climatol Assoc. 2013;124:46–60. | ||
Buccoliero R, Futerman AH. The roles of ceramide and complex sphingolipids in neuronal cell function. Pharmacol Res. 2003;47:409–419. | ||
http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=817892d1-ee12-4632-85fc-57ccdf16d7b8. Search word: miglustat. Accessed July 24, 2015. | ||
Schiffmann R, Fitzgibbon EJ, Harris C, et al. Randomized controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol. 2008;64:514–522. | ||
Futerman AH, Sussman JL, Horowitz M, Silman I, Zimran A. New directions in the treatment of Gaucher disease. Trends Pharmacol Sci. 2004;25:147–151. | ||
Futerman AH, Hannun YA. The complex life of simple sphingolipids. EMBO Rep. 2004;5:777–782. | ||
Lee L, Abe A, Shayman JA. Improved inhibitors of glucosylceramide synthase. J Biol Chem. 1999;274:14662–14669. | ||
McWachern KA, Fung J, Komarnitsky S, et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol Genet Metab. 2007;91:259–267. | ||
Marshall J, McEachern KA, Chuang WL, et al. Improved management of lysosomal glucosylceramide levels in a mouse model of type 1 Gaucher disease using enzyme and substrate reduction therapy. J Inherit Metab Dis. 2010;33:281–289. | ||
Pavlova EV, Archer J, Wang SZ, et al. Inhibition of UDP-glucosylceramide synthase in mice prevents Gaucher disease-associated B-cell malignancy. J Pathol. 2015;235:113–124. | ||
Cox TM. Eliglustat tartrate, an orally glucocerebroside synthase inhibitor for the potential treatment of Gaucher disease and other lysosomal storage diseases. Curr Opin Invest Drugs. 2010;11:1169–1181. | ||
US Food and Drug Administration. FDA approves new orphan drug to treat a form of Gaucher disease. August 19, 2014. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm302549.htm. Accessed July 12, 2015. | ||
Genzyme Corporation. Cerdelga (eliglustat). Available from: http://cerdelga.com/pdf/cerdelga_prescribing-information.pdf. Accessed June 23, 2015. | ||
Poole RM. Eliglustat: first global approval. Drugs. 2014;74:1829–1836. | ||
Peterschmitt MJ, Burke A, Blankstein L, et al. Safety, tolerability, and pharmacokinetics of eliglustat tartrate (Genz-112638) after single dose, multiple doses, and food in healthy volunteers. J Clin Pharmacol. 2011;51:695–705. | ||
Lukina E, Watman N, Arreguin EA, et al. A Phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood. 2010;116:893–899. | ||
Lukina E, Watman N, Arreguin EA, et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (GENZ-112638) treatment: 2-year results of a Phase 2 study. Blood. 2010;116:4095–4098. | ||
Lukina E, Watman N, Dragosky M, et al. Eliglustat, an investigational oral therapy for Gaucher disease type 1: Phase 2 trial results after 4 years of treatment. Blood Cells Mol Dis. 2014;53:274–276. | ||
Kamath RS, Lukina E, Watman N, et al. Skeletal improvement in patients with Gaucher disease type 1: a Phase 2 trial of oral eliglustat. Skeletal Radiol. 2014;43:1353–1360. | ||
Mistry PK, Lukina E, Turkia HB, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1. JAMA. 2015;313:695–706. | ||
Cox TM, Drelichman G, Cravo R, et al. Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilized on enzyme replacement therapy: a Phase 3, randomised, open-label non-inferiority trial. Lancet. 2015;385:2355–2362. | ||
Genzyme, A Sanofi Company. A study of eliglustat tartrate (Genz-112638) in patients with Gaucher disease (ENGAGE). Available from: http://www.clinicaltrials.gov/ct2/eliglusat. Accessed June 23, 2015. | ||
Wenstup RJ, Bailey L, Grabowski GA, et al. Gaucher disease: alendronate disodium improves bone mineral density in adults receiving enzyme therapy. Blood. 2004;104:1253–1257. | ||
Sanuel R, Katz K, Papapoulos SE, Yosipovitch Z, Zaizov R, Liberman UA. Amino hydroxy propylidene biphosphonate (APD) treatment improve the clinical skeletal manifestations of Gaucher’s disease. Pediatrics. 1994;94:385–389. | ||
Available from: http://www.goodrx.com/cerdelga. Accessed June 22, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.