")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Effect of renal impairment on the pharmacokinetics of levomilnacipran following a single oral dose of levomilnacipran extended-release capsule in humans
Authors Chen L, Greenberg W, Brand-Schieber E, Wangsa J, Periclou A, Ghahramani P
Received 25 March 2015
Accepted for publication 20 May 2015
Published 25 June 2015 Volume 2015:9 Pages 3293—3300
DOI https://doi.org/10.2147/DDDT.S85418
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Laishun Chen, William M Greenberg, Elimor Brand-Schieber, Julie Wangsa, Antonia Periclou, Parviz Ghahramani
Forest Research Institute, a subsidiary of Actavis Inc., Jersey City, NJ, USA
Purpose: Levomilnacipran extended-release (ER) is indicated for treatment of major depressive disorder in adults. We evaluated the pharmacokinetic and safety profile of levomilnacipran ER in individuals with impaired renal function.
Methods: A total of 32 individuals participated in four groups (eight in each group) with normal, mild, moderately, or severely impaired renal function. Each participant received one dose of levomilnacipran ER 40 mg. Blood and urine were assayed using liquid chromatography/tandem mass spectrometry. Results between normal and renally impaired groups were compared using analysis of variance. Safety measures included adverse events, laboratory evaluations, vital signs, suicidality, and electrocardiograms.
Results: Following administration of levomilnacipran, mean (standard deviation) maximum plasma concentration in participants with normal renal function, and mild, moderate, or severe renal impairment was 83.9 (21.0), 81.8 (23.4), 98.7 (18.1), and 122.1 (35.1) (ng/mL), respectively; area under the curve from time zero to infinity was 2,101.0 (516.9), 2,587.8 (649.9), 4,016.4 (995.4), and 5,900.8 (1,799.3) (h·ng/mL), respectively; terminal elimination half-life was 13.5 (2.8), 17.3 (3.5), 19.1 (4.6), and 27.7 (7.4) (hours), respectively; and renal clearance was 175.9 mL/min, 114.7 mL/min, 69.9 mL/min, and 28.6 mL/min, respectively. Levomilnacipran ER was generally well tolerated with no safety issues of concern identified.
Conclusion: Renal impairment was associated with increased plasma levels of levomilnacipran and prolonged half-life. No dose adjustment is required for individuals with mild renal impairment; the recommended maximum daily maintenance dose of levomilnacipran ER should not exceed 80 mg for individuals with moderate renal impairment and 40 mg for individuals with severe renal impairment.
Keywords: antidepressant, F2695, SNRI, pharmacokinetics, renal function, major depressive disorder
Introduction
Levomilnacipran (1S, 2R-milnacipran) is a serotonin and norepinephrine reuptake inhibitor (SNRI) with a greater potency for norepinephrine reuptake inhibition than for serotonin reuptake inhibition in vitro. An extended-release (ER) formulation of levomilnacipran was developed to allow for once-daily dosing. Levomilnacipran ER capsules (FETZIMA®) received US Food and Drug Administration (FDA) approval in July 2013 for the treatment of major depressive disorder in adults, with a recommended dose of 40–120 mg once daily.1
The pharmacokinetics of levomilnacipran are dose proportional (single-dose range of 25–120 mg and multiple-dose range of 25–300 mg/day), and steady-state pharmacokinetics are predictable from single-dose data.1 Levomilnacipran is well absorbed following administration of the ER capsule, with a bioavailability of 92% relative to oral solution; food has no effect on the bioavailability.1 Levomilnacipran has a mean apparent terminal elimination half-life (T1/2) of approximately 12 hours. On the basis of results from a human mass balance study using 14C-labeled levomilnacipran, 58% of the radioactivity of an oral dose was recovered in urine as unchanged levomilnacipran.1 The major metabolite of levomilnacipran recovered in urine is N-desethyl levomilnacipran, accounting for approximately 18% of the total administered radioactivity. Other metabolites excreted in the urine are levomilnacipran glucuronide (4%), desethyl levomilnacipran glucuronide (3%), p-hydroxy levomilnacipran glucuronide (1%), and p-hydroxy levomilnacipran (1%). None of the metabolites identified are pharmacologically active. Levomilnacipran is widely distributed with an apparent volume of distribution of 387–473 L; plasma protein binding is 22% over the concentration range of 10–1,000 ng/mL.1
As described above, urinary excretion was the predominant elimination pathway for levomilnacipran and its metabolites. Renal impairment can alter the pharmacokinetic profile of many drugs, particularly drugs with significant elimination via renal excretion. To investigate how renal impairment affects the pharmacokinetic behavior of levomilnacipran ER, a single-dose pharmacokinetic study was conducted in individuals with mild, moderate, or severe renal impairment, as well as in a cohort of sex-, age-, and weight-matched individuals with normal renal function.
Materials and methods
Participants and study design
A total of 32 men and women, 32–76 years of age, individuals with renal impairment and normal renal function, participated in this single-dose, open-label, parallel-group study. The study was conducted at two centers in the United States. Both centers received approval from independent ethics committees, and the study was conducted in accordance with the International Conference on Harmonisation Guidances on General Considerations for Clinical Trials, Nonclinical Safety Studies for the Conduct of Human Clinical Trials for Pharmaceuticals, and Good Clinical Practice guidelines. Signed informed consent was obtained from each participant before any study-related procedures were performed.
On day 1, participants were allocated into four groups (eight participants in each group) on the basis of estimated creatinine clearance (CLcr) values using the Cockcroft–Gault equation.2 The four groups were defined as normal renal function (CLcr ≥80 mL/min), mild renal impairment (CLcr ≥50 mL/min and <80 mL/min), moderate renal impairment (CLcr ≥30 mL/min and <50 mL/min), and severe renal impairment (CLcr ≥5 mL/min and <30 mL/min). Participants with normal renal function were recruited after those with impaired renal function had been enrolled so that individuals with normal renal function could be matched as closely as possible by sex, age range, and weight range to those with impaired renal function. The study that began in August 2009 was completed in April 2010, and was designed in accordance with the FDA guidance for industry.3
Key exclusion criteria were moderate or heavy cigarette use (≥10 cigarettes per day); sitting heart rate >100 or <50 beats per minute (bpm) at screening; pregnancy or not using adequate nonhormonal contraception (eg, nonhormonal intrauterine device, double-barrier methods); known hypersensitivity to SNRIs, selective serotonin reuptake inhibitors, or tricyclic antidepressants; clinically significant medical condition other than renal impairment; liver enzyme values greater than the upper limit of normal; clinically significant electrocardiogram (ECG) abnormalities; current or past (within last 5 years) alcohol/substance abuse or dependence; a history of narrow-angle glaucoma (controlled or uncontrolled); and a history of major depressive disorder or other psychiatric disorder except nongeneralized social phobia, as indicated by the Mini-International Neuropsychiatric Interview.
Concomitant medications were not permitted during the study, except for medications considered necessary to treat underlying disease states or medical conditions related to renal impairment. Participants requiring necessary treatment for renal impairment were instructed to take medications ≥2 hours before study drug was administered. Participants could also be excluded if a medication taken ≥14 days before the start of the study could potentially interfere with study outcomes, as judged by the investigator or study sponsor.
Levomilnacipran dosing and pharmacokinetic sample collection
All participants received a single 40 mg oral dose of levomilnacipran ER with 240 mL of water on day 1 under fasting conditions. Participants were required to drink 120 mL of water every hour until 4 hours after dose administration to facilitate urine production.
Blood and urine samples were collected over a 5-day period. Blood samples were collected at 0 hours (just before the dose was administered), and at 1 hour, 2 hours, 3 hours, 4 hours, 5 hours, 6 hours, 8 hours, 10 hours, 12 hours, 24 hours, 36 hours, 48 hours, 72 hours, and 96 hours after the dose was administered. A total of approximately 110 mL of blood was collected from each participant; 90 mL was used for analyses of levomilnacipran and N-desethyl levomilnacipran concentrations in plasma (15 samples, 6 mL each), and 20 mL was used for clinical laboratory analyses (before and after dosing, 10 mL each). Urine specimens were collected at −2 hours to 0 hours (just before the dose was administered), and at 0–4 hours, 4–8 hours, 8–12 hours, 12–16 hours, 16–32 hours, 32–48 hours, 48–72 hours, and 72–96 hours after the dose was administered.
Bioanalytical methods
Plasma and urine samples were assayed to determine levomilnacipran and N-desethyl levomilnacipran concentrations using liquid chromatography/tandem mass spectrometry methods that had been validated to demonstrate their accuracy, precision, linearity, and reproducibility.4 For both analytes, the plasma assay method was linear over the concentration range of 1–200 ng/mL and had a lower limit of quantification of 1 ng/mL in 100 μL of human plasma with K2EDTA as the anticoagulant. The urinalysis method was linear for both analytes over the concentration range of 100-5,000 ng/mL and had a lower limit of quantification of 100 ng/mL in 50 μL of human urine.
Safety assessments
Safety variables included adverse events (AEs), clinical laboratory values, vital signs (blood pressure and heart rate), ECG measurements, and suicidality (using the Columbia Suicide Severity Rating Scale). Treatment-emergent AEs (TEAEs) were defined as AEs that were not present at baseline or that were present at baseline but increased in severity during the treatment period, regardless of relationship to study drug.
Statistical analyses
Pharmacokinetic analysis
Pharmacokinetic parameters were calculated using noncompartmental analyses employing Phoenix WinNonlin software (version 6.1). Plasma and urine concentrations below the limit of quantification were treated as zero. Nominal sampling times were used to calculate pharmacokinetic parameters except when the nominal and actual sampling times were more than 5 minutes apart; in such cases, the actual sampling time was used. Pharmacokinetic parameters for levomilnacipran included the maximum plasma drug concentration (Cmax), area under the plasma concentration versus time curve from time zero to time t (AUC0–t) and from time zero to infinity (AUC0–∞), time of maximum plasma drug concentration (Tmax), T1/2, renal clearance (CLr), apparent total clearance from plasma after extravascular administration (CL/F), apparent volume of distribution at steady state after extravascular administration (Vss/F) and during the terminal phase after extravascular administration (Vz/F), cumulative amount of compound excreted unchanged into urine from time zero to time t (Ae0–t), and percentage of compound excreted unchanged in urine relative to administered dose (fraction of dose). For the inactive metabolite, N-desethyl levomilnacipran, the following pharmacokinetic parameters were determined: Cmax, Tmax, AUC0–t, AUC0–∞, T1/2, and Ae0–t.
Descriptive statistics were used to summarize all pharmacokinetic and safety data. Log-transformed Cmax, AUC0–t, and AUC0–∞ for levomilnacipran and N-desethyl levomilnacipran, if applicable, were compared between each of the impaired renal function groups and the normal renal function group using analysis of variance with group as a fixed effect in Phoenix WinNonlin software. Point estimates and 90% confidence intervals for differences between normal and impaired renal function groups on the log scale were exponentiated to obtain estimates for ratios of geometric means of Cmax, AUC0–t, and AUC0–∞ on the original scale.
For levomilnacipran, the relationship between CL/F versus CLcr and between CLr versus CLcr was evaluated by a linear regression model Y = a + b·X using SigmaPlot software (version 12.0). Dosing recommendations were proposed on the basis of the relationship between CL/F and CLcr established in this study, overall safety profile of levomilnacipran, and available dosing strengths of the product (20 mg, 40 mg, 80 mg, and 120 mg). For the purpose of dosing recommendations, classification into a renal function group was based on CLcr values as provided in the recent draft FDA guidance (2010)5 that was released after initiation of this study: control (CLcr ≥90 mL/min), mild renal impairment (CLcr 60–89 mL/min), moderate renal impairment (CLcr 30–59 mL/min), and severe renal impairment (CLcr 15–29 mL/min). Based on the regression line between CL/F and CLcr, CL/F values were determined for the midpoint CLcr value of each renal impairment group, ie, 74.5 mL/min, 44.5 mL/min, and 22 mL/min for mild, moderate, and severe renal impairment, respectively. For the purpose of comparing the CL/F value of each renally impaired group to the normal group using the linear regression model, the range of CLcr for the normal renal group was assumed to be 90–120 mL/min with a midpoint of 105 mL/min. Based on the change in the predicted CL/F value at the midpoint CLcr for each renally impaired group relative to the CL/F for the normal renal function group, an appropriate dose was determined for each renally impaired group, assuming a dose of 120 mg in the normal group.
Safety analysis
The safety population included all study participants who received the single dose of study drug. Safety data were analyzed using SAS software (version 9.1.3). Incidences were determined using the total number of study participants in each group. For calculation of mean changes from baseline to end of study, baseline was defined as the last assessment before administration of the study drug (ie, screening value).
Results
Disposition and demographics
All 32 participants completed the study and were included in the pharmacokinetic and safety analyses. No notable demographic differences were found among the subgroups, except for a lower percentage of women in the group with moderate renal impairment (37.5% versus >60% for other groups) (Table 1).
 | Table 1 Demographic characteristics by renal function |
Pharmacokinetics
With the exception of Cmax for levomilnacipran in participants with mild renal impairment, the Cmax, AUC0–t, and AUC0–∞ values for both levomilnacipran and N-desethyl levomilnacipran were higher in participants with renal impairment than in healthy subjects (Table 2; Figure 1). In individuals with mild, moderate, and severe renal impairment, levomilnacipran Cmax was 4% lower, and 19% and 44% higher, respectively, and AUC0–∞ was 23%, 93%, and 180% higher, respectively, relative to participants with normal renal function (Table 3). In participants with mild, moderate, and severe renal impairment, N-desethyl levomilnacipran Cmax was 34%, 77%, and 236% higher, respectively, and AUC0–t was 101%, 309%, and 898% higher, respectively, compared with the respective values obtained in participants with normal renal function (Table 3). In addition, the 90% confidence intervals of the geometric mean ratios were outside the upper boundary of 80%–125%. AUC0–∞ of N-desethyl levomilnacipran was not compared between normal and renal impairment groups because the AUC0–∞ in participants with severe renal impairment could not be determined reliably.
 | Table 2 Mean (SD) pharmacokinetic parameters following a single dose of 40 mg levomilnacipran ER by renal function |
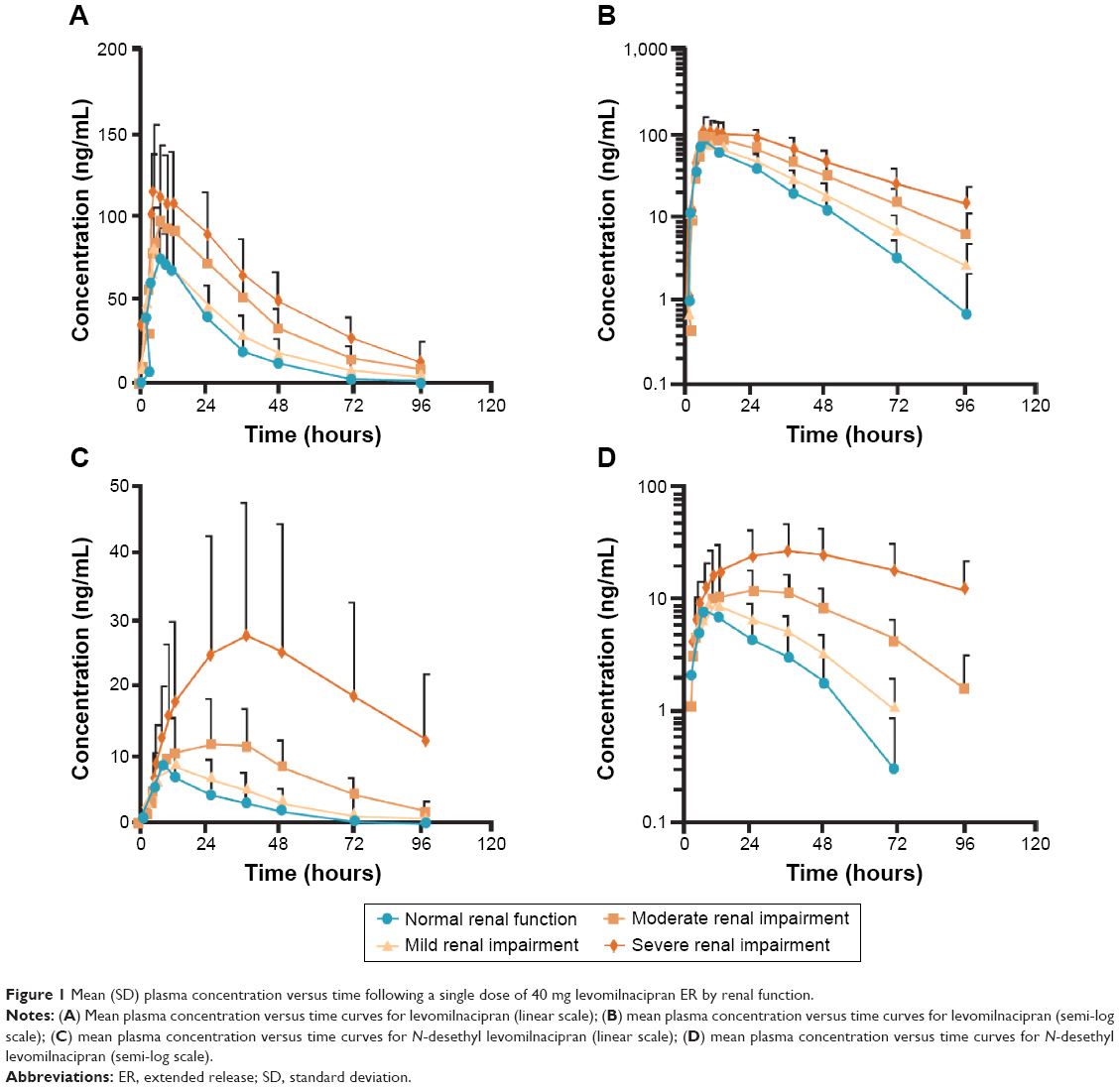 | Figure 1 Mean (SD) plasma concentration versus time following a single dose of 40 mg levomilnacipran ER by renal function. |
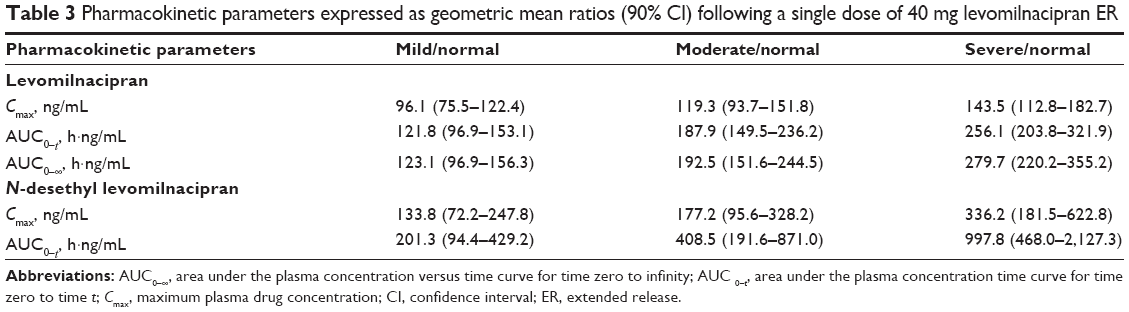 | Table 3 Pharmacokinetic parameters expressed as geometric mean ratios (90% CI) following a single dose of 40 mg levomilnacipran ER |
The T1/2 of levomilnacipran was prolonged as severity of renal impairment increased, averaging 27.7 hours in participants with severe renal impairment compared with 13.5 hours in participants with normal renal function (Table 2). Prolongation of the T1/2 was also found for N-desethyl levomilnacipran in participants with renal impairment, averaging 45.8 hours in participants with severe renal impairment compared with 16.1 hours in participants with normal renal function.
Mean CL/F and CLr of levomilnacipran decreased with increased renal impairment (Table 2). Mean Vss/F and Vz/F were lower in the severe and moderate renal impairment groups than in the other groups (Table 2). Linear correlations were found between CL/F and CLcr (R=0.8043) and between CLr and CLcr (R=0.9152) (Table 4; Figure 2).
 | Table 4 Slope (b) and intercept (a) estimates (± standard error) for regression analyses |
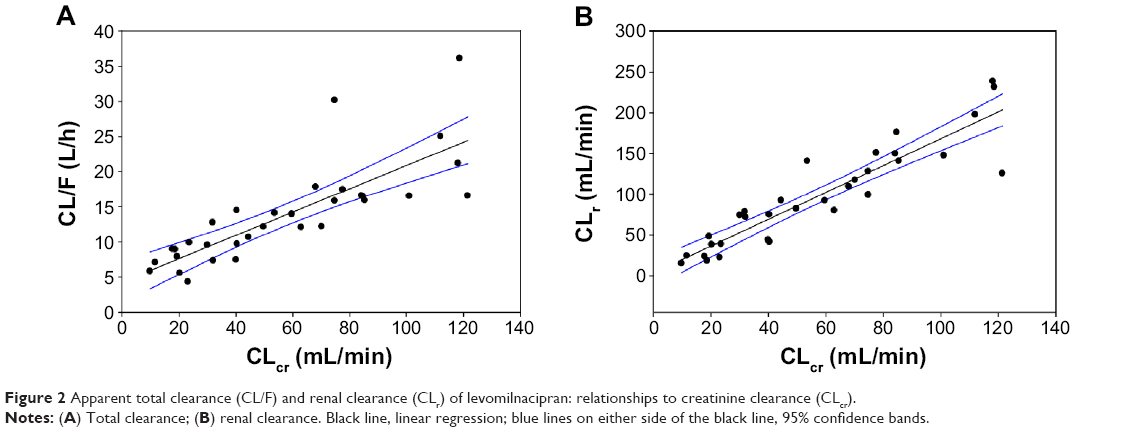 | Figure 2 Apparent total clearance (CL/F) and renal clearance (CLr) of levomilnacipran: relationships to creatinine clearance (CLcr). |
CL/F values derived from the linear regression analysis were 16.7 L/h, 11.7 L/h, and 8.0 L/h for an individual with mild, moderate, or severe renal impairment, respectively (Table 5). On the basis of the change in predicted CL/F for each renally impaired group compared with the normal renal function group, levomilnacipran ER doses of 120 mg, 80 mg, and 40 mg were recommended for the mild, moderate, and severe renal impairment groups, respectively.
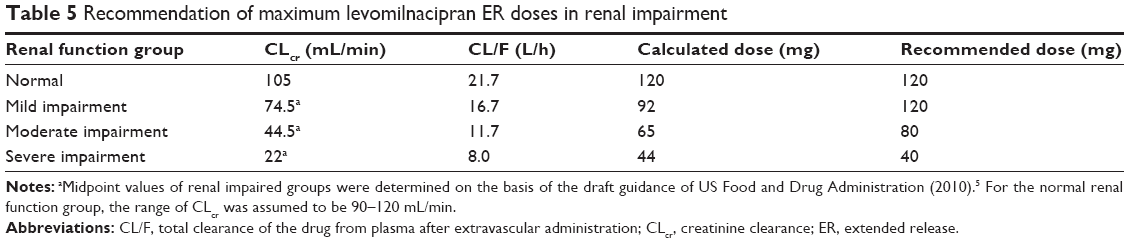 | Table 5 Recommendation of maximum levomilnacipran ER doses in renal impairment |
Safety and tolerability
No deaths, serious AEs, or discontinuations as a result of AEs or otherwise occurred during the study. Overall, 40.6% (13/32) of all participants experienced at least one TEAE. TEAEs that occurred in ≥5% of all study participants were as follows: nausea (15.6%), vomiting (15.6%), headache (12.5%), and diarrhea (9.4%). TEAEs did not occur more frequently in participants with moderate or severe renal impairment than in participants with normal renal function.
Increases in mean heart rate were observed in all groups (range, 7.0 bpm in participants with normal renal function to 10.9 bpm in participants with moderate renal impairment). Increases in mean systolic (3.0 mmHg) and diastolic blood pressure (4.1 mmHg) occurred in the group with moderate renal impairment. During the pharmacokinetic profiling period (time between dosing and end of study), one participant with mild renal impairment had a potentially clinically significant high diastolic blood pressure (ie, ≥105 mmHg and ≥15 mmHg increase from the value at screening) on day 1 at 12 hours postdose. At the end of study, no participant had any potentially clinically significant changes in blood pressure or heart rate.
Except for increases in mean heart rate that were comparable among all groups, there were no notable changes in ECG measurements. No notable changes in mean laboratory values were detected during the study in any groups. No participant exhibited suicidal ideation or behavior before or during the study.
Discussion
The major finding of this study was that exposure to levomilnacipran is increased as a function of the severity of renal impairment. Previous studies have established that levomilnacipran has linear pharmacokinetics and that steady-state exposure is predictable from single-dose data.1 Therefore, the dose adjustment recommendations given in this article (ie, based on this single-dose study) are applicable to patients receiving a multiple-dose regimen of levomilnacipran ER at all therapeutic doses ranging from 40 mg/day to 120 mg/day.
About 50% of the administered levomilnacipran dose was recovered unchanged in the urine from participants with normal renal function in this study (Table 2), which is in agreement with observations from other studies,1 indicating that urinary excretion of levomilnacipran is the primary elimination pathway. Comparing the CLr of levomilnacipran (175.9 mL/min) to the total clearance (CL/F) of levomilnacipran (341.7 mL/min) in participants with normal renal function indicates that 51% elimination of levomilnacipran is due to renal excretion (Table 2).
Only a modest increase (1.2-fold) in the AUC0–∞ value was observed for patients with mild renal impairment compared with individuals with normal renal function. Thus, no dose adjustment is recommended in patients with mild renal impairment. The pharmacokinetic analysis showed that moderate and severe renal impairment resulted in an approximately 2-fold and 3-fold increase in levomilnacipran AUC0–∞, respectively. Levomilnacipran shows greater exposure and longer T1/2 in patients with moderate and severe renal impairment; therefore, dosing modifications of levomilnacipran ER are warranted in these patients. Based on the observed relationship between CL/F and CLcr and considering the overall safety profile of levomilnacipran ER in clinical trials6–10 and the available capsule strengths of 20 mg, 40 mg, 80 mg, and 120 mg, a maximum dose of 80 mg/day and 40 mg/day is recommended for moderate and severe renal impairment, respectively (Table 5). This dose adjustment will result in exposures similar to those seen in individuals with normal renal function.
The increases in Cmax and AUC values of N-desethyl levomilnacipran, the inactive metabolite of levomilnacipran, as a result of renal impairment were greater than those observed for levomilnacipran. Up to a 9-fold increase in AUC0–t of N-desethyl levomilnacipran was observed in patients with severe renal impairment compared with individuals with normal renal function. Because CLr of N-desethyl levomilnacipran is approximately twice that of levomilnacipran, renal function may play a more important role in removing N-desethyl levomilnacipran from the body than it does for levomilnacipran. This metabolite is pharmacologically inactive and its formation in the body is relatively low (~18% of the levomilnacipran dose administered).1 Furthermore, based on preclinical toxicology studies, the plasma concentrations of this metabolite following doses that were found to be safe in rat and monkey were higher than those observed at the recommended therapeutic doses in humans (data on file at Forest).
One demographic difference was noted among the renal groups evaluated in this study. The moderate renal impairment group had a lower percentage of women than the other groups (37.5% versus >60% for the other groups). This imbalance of female study participants among the study groups is not expected to have any impact on levomilnacipran ER dosing recommendations for renal impairment because there are no differences in levomilnacipran pharmacokinetics between males and females.1
Results of a separate, multiple-dose study (data on file at Forest), which compared the pharmacokinetics of levomilnacipran in elderly adults (>65 years old) with those of younger adults (18–45 years old) indicated that age does not have a significant impact on the AUC and Cmax of levomilnacipran ER. Therefore, dose adjustments are not recommended in the elderly, unless renal function is impaired.
Levomilnacipran ER was generally well tolerated by study participants with normal renal function as well as by patients with mild, moderate, and severe renal impairment. AEs were consistent with the known safety profile derived from both the large Phase I program and the Phase II/III placebo-controlled studies.6–10 The presence and severity of renal impairment had no apparent impact on the incidence of AEs.
In summary, the results of this single-dose, open-label parallel-group study in patients with renal impairment show that renal impairment significantly increased exposure to levomilnacipran. Although no dose adjustment of levomilnacipran is needed for patients with mild renal impairment, dosing adjustments are recommended in patients with moderate or severe renal impairment. The recommended maximum maintenance dose of levomilnacipran ER is 80 mg once daily for patients with moderate renal impairment and 40 mg once daily for patients with severe renal impairment.
Acknowledgments
Medical writing and editorial support were provided by Dana L Randall, PharmD, and Joann Hettasch, PhD, of Arbor Communications, Inc., Ann Arbor, MI, USA, and funded by Forest Research Institute, Inc., a subsidiary of Actavis, Inc., Jersey City, NJ, USA.
Funding source
The study was supported by funding from Forest Research Institute, a subsidiary of Actavis, Inc., Jersey City, NJ, USA, and Pierre Fabre Médicament, Boulogne, France. Forest Laboratories was involved in the study design, data collection, data analysis and interpretation, and the decision to publish these results.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
L Chen, A Periclou, and P Ghahramani are employees of Forest Research Institute and may own company shares. WM Greenberg, E Brand-Schieber, and J Wangsa were employees of Forest Research Institute at the time this research was conducted. The authors report no other conflicts of interest in this work.
References
Forest Pharmaceuticals, Inc. FETZIMA™ (levomilnacipran) extended-release capsules [package insert]. St Louis, MO: Forest Pharmaceuticals, Inc.; 2014. | ||
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. | ||
US Food and Drug Administration. Guidance for industry: pharmacokinetics in patients with impaired renal function – study design, data analysis, and impact on dosing and labeling; 1998. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072127.pdf. Accessed August 22, 2013. | ||
Chen L, Boinpally R, Greenberg WM, Wangsa J, Periclou A, Ghahramani P. Effect of hepatic impairment on the pharmacokinetics of levomilnacipran following a single oral dose of a levomilnacipran extended-release capsule in human participants. Clin Drug Investig. 2014;34:351–359. | ||
US Food and Drug Administration. Guidance for industry: pharmacokinetics in patients with impaired renal function – study design, data analysis, and impact on dosing and labeling. Draft guidance; 2010. Available from: http://www.fda.gov/downloads/Drugs/Guidances/UCM204959.pdf. Accessed August 22, 2013. | ||
Asnis G, Bose A, Gommoll C, Chen C, Greenberg WM. The efficacy and safety of levomilnacipran SR 40 mg, 80 mg, or 120 mg in major depressive disorder: a phase III, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74:242–248. | ||
Bakish D, Bose A, Gommoll C, et al. Levomilnacipran ER 40 mg and 80 mg in patients with major depressive disorder: a phase III, randomized, double-blind, fixed-dose, placebo-controlled study. J Psychiatry Neurosci. 2014;39:40–49. | ||
Montgomery S, Mansuy L, Ruth A, Bose A, Li H, Li D. The efficacy and safety of levomilnacipran SR in major depressive disorder: a randomized, double-blind, placebo-controlled, proof-of-concept study. J Clin Psychiatry. 2013;74:363–369. | ||
Sambunaris A, Bose A, Gommoll CP, Chen C, Greenberg WM, Sheehan DV. A phase III, double-blind, placebo-controlled, flexible-dose study of levomilnacipran extended-release in patients with major depressive disorder. J Clin Psychopharmacol. 2014;34:47–56. | ||
Gommoll CP, Greenberg WM, Chen C. A randomized, double-blind, placebo-controlled study of flexible doses of levomilnacipran ER (40–120 mg/day) in patients with major depressive disorder. J Drug Assess. 2014;3:10–19. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.