")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Efficacy of the dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with imatinib mesylate against chronic myelogenous leukemia cell lines
Authors Xin P, Li C, Zheng Y, Peng Q, Xiao H, Huang Y, Zhu X
Received 10 January 2017
Accepted for publication 10 February 2017
Published 3 April 2017 Volume 2017:11 Pages 1115—1126
DOI https://doi.org/10.2147/DDDT.S132092
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Pengliang Xin, Chuntuan Li, Yan Zheng, Qunyi Peng, Huifang Xiao, Yuanling Huang, Xiongpeng Zhu
Department of Haematology, First Hospital of Quanzhou Affiliated to Fujian Medical University, Licheng, Quanzhou, Fujian Province, China
Background: Phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway is a therapy target of cancer. We aimed to confirm the effect of dual PI3K/mTOR inhibitor NVP-BEZ235 on proliferation, apoptosis, and autophagy of chronic myelogenous leukemia (CML) cells and sensitivity of tyrosine kinase inhibitor in vitro.
Methods: Two human CML cell lines, K562 and KBM7R (T315I mutant strain), were used. The proliferation of CML cells was detected by MTS (Owen’s reagent) assay. Cell cycle and apoptosis assay were examined by flow cytometric analysis. The phosphorylation levels and the expression levels were both evaluated by Western blot analysis. NVP-BEZ235 in combination with imatinib was also used to reveal the effect on proliferation and apoptosis.
Results: NVP-BEZ235 significantly inhibited the proliferation in a time- and dose-dependent manner, and the half-maximal inhibitory concentration values of NVP-BEZ235 inhibiting the proliferation of K562 and KBM7R were 0.37±0.21 and 0.43±0.27 µmol/L, respectively, after 48 h. Cell apoptosis assay showed that NVP-BEZ235 significantly increased the late apoptotic cells. Cell cycle analysis indicated that the cells were mostly arrested in G1/G0 phase after treatment by NVP-BEZ235. In addition, results also found that, after treatment by NVP-BEZ235, phosphorylation levels of Akt kinase and S6K kinase significantly reduced, and the expression levels of cleaved caspase-3 significantly increased; meanwhile, the expression levels of caspase-3, B-cell lymphoma-2, cyclin D1, and cyclin D2 significantly decreased, and the ratio of LC3II/LC3I was significantly increased with increased LC3II expression level. Moreover, imatinib in combination with NVP-BEZ235 induced a more pronounced colony growth inhibition than imatinib alone.
Conclusion: NVP-BEZ235 effectively inhibited cell proliferation by G0/G1 cell cycle arrest and induced apoptosis through deregulating PI3K/Akt/mTOR pathway in CML cells; in addition, NVP-BEZ235 can enhance cell autophagy, and is conducive to raising CML cell sensitivity to imatinib to inhibit the growth of imatinib-resistant cells.
Keywords: chronic myelogenous leukemia, NVP-BEZ235, phosphatidylinositol 3-kinase/Akt/mammalian pathway, imatinib, apoptosis, autophagy
Background
Chronic myelogenous leukemia (CML) is a myeloproliferative disorder of hematopoietic stem cells, characterized by the presence of the Philadelphia chromosome (Ph),1,2 and the expression of BCR–ABL fusion oncoprotein. Even though conventional chemotherapy kills a majority of CML cells, it does not eliminate CMLs thoroughly. Although allogeneic hematopoietic stem cell transplantation (Allo-SCT) can affect a radical cure on CML, it suffers from the potential disadvantage of difficulty in finding a suitable donor, as well as a high death risk and a low cure rate. Therefore, either conventional chemotherapy or Allo-SCT is not the most effective therapeutic schedule.
However, the ABL proto-oncogene (ABL) tyrosine kinase inhibitor (TKI) imatinib failed to completely cure all the variants that present strong resistance to BCR–ABL-targeting medicines because of the survival extension of CML patients. Therefore, the second-generation products, such as nilotinib, bosutinib, and dasatinib, arise at the historic moment for clinical application. Originally, the majority of imatinib-resistant or imatinib-intolerant patients responded to treatment with these second-generation BCR–ABL kinase inhibitors.3 However, therapy with second-generation BCR–ABL kinase inhibitors or transplantation is less successful in patients with advanced or blast-phase CML than in those with chronic phase.4 Furthermore, if quiescent leukemia stem cells are not eradicated, current BCR–ABL TKI will not cure the disease.5 Alternative treatment modalities, such as BCR–ABL-targeting TKIs, combined with tolerated agents that target pathways downstream of BCR–ABL could prevent the emergence of resistant clones. Therefore, novel and effective treatments are needed to improve clinical outcomes in the treatment of CML patients.
The phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway deregulation is a common event in human cancer and associated with tumor cell proliferation, growth, and apoptosis.6,7 As the PI3K/Akt/mTOR pathway is highly activated in most human cancers, this pathway has become a favorable therapy target of cancer currently.7–9 Previously, it was established that the upregulation and activation of PI3K/Akt/mTOR signaling were important for conferring a growth advantage to leukemia cells, including CML.10 NVP-BEZ235, which is a dual inhibitor of PI3K and mTOR, is a synthetic compound of belonging to the class of imidazoquinolines, and inhibits PI3K and mTOR catalytic activity by competitively binding to the ATP-binding cleft.11 Therefore, we speculate that NVP-BEZ235 may play an inhibitory effect on CMLs for its dual inhibitor of PI3K and mTOR, and it has shown antitumor activity against various tumor types.
Furthermore, as the PI3K/Akt/mTOR pathway is also implicated in the control of autophagy, inhibition of autophagy was used as a strategy to enhance the efficacy of PI3K/Akt/mTOR inhibitors. Therefore, we performed our study using two CML cell lines (K562 and KBM7R) to further assess and confirm the effects of NVP-BEZ235 on proliferation, apoptosis, autophagy, and cell cycle, then discuss the effects of NVP-BEZ23 on imatinib.
Materials and methods
Cell lines and reagents
A BCR–ABL-positive cell line K562 was obtained from the Institute of Hematology of Fujian Province, and cell line KBM7R was from Institute of Hematology of Haerbin Province. This research had ethical review approval of Ethics Committee of the First Hospital of Quanzhou Affiliated Fujian Medical University. NVP-BEZ235 and imatinib were both purchased from Selleckchem (Houston, TX, USA). NVP-BEZ235 was dissolved in dimethylsulfoxide (DMSO), and imatinib was dissolved in distilled water prior to dilution to the desired concentration in the grown medium. Before experiment, NVP-BEZ235 was stored at −20°C. In the following experiments, it would be further diluted to an appropriate final concentration.
Cell culture and treatment
Cell lines K562 and KBM7R were grown in Roswell Park Memorial Institute Medium 1640 supplemented with 10% fetal bovine serum (FBS) (Gibco, Waltham, MA, USA) and were maintained in a 5% CO2 humidified atmosphere at 37°C.
Cell proliferation assay
The cells were seeded in 96-well plates at a density of 2×104 cells/well. The cells were treated with NVP-BEZ235 at various concentrations for 24 and 48 h. A control group, cells incubated with equal volume of DMSO instead of NVP-BEZ235, and a zero adjustment group were also included. The cell viability and survival from various concentrations of NVP-BEZ235 were evaluated using MTS.
Cell apoptosis assay
The cells were seeded in six-well plates with 10% FBS at a density of 5×105 cells/well, and, respectively, treated with various concentrations (0.01, 0.1, 1.0, and 10 μmol/L) of NVP-BEZ235 for 24 and 48 h. We set the experimental group by adding 200 μL suspended cells at a concentration of 5×105/mL suspended with 1× binding buffer to a 1.5-mL EP tube, then adding 5 μL annexin V fluorescein isothiocyanate (FITC) and 5 μL propidium iodide (PI). Meanwhile, the cells incubated with equal volume of DMSO instead of NVP-BEZ235 were used as control. After incubation period, cells were harvested through centrifugation. Subsequently, apoptosis was assayed using the FITC annexin V Apoptosis Detection Kit I (BD Biosciences, San Jose, CA, USA), following the manufacturer’s instructions. The cells were assessed using flow cytometric analysis. Viable cells were FITC annexin V and PI-negative cells; cells that were FITC annexin V-positive and PI-negative were considered as being in early apoptosis, whereas necrotic cells were FITC annexin V-negative/low PI-positive; cells that were both FITC annexin V- and PI-positive were considered as being in late apoptosis.
Cell cycle analysis
The cells were seeded in six-well plates with 10% FBS at a density of 5×105 cells/well, and, respectively, treated with various concentrations of NVP-BEZ235 for 24 and 48 h. Meanwhile, the cells incubated with equal volume of DMSO instead of NVP-BEZ235 were used as control. After incubation period, cells were harvested through centrifugation and washed twice with ice-cold phosphate-buffered saline (PBS). Afterwards, the cells were fixed using 1 mL ice-cold PBS and 3 mL 70% ethanol at −20°C overnight. After fixation, the cells were washed once again with ice-cold PBS, and then incubated with 30 μg/mL of PI (Sigma-Aldrich, St Louis, MO, USA) and 40 μg/mL of RNase (Sigma-Aldrich) for 30 min at room temperature in a dark room.
Western blot analysis
The cells were seeded in six-well plates with 10% FBS at a density of 5×105 cells/well and, respectively, treated with various concentrations of NVP-BEZ235 for 24 and 48 h. Meanwhile, the cells incubated with equal volume of DMSO instead of NVP-BEZ235 were used as control. Western blotting was performed as described previously.12 Briefly, after washing twice by PBS, cells were lysed with radio immunoprecipitation assay lysis buffer (Solarbio, Beijing, China), which contained 1 mmol/L phenylmethanesulfonyl fluoride. The protein concentrations of the cell lysates were determined using the Enhanced BCA Protein Assay kit (Beyotime, Haimen, China). Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to nylon membranes. The membranes were blocked in tris-buffered saline with Tween 20 (TBS-T) buffer solution containing 5% nonfat dry milk at 4°C overnight, and subsequently washed by TBS-T 3 times. Then they were incubated with horseradish peroxidase–conjugated secondary antibody (anti-rabbit and anti-mouse) at 37°C for 2 h, and washed by TBS-T 3 times again. Immunoblotting was performed using rabbit polyclonal antibodies against Akt (1:100 dilution), phosphorylated Akt (Ser473 or Thr308, both 1:100 dilution), extracellular signal-regulated kinase (ERK), phosphorylated ERK, STAT5, phosphorylated STAT5, B-cell lymphoma-2 (Bcl-2), cleaved caspase-3, LC3II/LC3I, and β-actin. All antibodies were purchased from Abcam and Santa Cruz. Band density was imaged and the levels of protein expression quantified using Molecular Imager VersaDoc MP 4000 system (Bio-Rad) after electrochemiluminescence and normalized to the β-actin levels. The phosphorylation levels of proteins were assessed based on the ratios of phosphorylated proteins to the corresponding total proteins.
Statistical analysis
Data for comparison of multiple groups are presented as mean ± standard deviation and were analyzed by analysis of variance. P<0.05 was considered statistically significant. Statistical analyses were performed using SPSS 17.0 software (SPSS, Inc., Chicago, USA). Western blotting analyses were performed using Quantity one.
Results
NVP-BEZ235 inhibited K562 and KBM7R cell proliferation
To examine the inhibitory effect of NVP-BEZ235 on the proliferation of CML cells, we evaluated the growth of K562 and KBM7R cells before and after NVP-BEZ235. As shown in Figure 1A and B, the inhibition rates of two CML cell lines were significantly increased with the increasing incubation time (24, 48, and 72 h) and concentration (0.01, 0.10, 0.50, 2.00, 5.00, and 10.00 μM) of NVP-BEZ235 (P<0.05), indicating that NVP-BEZ235 could inhibit the proliferation of CML cells and this antiproliferative effect was time and dose dependent. The half-maximal inhibitory concentration (IC50) of NVP-BEZ235 was 0.37±0.21 and 0.43±0.27 μM, respectively.
 | Figure 1 BEZ235 inhibits the proliferation of CML cells and enhances the sensitivity of imatinib. |
Cotreatment with NVP-BEZ235 and imatinib inhibited the growth of K562 and KBM7R cells
To examine the inhibitory effect of NVP-BEZ235 and imatinib on the proliferation of CML cells, we evaluated the cell viability of K562 and KBM7R by treatment with imatinib alone or cotreatment with NVP-BEZ235. As indicated in Figure 1C and D, compared to treatment with imatinib alone, the cell viability of K562 and KBM7R decreased after cotreatment with NVP-BEZ235 (0.2 μM) and imatinib (0.01, 0.10, 0.50, 2.00, 5.00, and 10.00 μM) in 48 h. The IC50 values of imatinib inhibiting growth of K562 before and after cotreatment with NVP-BEZ235 (0.2 μM) were 0.14±0.05 and 0.09±0.04 μM (P<0.05), respectively, in 48 h; meanwhile, the IC50 values were 3.93±2.29 and 0.44±0.22 μM (P<0.05), respectively, in KBM7R.
NVP-BEZ235 inhibited PI3K/Akt/mTOR pathway in CML cells
Figure 2A showed the Western blotting results. The phosphorylation levels of Akt and RPS6K were modestly inhibited by NVP-BEZ235 in both K562 and KBM7R cells (P<0.05). Moreover, the inhibition effect was significantly enhanced with the increasing dose of NVP-BEZ235, indicating that this inhibition effect was dose dependent (Figure 2B). Meanwhile, we found that there was no significant difference in the phosphorylation levels of ABL and STAT5, and the expression levels of c-ABL, ERK, and STAT5 with increasing dose of NVP-BEZ235.
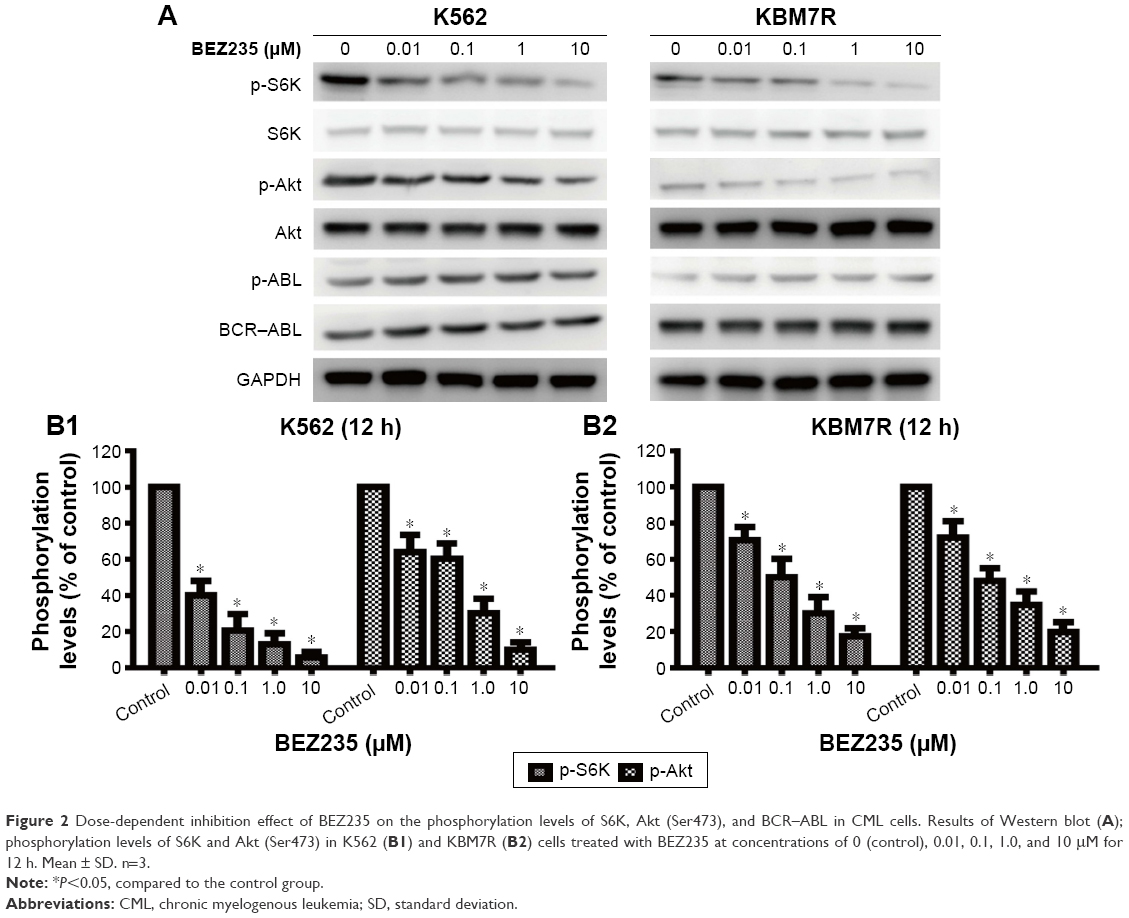 | Figure 2 Dose-dependent inhibition effect of BEZ235 on the phosphorylation levels of S6K, Akt (Ser473), and BCR–ABL in CML cells. Results of Western blot (A); phosphorylation levels of S6K and Akt (Ser473) in K562 (B1) and KBM7R (B2) cells treated with BEZ235 at concentrations of 0 (control), 0.01, 0.1, 1.0, and 10 μM for 12 h. Mean ± SD. n=3. |
NVP-BEZ235 induces apoptosis in K562 and KBM7R cells
We next examined whether NVP-BEZ235 induced apoptosis in K562 and KBM7R cells. Both K562 and KBM7R cells were incubated for 24 and 48 h with increasing concentrations of NVP-BEZ235 (Figures 3 and 4). The percentage of apoptotic K562 and KBM7R cells increased in a time- and dose-dependent manner after NVP-BEZ235 treatment (Figure 3A and B). Moreover, we found that apoptotic cells significantly increased by cotreatment with NVP-BEZ235 and imatinib compared with treatment with NVP-BEZ235 or imatinib alone (P<0.05). After being incubated for 24 h with increasing concentrations of NVP-BEZ235, the expression level of cleaved caspase-3 of K562 (Figure 5A1 and B1) and KBM7R (Figure 5A2 and B2) cells both upregulated in a time- and dose-dependent manner, while Bcl-2 and caspase-3 downregulated with increasing concentrations of NVP-BEZ235 in immunoblot analysis (P<0.05).
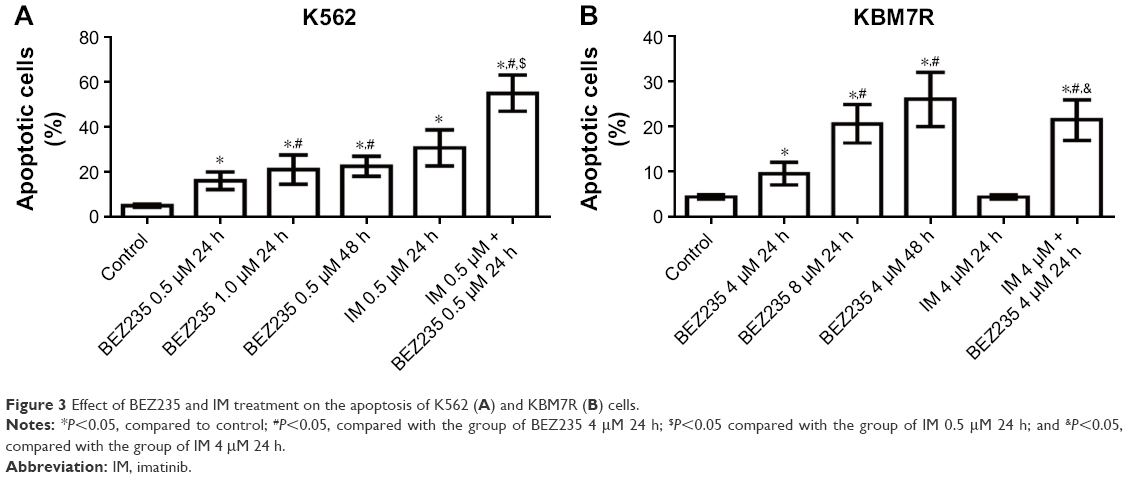 | Figure 3 Effect of BEZ235 and IM treatment on the apoptosis of K562 (A) and KBM7R (B) cells. |
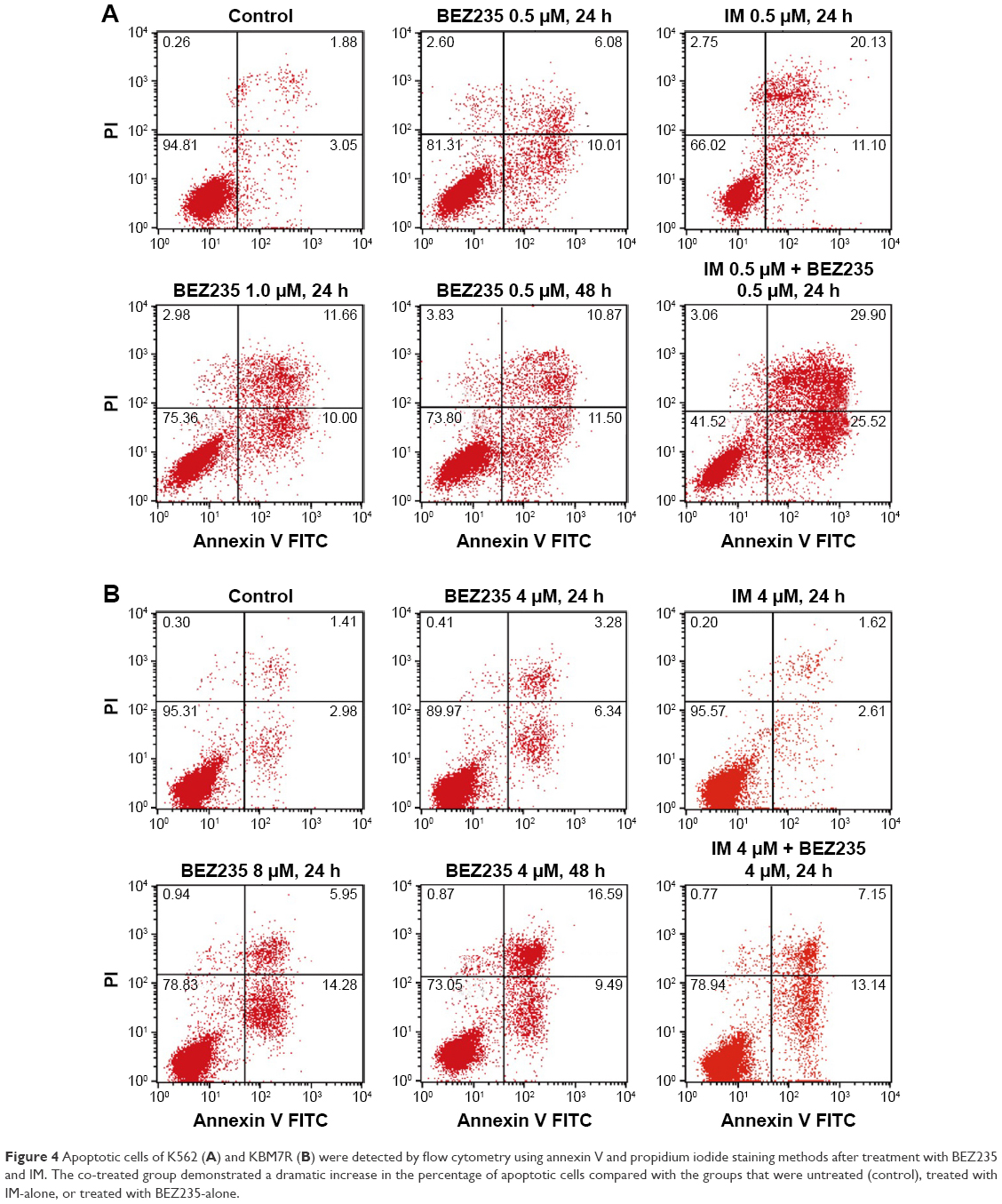 | Figure 4 Apoptotic cells of K562 (A) and KBM7R (B) were detected by flow cytometry using annexin V and propidium iodide staining methods after treatment with BEZ235 and IM. The co-treated group demonstrated a dramatic increase in the percentage of apoptotic cells compared with the groups that were untreated (control), treated with IM-alone, or treated with BEZ235-alone. |
 | Figure 5 Effect of BEZ235 on the expression of B-cell lymphoma-2 (Bcl-2), caspase-3, and cleaved caspase-3. K562 (A1) and KBM7R (A2) cells were treated with BEZ235 for 24 h; total protein lysates were immunoblotted with antibodies for Bcl-2, caspase-3, and cleaved caspase-3. Percentage of apoptotic protein expression of K562 (B1) and KBM7R (B2) cells compared to control. |
NVP-BEZ235 induced cell cycle arrest in G1/G0 phase in K562 and KBM7R cells
Both CML cells were incubated with 0.5 and 1.0 μM of NVP-BEZ235 for 48 h or 1.0 μM of NVP-BEZ235 for 24 and 48 h. We analyzed cycle distribution by flow cytometry (Figure 6C1 and C2), and we found that different concentrations of NVP-BEZ235 induced cell cycle arrest in G1/G0 phase in K562 (Figure 6A1) and KBM7R (Figure 6A2) cell lines. Compared with control, K562 and KBM7R cells in the G1/G0 phase were significantly increased (P<0.05), and in G2/M and S phases were significantly reduced in cell cycle test after 48 h treatment by 0.5 or 1.0 μM of NVP-BEZ235 (P<0.05). Moreover, compared with cells treated with 0.5 μM NVPBEZ235, there were significantly more cells in G1/G0 phase when cells were treated with 1.0 μM NVPBEZ235 (P<0.05). In addition, when treated with 1.0 μM of NVP-BEZ235 for 24 and 48 h, cells in the G1/G0 phase also significantly increased compared to control (P<0.05, Figure 6B1 and B2), and compared with cells treated for 24 h, there were significantly more cells in G1/G0 phase when treated for 48 h.
 | Figure 6 BEZ235 blocked the cell cycle of K562 and KBM7R cells at the G1/G0 phase. BEZ235 blocked the cell cycle of K562 (A1) and KBM7R (A2) cells at the G1/G0 phase with increasing concentration; BEZ235 blocked the cell cycle of K562 (B1) and KBM7R (B2) cells at the G1/G0 phase with increasing treatment time; cell cycle distributions of K562 (C1) and KBM7R (C2) were analyzed by flow cytometry. Cellular DNA content was determined by staining with a hypotonic propidium iodide solution by flow cytometry. |
Meanwhile, expression levels of related cell cycle regulatory proteins cyclin D1 and cyclin D2 of K562 (Figure 7A1 and B1) and KBM7R (Figure 7A2 and B2) cells downregulated with increasing concentrations of NVP-BEZ235 for 24 h in the immunoblot analysis (P<0.05).
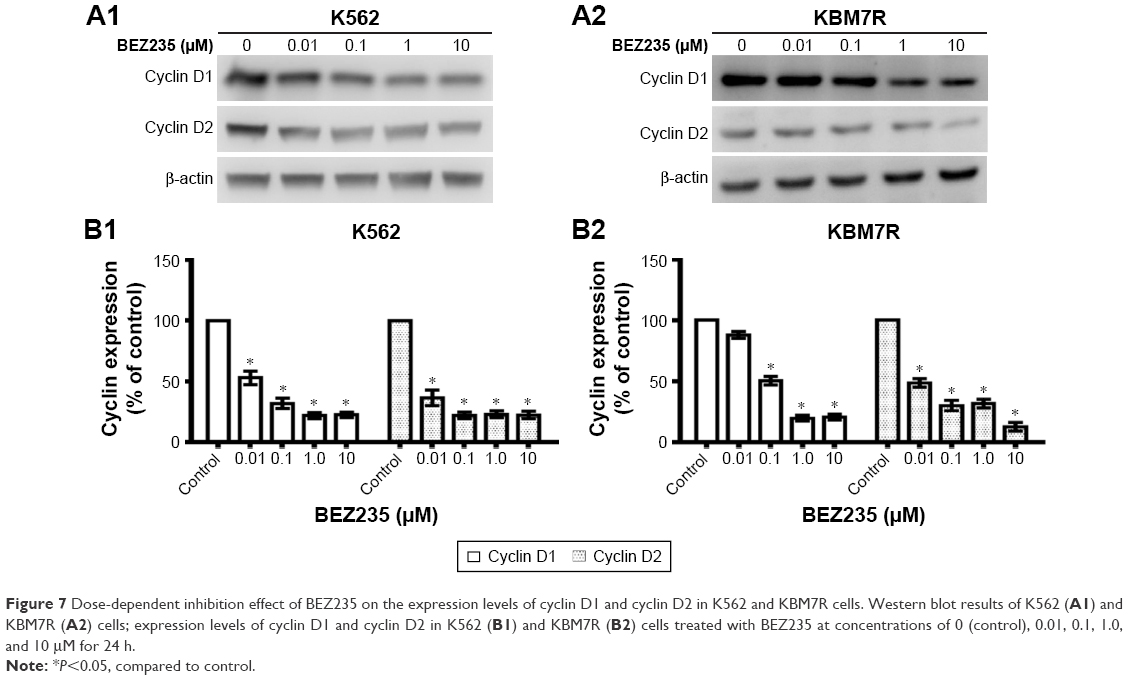 | Figure 7 Dose-dependent inhibition effect of BEZ235 on the expression levels of cyclin D1 and cyclin D2 in K562 and KBM7R cells. Western blot results of K562 (A1) and KBM7R (A2) cells; expression levels of cyclin D1 and cyclin D2 in K562 (B1) and KBM7R (B2) cells treated with BEZ235 at concentrations of 0 (control), 0.01, 0.1, 1.0, and 10 μM for 24 h. |
NVP-BEZ235 enhanced the expression level of LC3II
Both K562 and KBM7R cells were incubated for 0, 6, 12, 24, and 48 h with 1.0 μM concentration of NVP-BEZ235 or for 24 h with 0, 0.01, 0.1, 1.0, and 10 μM NVP-BEZ235. In the immunoblot analysis (Figure 8A and B), we found that NVP-BEZ235 increased the expression level of LC3II of both CML cells in a time- and dose-dependent manner, and also increased the ratio of LC3II/LC3I (P<0.05, Figure 8C and D), though there was no obvious enhancement effect of NVP-BEZ235 on the expression level of LC3.
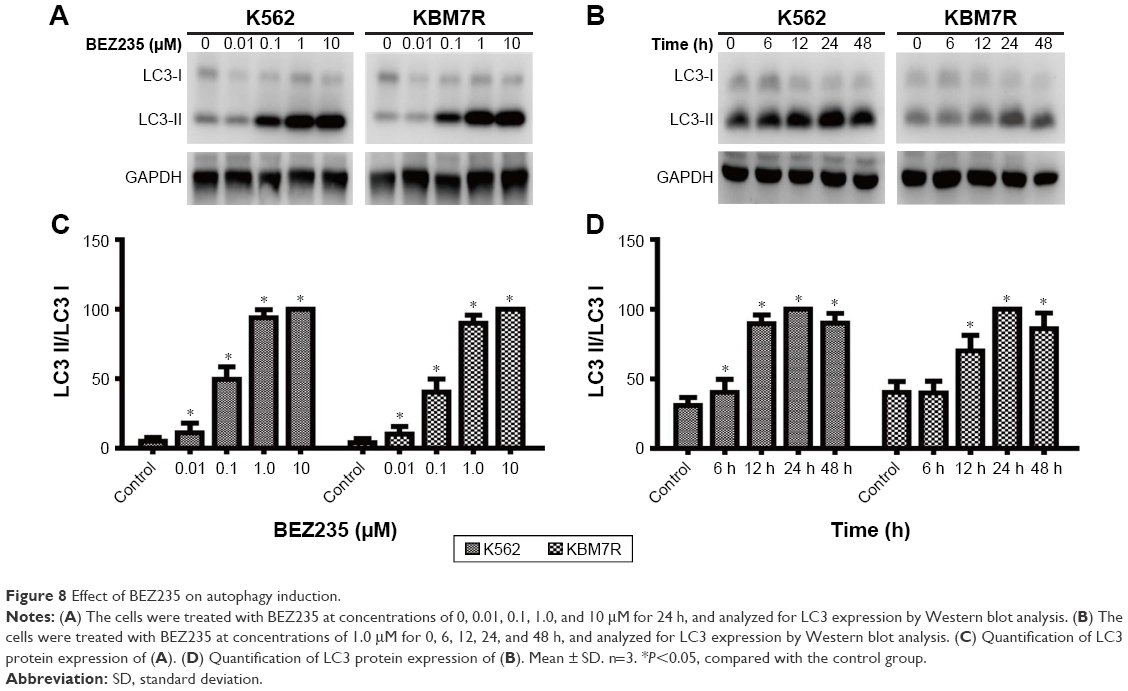 | Figure 8 Effect of BEZ235 on autophagy induction. |
Discussion
The PI3K/Akt/mTOR pathway deregulation is a common event in human cancer and associated with the tumor cell proliferation, growth, and apoptosis.6,7 Till now, it has been established that the activation of mTOR was closely related with the proliferation, transfer, and differentiation of breast cancer cells, lung cancer cells, and leukemia cells.13–16 Therefore, the targeting of the PI3K/Akt/mTOR signaling pathway by specific kinase inhibitors has been the focus of extensive treatment for CML cells, and a number of PI3K/Akt/mTOR signaling inhibitors are under investigation. Among these, NVP-BEZ235 has shown antitumor activity against various tumor types.17–19
Regarded as one of the most representative cell lines in CML research, K562 has been studied frequently. That is why we selected K562 cell lines as research subject to reveal the effect of NVP-BEZ235 on sensitivity of TKI. And for KBM7R (T315I mutant strain), T315I mutant would commonly cause ABL kinase domain mutation that could lead to TKI resistance, and what is more important, clinical scientists were never able to overcome TKI resistance caused by T315I mutant so far.
Rapamycin has been shown to be effective in vitro against imatinib-resistant CML cells.20 In our study, we found that NVP-BEZ235 showed the similar efficacy to rapamycin in inhibiting mTOR pathway of K562,20,21 inducing cell cycle arrest in G1/G0 phase in both CML cell lines, which might be caused by the inhibition of PI3K/Akt/mTOR pathway by decreasing the expression of p-Akt and p-S6K.
However, as rapamycin is just a single inhibitor of mTOR, we chose NVP-BEZ235, the dual inhibitor of PI3K and mTOR, as investigational drug to study its effect on the PI3K/Akt/mTOR pathway of K562 and KBM7R cells. The significantly reduced phosphorylation levels of Akt and S6K indicated that NVP-BEZ235 worked by inhibiting PI3K/Akt/mTOR pathway. NVP-BEZ235 inhibited growth of CML cells by inducing cell cycle arrest in G1/G0 phase, decreasing expression levels of cyclin D1 and cyclin D2.22 Moreover, it also induces apoptosis in CML cells by changing the expression levels of cleaved caspase-3 and Bcl-2, showing a powerful antileukemia activity.13,23,24
The current studies suggested that autophagy was closely related with PI3K/Akt/mTOR pathway, especially with target point of PI3K and mTOR. Therefore, it is necessary to find feasible and effective drug targeting this pathway in the clinical treatment of CML cells. The dual inhibitor of PI3K and mTOR NVP-BEZ235 inhibits PI3K and mTOR catalytic activity, affecting the activity of K562 cell lines of autophagy.
We all know that the formation of autophagic vacuole is closely related with the modification process of LC3. Either the content of LC3II or the ratio of LC3II/LC3I is positively correlated with the quantity of autophagic vacuole. In this study, treated with different incubation time and concentrations of NVP-BEZ235, the increasing tendency of the expression levels of LC3II and the ratio of LC3-II/LC3-I in CML cells showed a positive role of NVP-BEZ235 in autophagy of K562 cells. However, the relation between autophagy and apoptosis still remains unclear and needs further studies and verification.25–28
Previous studies showed that NVP-BEZ235 in combination with TKIs is an efficacious treatment of Ph-positive leukemia.13,29,30 In this study, NVP-BEZ235 in combination with imatinib inhibited the growth of K562 cell line, and it even showed great inhibitory effect on the growth of primary T315I mutant cells KBM7R. Therefore, NVP-BEZ235 in combination with imatinib was contributed to overcoming TKI-resistant mutant cells and improving the curative effect on CML cells.
NVP-BEZ235 has also shown in vivo anticancer efficacy in models of lung cancer, breast cancer, glioma, and myeloma19,26,31,32; thus, we would establish animal models to verify the efficacy and reliability of NVP-BEZ235 for CML.
In summary, the experimental data presented in this report suggest that NVP-BEZ235, a PI3K/Akt/mTOR inhibitor, potentiates imatinib-induced apoptosis of CML cells, enhances cell autophagy, and raises CML cell sensitivity to imatinib to inhibit the growth of imatinib-resistant cells in vitro. This in vitro model will provide useful information for development of therapeutic strategies against CML cells.
Acknowledgments
This work was supported by the Medical Innovation Program of Fujian Province (No 2014-CX-34) and the Natural Science Foundation of Fujian Province of China (No 2015 J01511).
Disclosure
The author reports no conflicts of interest in this work.
References
NOWELL PC. The minute chromosome (Phl) in chronic granulocytic leukemia. Blut. 1962;8:65–66. | ||
Daley GQ. Chronic myeloid leukemia: proving ground for cancer stem cells. Cell. 2004;119(3):314–316. | ||
Baccarani M, Cortes J, Pane F, et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol. 2009;27(35):6041–6051. | ||
Garg RJ, Kantarjian H, O’Brien S, et al. The use of nilotinib or dasatinib after failure to 2 prior tyrosine kinase inhibitors: long-term follow-up. Blood. 2009;114(20):4361–4368. | ||
Apperley JF. Chronic myeloid leukaemia. The Lancet. 2015;385(9976):1447–1459. | ||
Sharp ZD, Bartke A, Smith JR. Evidence for down-regulation of phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR)-dependent translation regulatory signaling pathways in Ames dwarf mice. J Gerontol A Biol Sci Med Sci. 2005;60(3):293–300. | ||
Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs. 2005;16(8):797–803. | ||
Brown RE. Morphoproteomics: exposing protein circuitries in tumors to identify potential therapeutic targets in cancer patients. Expert Rev Proteomics. 2005;2(3):337–348. | ||
Yip PY. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl Lung Cancer Res. 2015;4(2):165–176. | ||
LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11(1–2):32–50. | ||
Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–1863. | ||
Li C, Xin P, Xiao H, Zheng Y, Huang Y, Zhu X. The dual PI3K/mTOR inhibitor NVP-BEZ235 inhibits proliferation and induces apoptosis of burkitt lymphoma cells. Cancer Cell Int. 2015;15:65. | ||
Okabe S, Tauchi T, Tanaka Y, et al. Efficacy of the dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with nilotinib against BCR-ABL-positive leukemia cells involves the ABL kinase domain mutation. Cancer Biol Ther. 2014;15(2):207–215. | ||
Sun Z, Li Q, Zhang S, et al. NVP-BEZ235 overcomes gefitinib-acquired resistance by down-regulating PI3K/AKT/mTOR phosphorylation. Onco Targets Ther. 2015;8:269–277. | ||
Kuger S, Corek E, Polat B, Kammerer U, Flentje M, Djuzenova CS. Novel PI3K and mTOR inhibitor NVP-BEZ235 radiosensitizes breast cancer cell lines under normoxic and hypoxic conditions. Breast Cancer (Auckl). 2014;8:39–49. | ||
Issa A, Gill JW, Heideman MR, et al. Combinatorial targeting of FGF and ErbB receptors blocks growth and metastatic spread of breast cancer models. Breast Cancer Res. 2013;15(1):R8. | ||
Schnell CR, Stauffer F, Allegrini PR, et al. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res. 2008;68(16):6598–6607. | ||
Serra V, Markman B, Scaltriti M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68(19):8022–8030. | ||
Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–1356. | ||
Sillaber C, Mayerhofer M, Bohm A, et al. Evaluation of antileukaemic effects of rapamycin in patients with imatinib-resistant chronic myeloid leukaemia. Eur J Clin Invest. 2008;38(1):43–52. | ||
Li J, Xue L, Hao H, Li R, Luo J. Rapamycin combined with celecoxib enhanced antitumor effects of mono treatment on chronic myelogenous leukemia cells through downregulating mTOR pathway. Tumour Biol. 2014;35(7):6467–6474. | ||
Pellicano F, Scott MT, Helgason GV, et al. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells. 2014;32(9):2324–2337. | ||
Wohrle FU, Halbach S, Aumann K, et al. Gab2 signaling in chronic myeloid leukemia cells confers resistance to multiple Bcr-Abl inhibitors. Leukemia. 2013;27(1):118–129. | ||
Seitz C, Hugle M, Cristofanon S, Tchoghandjian A, Fulda S. The dual PI3K/mTOR inhibitor NVP-BEZ235 and chloroquine synergize to trigger apoptosis via mitochondrial-lysosomal cross-talk. Int J Cancer. 2013;132(11):2682–2693. | ||
Cerniglia GJ, Karar J, Tyagi S, et al. Inhibition of autophagy as a strategy to augment radiosensitization by the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Mol Pharmacol. 2012;82(6):1230–1240. | ||
Liu TJ, Koul D, LaFortune T, et al. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther. 2009;8(8):2204–2210. | ||
Shen S, Kepp O, Kroemer G. The end of autophagic cell death? Autophagy. 2012;8(1):1–3. | ||
Clarke PGH, Puyal J. Autophagic cell death exists. Autophagy. 2012;8(6):867–869. | ||
Sinclair A, Latif AL, Holyoake TL. Targeting survival pathways in chronic myeloid leukaemia stem cells. Br J Pharmacol. 2013;169(8):1693–1707. | ||
Fenouille N, Puissant A, Dufies M, et al. Persistent activation of the Fyn/ERK kinase signaling axis mediates imatinib resistance in chronic myelogenous leukemia cells through upregulation of intracellular SPARC. Cancer Res. 2010;70(23):9659–9670. | ||
Crowder RJ, Phommaly C, Tao Y, et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009;69(9):3955–3962. | ||
McMillin DW, Ooi M, Delmore J, et al. Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer Res. 2009;69(14):5835–5842. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.