")
Back to Journals » International Journal of Nanomedicine » Volume 13
Effect of CCR2 inhibitor-loaded lipid micelles on inflammatory cell migration and cardiac function after myocardial infarction
Authors Wang J , Seo MJ, Deci MB, Weil BR, Canty JM, Nguyen J
Received 29 June 2018
Accepted for publication 30 August 2018
Published 15 October 2018 Volume 2018:13 Pages 6441—6451
DOI https://doi.org/10.2147/IJN.S178650
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Jinli Wang,1,2,* Min Jeong Seo,1,* Michael B Deci,1 Brian R Weil,3 John M Canty,3,4 Juliane Nguyen1,2
1Department of Pharmaceutical Sciences, School of Pharmacy, University at Buffalo, The State University of New York, Buffalo, NY, USA; 2Department of Biomedical Engineering, University at Buffalo, The State University of New York, Buffalo, NY, USA; 3Department of Medicine, Department of Physiology and Biophysics, Department of Biomedical Engineering, The Clinical and Translational Research Center, University at Buffalo, Buffalo, NY, USA; 4VA Western New York Healthcare System, Buffalo, NY, USA
*These authors contributed equally to this work
Background: After myocardial infarction (MI), inflammatory cells infiltrate the infarcted heart in response to secreted stimuli. Monocytes are recruited to the infarct via CCR2 chemokine receptors along a CCL2 concentration gradient. While infiltration of injured tissue with monocytes is an important component of the reparatory response, excessive or prolonged inflammation can adversely affect left ventricular remodeling and worsen clinical outcomes.
Materials and methods: Here, we developed poly(ethylene glycol) (PEG)-distearoylphosphatidylethanolamine (PEG-DSPE) micelles loaded with a small molecule CCR2 antagonist to inhibit monocyte recruitment to the infarcted myocardium. To specifically target CCR2-expressing cells, PEG-DSPE micelles were further surface decorated with an anti-CCR2 antibody.
Results: Targeted PEG-DSPE micelles showed eight-fold greater binding to CCR2-expressing RAW 264.7 monocytes than plain, non-targeted PEG-DSPE micelles. In a mouse model of MI, CCR2-targeting PEG-DSPE micelles loaded with a CCR2 small molecule antagonist significantly decreased the number of Ly6Chigh inflammatory cells to 3% of total compared with PBS-treated controls. Furthermore, CCR2-targeting PEG-DSPE micelles significantly reduced the infarct size based on epicardial and endocardial infarct arc lengths.
Conclusion: Both non-targeted and CCR2-targeting PEG-DSPE micelles showed a trend toward improving cardiac function. As such, PEG-DSPE micelles represent a promising cardiac therapeutic platform.
Keywords: CCR2, inflammatory monocytes, micelles, myocardial infarction
Introduction
Ischemic heart disease, including myocardial infarction (MI), accounted for ~10 million deaths in 2016 and is a major cause of morbidity throughout the world.1,2 The contemporary treatment of MI requires rapid coronary reperfusion using percutaneous coronary intervention (PCI). Early reperfusion coupled with chronic medical therapy, including beta blockers, angiotensin inhibition, statins, and antiplatelet therapy have led to significant improvements in survival. Nevertheless, a significant number of those who survive the acute event develop large infarctions and postinfarction left ventricular (LV) remodeling.3 Many others present too late to be considered candidates for reperfusion with acute PCI.4 As a result, a significant number of patients continue to be at high risk of late complications, such as lethal ventricular arrhythmias and congestive heart failure.5 Thus, there is an urgent need for new therapeutics that can modify the course of disease when administered after reperfusion and improve long-term cardiac repair and restore myocardial function.6–9 After MI, there is a dynamic cascade of host inflammatory cells that infiltrate the heart in response to paracrine stimuli secreted by the damaged tissue.10,11 Monocytes are recruited to the infarct via the chemokine receptor CCR2 along a CCL2 concentration gradient. While monocyte infiltration early after MI is important, excessive or prolonged inflammation can adversely affect LV remodeling and impact clinical outcomes.12
Lipid micelles composed of poly(ethylene glycol) (PEG)-distearoylphosphatidylethanolamine (PEG-DSPE) are an attractive class of nano-sized carrier because PEG-DSPE has high biocompatibility and is an US Food and Drug Administration-approved excipient.13 Furthermore, due to their small size, PEG-DSPE micelles can extravasate from leaky vasculature and accumulate in the heart after MI.14,15 By virtue of their hydrophobic core, lipid micelles are ideal vehicles for poorly water-soluble drugs, which often make suboptimal therapeutics. While organic co-solvents and surfactants are frequently used to increase drug solubility, they can increase toxicity and have other undesirable effects.16 Aside from their high biocompatibility and ability to solubilize hydrophobic drugs, PEG-DSPE micelles are attractive delivery vehicles because they are amenable to surface modification with different targeting ligands that could enable multivalent interactions with disease targets and enhance binding specificity and/or affinity.14,17
Here, we assessed if lipid micelles loaded with a poorly water-soluble CCR2 small molecule antagonist and surface decorated with a CCR2-targeting antibody had therapeutic utility in a pre-clinical model of MI. First, we tested the ability of CCR2-targeting lipid micelles to bind to CCR2-expressing macrophages in vitro. Then, we assessed the ability of the CCR2-targeting, loaded lipid micelles to reduce the number of inflammatory cells at the infarct site in a mouse model of MI. Finally, the therapeutic efficacy was tested by measuring cardiac function and infarct size in vivo.
Materials and methods
Reagents
mPEG-DSPE and NHS-PEG-DSPE were obtained from Avanti Polar Lipids (Alabaster, AL, USA). Mouse CCR2 antibody (Clone # 475301) was purchased from R&D Systems, Inc. (Minneapolis, MN, USA) and had a molecular weight of 593.66 Da. Both 1,1-dioctadecyl-3,3,3,3,-tetramethylindodicarbocyanide (DiD) and DAPI were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). The CCR2 antagonist BMS CCR2 22 was purchased from Tocris Bioscience (Bristol, UK). Anti-NK1.1-biotin, anti-CD49b-biotin, anti-Ly6G-biotin, anti-MHC II-biotin, anti-CD90.2-biotin, anti-CD45R (B220)-biotin, anti-F4/80-APC, anti-CD11c-PE, and anti-Ly6C-fluorescein isothiocyanate were purchased from Miltenyi Biotec (Auburn, CA, USA). Anti-CD11b-BV421 and V500 streptavidin beads were purchased from BD Biosciences (San Jose, CA, USA), collagenase II was purchased from Worthington (Lakewood, NJ, USA), and DNase I was from Thermo Fisher Scientific (Waltham, MA, USA). The RAW 264.7 cells were purchased from American Type Culture Collection. All other chemicals and solvents were purchased from Thermo Fisher Scientific unless otherwise stated.
Micelle synthesis
Plain and CCR2-targeting micelles were prepared as follows: mPEG-DSPE/NHS-PEG-DSPE/DiD (98/2/0.5) were dissolved in chloroform and mixed with the CCR2 antagonist dissolved in methanol.14,18,19 The small molecule antagonist CCR2 was loaded into the lipid micelles at 1:22 molar ratio, which corresponds to a loading of 4.5% w/w. The solvent was removed by rotary evaporation to yield a lipid film. The dried film was rehydrated with PBS. The micelles contain the far-red fluorescent, lipophilic DiD, which incorporates into the lipophilic region of the micelles upon rehydration. The mPEG-DSPE film and the mPEG-DSPE/NHS-PEG-DSPE mixture were then sonicated for 5 minutes at room temperature. The mPEG-DSPE/NHS-PEG-DSPE mixture was incubated for 4 hours with an anti-CCR2 antibody for conjugation to the NHS-PEG-DSPE lipid. The PEG-DSPE micelles contained 0.3 mol% of anti-CCR2 antibody. The particle size and zeta potential of the plain and CCR2-targeted micelles were measured by dynamic light scattering (DLS) using the Nanobrook Omni (Brookhaven Instruments, Holtsville, NY, USA).
Binding of CCR2-antibody conjugated micelles to CCR2-expressing cells
The binding and internalization of DiD-labeled CCR2-targeting and plain micelles to RAW 264.7 cells were measured by flow cytometry. A total of 106 cells were incubated with the micelles for 1 hour at 4°C and washed three times before measurement. MACSQuant from Miltenyi Biotec was used for flow cytometry at ex/em wavelengths of 644/663 nm. Data were analyzed with FCS Express 5 (De Novo Software, Glendale, CA, USA). All experiments were performed in triplicate, and data are represented as mean ± SD.
MTT assay
MTT assay was performed to determine the cytotoxicity of the non-targeted and CCR2-targeting micelles loaded with the CCR2 small molecule antagonist. RAW 264.7 cells were plated into 96-well plates at a density of 8,000 cells/well. RAW 264.7 cells were incubated with increasing micelle concentrations for 24 hours. The media was replaced and the MTS substrate (Sigma Aldrich, St Louis, MO, USA) was added to the wells and incubated for 2 hours at 37°C. The formazan dye produced by the viable cells was measured using a SpectraMax i3 plate reader at 490 nm.
In vitro migration assays
Transwell migration assays were performed using a chemotaxis cell migration assay with a membrane pore size of 5 μm from MilliporeSigma (Burlington, MA, USA). RAW 264.7 cells were serum starved for 18–24 hours prior to assaying and then plated onto the transwells. CCL2, a chemokine ligand of the CCR2 receptor, was loaded into the basolateral chamber. The cells were incubated for 4 hours at 37°C in a humidified 5% CO2 incubator. Cells were pretreated with the CCR2 antagonist for 30 minutes. Cells that migrated to the feeder tray were lysed with lysis buffer and fluorescently labeled with CyQuant GR Dye for quantification. A plate reader was used to measure fluorescence at ex/em wavelengths of 480/520 nm. Measurements were performed in triplicate, and data are represented as mean ± SD.
Mouse model of MI
Male C57BL/6 mice were used (10–12 weeks) for the in vivo studies. All animal procedures were approved by the University at Buffalo – State University of New York Institutional Animal Care and Use Committee. All methods and experiments were performed in accordance with the US National Institute of Health Guide for Care and Use of Laboratory Animals. Mice were anesthetized with isoflurane via inhalation (1%–3%). The body temperature was maintained with a heating pad. A small incision under the mandible was made to visualize the trachea. Mice were intubated with a 20 gauge blunt needle and connected to a ventilator (Harvard Rodent Ventilator; Harvard Apparatus, Cambridge, MA, USA). A left lateral thoracotomy was made by use of a micro-dissecting microscope (Leica Biosystems, Wetzlar, Germany) to expose the heart. The left anterior descending (LAD) artery was identified and occluded permanently with an 8–0 nylon suture. Successful LAD ligation was confirmed by blanching and LV dysfunction. The thorax was closed in layers (ribs, muscles, and skin). Analgesia (buprenorphine and carprofen) was provided according to the protocol, and mice were monitored carefully. In parallel subgroups, the mice had a similar surgical procedure without ligating the suture around the LAD (sham group). The survival rate after MI was 84%, with no significant differences between treatment groups.
Analysis of Ly-6Chigh monocyte accumulation in the infarcted heart and spleen of C57BL/6 mice by flow cytometry – Group I
To determine the accumulation of inflammatory Ly-6Chigh monocytes in the infarcted heart and spleen (Group I), animals were treated with plain and CCR2-targeting micelles 48 and 72 hours after MI. Mice subjected to permanent LAD occlusion were randomized to receive PBS (n=5), plain micelles (33 mg/kg, n=5), or CCR2-targeting micelles (33 mg/kg, n=5) via tail vein injections 48 hours and 72 hours after inducing MI. Both micelles were loaded with the CCR2 small molecule antagonist at a dose of 1.5 mg/kg. A total of 1.5 mg/kg of CCR2 small antagonist in 33 mg/kg micelles was administered because from the three doses tested (1 mg/kg, 1.5 mg/kg, and 2 mg/kg of CCR2), it proved to be the maximum tolerated dose without causing fur ruffling in mice. Twenty-four hours after the final injection (96 hours post-infarction), animals were euthanized and hearts and spleens were isolated and digested to quantify inflammatory monocytes by flow cytometry. Spleens were removed, rinsed in Hanks’ balanced salt solution (HBSS) at 4°C, digested, and filtered through a 40 micron cell strainer. Hearts were removed, rinsed in HBSS, minced with fine scissors, and placed into a cocktail of 600 U/mL collagenase I and 60 U/mL DNase I for 1 hour at 37°C with 400 rpm shaking. Single-cell suspensions were filtered through a 40 micron cell strainer and counted. Monocytes were enriched by CD11b microbeads using MACS LS columns (Miltenyi). Cell suspensions were incubated with a cocktail of monoclonal antibodies for 1 hour at 4°C. For the last 15 minutes of incubation, V500 streptavidin beads were added. A total of 15,000 cells were counted for spleen samples and 10,000 cells for heart samples. Monocytes were identified as CD11bhi (CD90/B220/CD49b/NK1.1/Ly-6G/I-Ab)lo (F4/80b/CD11c)lo Ly-6Chi. Reported cell percentages were calculated as the percent of live cells within the monocyte gate. Data were acquired on a MACSQuant Analyzer 10 (Miltenyi) and analyzed with FCS Express 5 (De Novo Software).
Micelle biodistribution – Group II
To assess micelle biodistribution (Group II), C57BL/6 mice were administered plain micelles (33 mg/kg, n=3) and CCR2-targeting micelles (33 mg/kg, n=3) via tail vein injection 48 hours after inducing MI (Figure S1). Mice were sacrificed 4 hours after injection, and organs (lung, spleen, heart, liver, and kidneys) were collected for in vivo imaging using the IVIS Spectrum system (PerkinElmer, Waltham, MA, USA) (n=3). Whole blood was centrifuged at 2,000 rpm for 10 minutes to obtain serum. The percentage of the injected dose was determined using a standard curve.
Measurement of cardiac function and infarct size 12 days after inducing MI – Group III
To assess longer term functional effects of two boluses of the CCR2 antagonist-loaded micelles, cardiac function was assessed by echocardiography and infarct size determined by Masson trichrome staining. Mice subjected to permanent LAD occlusion were randomized to receive PBS, plain micelles (33 mg/kg), or CCR2-targeting micelles (33 mg/kg) via tail vein injections 48 and 72 hours after inducing MI. Both micelles were loaded with the CCR2 small molecule antagonist at a dose of 1.5 mg/kg. Cardiac function and infarct size were determined 10 days after the final injection (sham [n=6], PBS treated [n=6], plain micelle groups [n=6], and CCR2-targeting micelle groups [n=4]). Under isoflurane anesthesia (1%–3%), echocardiograms were performed with a Vivid 7 system (GE Healthcare, Little Chalfont, UK) equipped with an i13L probe. Two-dimensional mode parasternal long- and short-axis views were recorded. The LV dimensions and wall thicknesses were determined from the M-mode images at the mid-papillary muscle level. LV stroke volume was calculated as the difference between the end diastolic volume (EDV) and end systolic volume (ESV). LV ejection fraction (EF) was calculated as ([EDV-ESV]/EDV)*100. All measurements were performed with investigators blinded to experimental groups.
Mice were subsequently euthanized under anesthesia using potassium chloride injected into the left ventricle to arrest the heart in diastole. Heart samples were embedded in Tissue-Tek® O.C.T.™ compound and sliced transversely from the apex to base at intervals of 900 μm between each section. Heart sections (10 μm) were fixed with 10% formalin solution and stained with the Masson Trichrome Stain Kit (Richard-Allan Scientific, Thermo Fisher Scientific). Whole heart imaging was performed using a Zeiss AxioImager Microscope. The infarct size was measured with ImageJ and calculated as infarct size = (sum of infarct areas from all sections)/(sum of left ventricle areas from all sections) × 100. The infarct size based on length measurement was quantified as published by Takagawa et al.20 All measurements were performed with investigators blinded to treatment groups.
Results
Physicochemical characterization of CCR2-loaded plain and targeted PEG-DSPE micelles
Schematics of plain and CCR2-targeting PEG-DSPE micelles encapsulating the small molecule CCR2 antagonist in their hydrophobic core are shown in Figure 1. The CCR2-targeting PEG-DSPE micelles were surface modified to carry an anti-CCR2 antibody to bind inflammatory CCR2-positive cells. Plain PEG-DSPE micelles were 21.1±4.7 nm in hydrodynamic diameter as measured by DLS. CCR2-targeting PEG-DSPE micelles were slightly larger in size (34.7±14 nm) due to the addition of the CCR2-antibody. The hydrodynamic diameter of the antibody alone was 11.2±4.7 nm with a zeta potential of −1.5±1.0 mV. The zeta potential of plain micelles was approximately −9.2±1.0 mV and for CCR2-targeting micelles −10.9±1.2 mV.
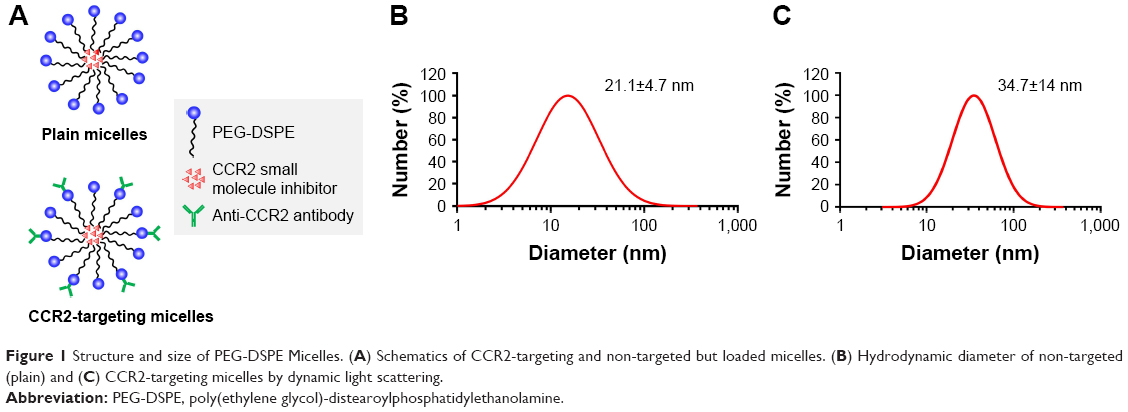 | Figure 1 Structure and size of PEG-DSPE Micelles. (A) Schematics of CCR2-targeting and non-targeted but loaded micelles. (B) Hydrodynamic diameter of non-targeted (plain) and (C) CCR2-targeting micelles by dynamic light scattering. |
CCR2 small molecule antagonist inhibit macrophage migration
The CCR2 inhibitory effect of the small molecule antagonist was first assessed in a transwell cellular migration assay. Monocytes and macrophages are known to express CCR2 and migrate toward high local concentrations of cytokines, such as CCL2.21 RAW 267.4 cells were incubated with increasing concentrations of the CCR2 small molecule antagonist (Figure 2A). The CCR2 small molecule antagonist (Figure 2B) inhibited macrophage migration with an IC50 of 8 nM.
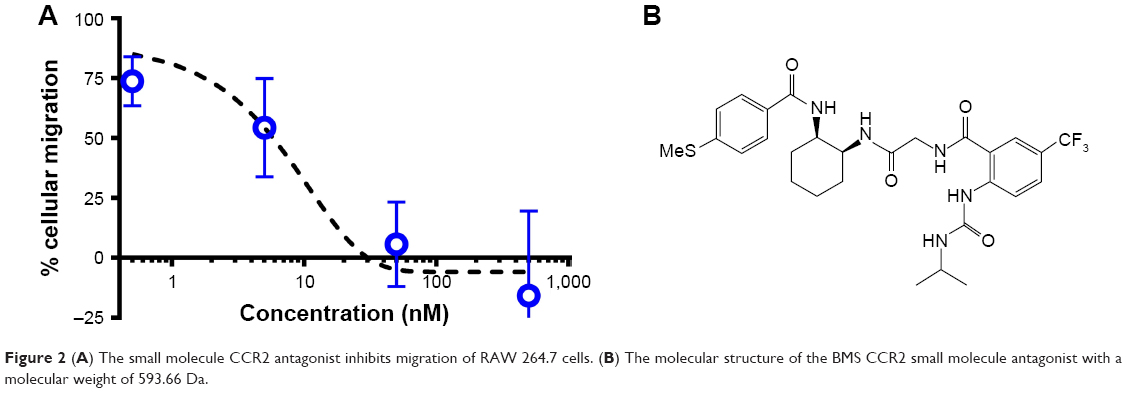 | Figure 2 (A) The small molecule CCR2 antagonist inhibits migration of RAW 264.7 cells. (B) The molecular structure of the BMS CCR2 small molecule antagonist with a molecular weight of 593.66 Da. |
Binding to CCR2-expressing RAW 264.7 cells
We next assessed if lipid micelles decorated with a CCR2-targeting antibody bound to CCR2-expressing RAW 264.7 macrophages. RAW 264.7 CCR2 overexpressing cells were incubated with plain and CCR2-targeting micelles encapsulating the CCR2 inhibitor. CCR2-targeting micelles showed significantly greater binding to RAW 264.7 cells than plain micelles. RAW 264.7 cells incubated with antibody-coated targeting micelles showed an eight-fold increase in peak MFI (6483) compared with the plain, non-targeted micelles (821 MFI) (Figure 3A and B). Enhanced binding of CCR2-targeting micelles to RAW 264.7 cells compared with non-targeted micelles were further confirmed by fluorescence microscopy (Figure 3C and D). RAW 264.7 cells showed high binding of CCR2-targeting micelles, visualized in red, while binding of non-targeted micelles to RAW 264.7 cells was minimal. Both, non-targeted and CCR2-targeting micelles loaded with the CCR2 small molecule antagonist displayed high biocompatibility. Even at very high concentrations (1,000 nM), no toxic effects were observed (Figure 3B).
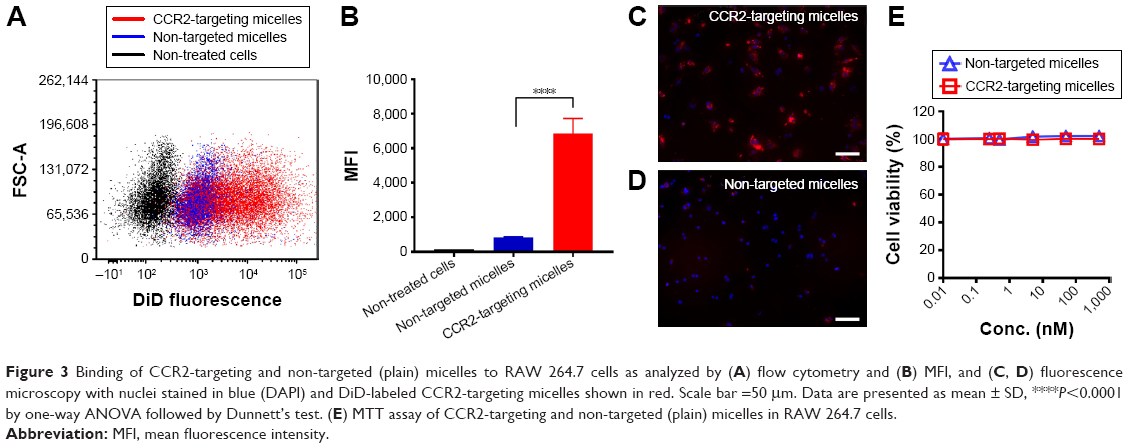 | Figure 3 Binding of CCR2-targeting and non-targeted (plain) micelles to RAW 264.7 cells as analyzed by (A) flow cytometry and (B) MFI, and (C, D) fluorescence microscopy with nuclei stained in blue (DAPI) and DiD-labeled CCR2-targeting micelles shown in red. Scale bar =50 μm. Data are presented as mean ± SD, ****P<0.0001 by one-way ANOVA followed by Dunnett’s test. (E) MTT assay of CCR2-targeting and non-targeted (plain) micelles in RAW 264.7 cells. |
Targeted CCR2-loaded micelles decrease Ly-6Chigh monocyte accumulation in the infarcted heart and spleen
Next, we examined whether CCR2-targeting of loaded lipid micelles inhibited migration of inflammatory monocytes to the heart after MI. The small antagonist CCR2 was loaded into the lipid micelles at 1:22 ratio, which corresponds to a loading of 4.5% w/w. Myocardial infarcts were induced in mice followed by intravenous injection of plain and CCR2-targeting lipid micelles at post-operative days 2 and 3. Both micelle formulations contained the CCR2 small molecule antagonist. The splenic reservoir has recently been reported to be the main contributor of Ly-6Chigh inflammatory monocytes, which are recruited to heart tissue post-MI mediated by CCR2 to give rise to classical macrophages and promote inflammatory disease progression.22 Thus, we hypothesized that therapeutic treatment with a CCR2 small molecule antagonist would decrease the number of infiltrating, inflammatory monocytes. As shown in Figure 4, systemic administration of non-targeted and CCR-targeting micelles reduced the number of monocytes identified as CD11bhigh (CD90/B220/CD49b/NK1.1/Ly-6G/I-Ab)low (F4/80b/CD11c)low Ly-6Chigh. The number of inflammatory Ly-6Chigh monocytes decreased from 10.2±5.7% in PBS-treated animals to 7.0±1.6% in non-targeted and 3.2±2.2% in CCR2-targeting micelles in the heart. In the spleen, these fell from 11.2±2.8% in PBS-treated animals to 6.9±2.9% in non-targeted and 3.2±2.2% in CCR2-targeting micelles. Considering that the CCR2-targeting and plain micelles showed similar deposition in the heart (8.1±1.2% vs 6.3±1.2% ID) and spleen (2.7±2.1% vs 2.6±2% ID) as shown in Figure S1, the ability of the CCR2-targeting micelles to mediate significant reductions of the cardiac and splenic inflammatory Ly-6Chigh population is most likely the result of their ability to specifically target CCR2-positive cells (Figure 3). Furthermore, based on our immunohistochemical analyses, CCR2-targeting micelles showed significantly higher colocalization with CCR2-positive cells in the infarcted myocardium than non-targeted micelles (P=0.0004, Figure S2).
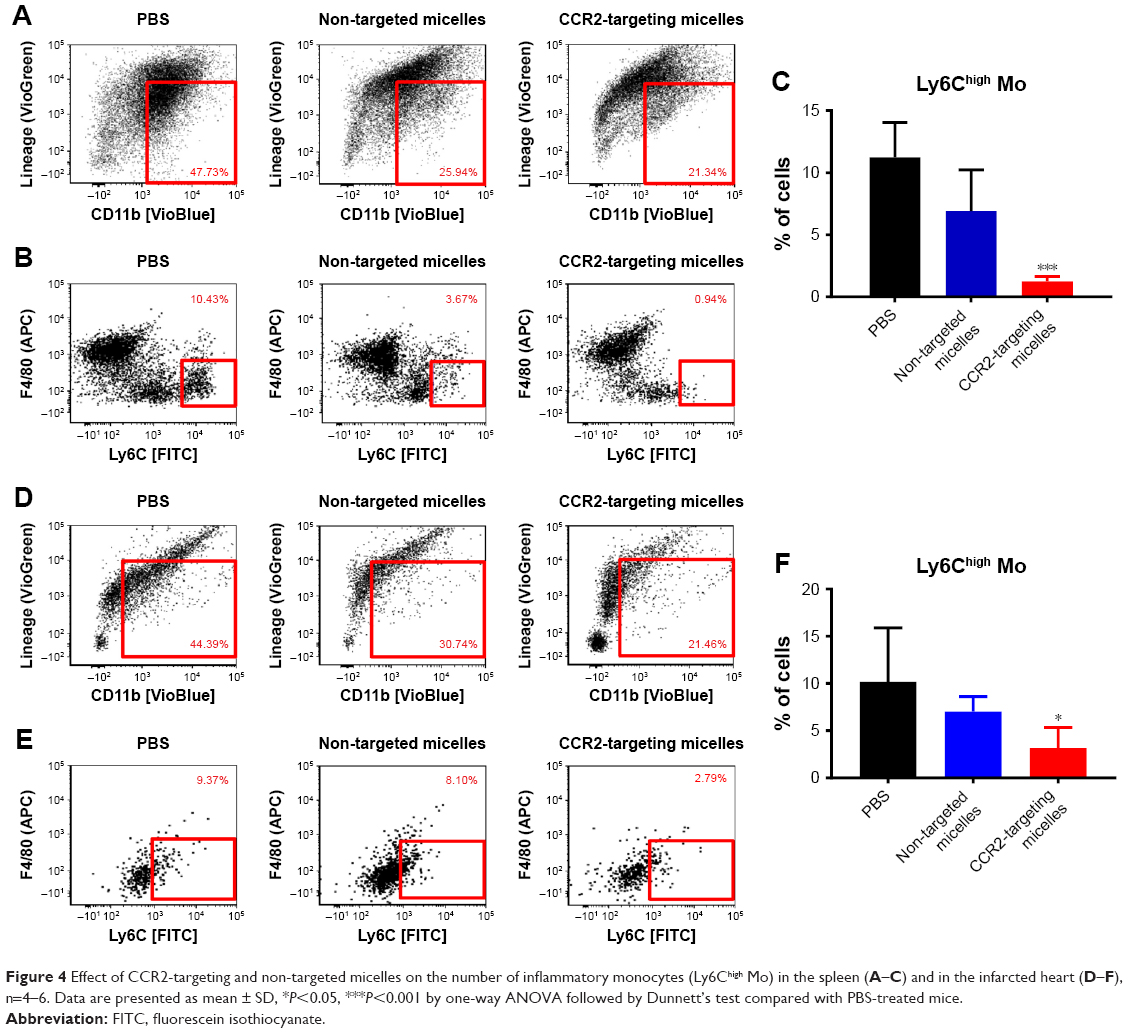 | Figure 4 Effect of CCR2-targeting and non-targeted micelles on the number of inflammatory monocytes (Ly6Chigh Mo) in the spleen (A–C) and in the infarcted heart (D–F), n=4–6. Data are presented as mean ± SD, *P<0.05, ***P<0.001 by one-way ANOVA followed by Dunnett’s test compared with PBS-treated mice. |
Effect of CCR2-loaded micelles on cardiac function and infarct size
To assess the therapeutic efficacy of targeted and plain CCR2-loaded micelles, we determined the infarct size and cardiac function of mice with induced MI in a pilot study (Figure 5). Masson trichrome staining was performed on heart sections 12 days after inducing MI to assess the infarct size. Sequential heart sections from the apex to base are shown (Figure 5D). Using this method, fibrotic tissue appears blue–violet. Infarct areas were determined based on epicardial and endocardial infarct arc lengths20 and area measurements expressed as infarct area/LV area (%) (Figure 5B and C).20 When the length measurement was used for analysis, the CCR2-targeting micelles significantly decreased the infarct size from 34.9±5.6% in PBS controls vs 28.0±8.2% in non-targeted micelles and 22.4±6.9% in CCR2-targeting micelles (Figure 5B). The infarct area of mice treated with non-targeted micelles (18.0±3.6%) and CCR2-targeting micelles (17.2±4.6%) tended to decrease vs PBS controls (21.6±4.9%) but did not reach statistical significance (Figure 5C). Likewise, EF in PBS-treated mice averaged 37.3±3.1% while it demonstrated a trend to be higher after treatment with plain (47.5±5.8%) and CCR2-targeting loaded micelles (44.2±7.3%) (Figure 5E). Similar changes were seen for fractional shortening (Figure 5E).
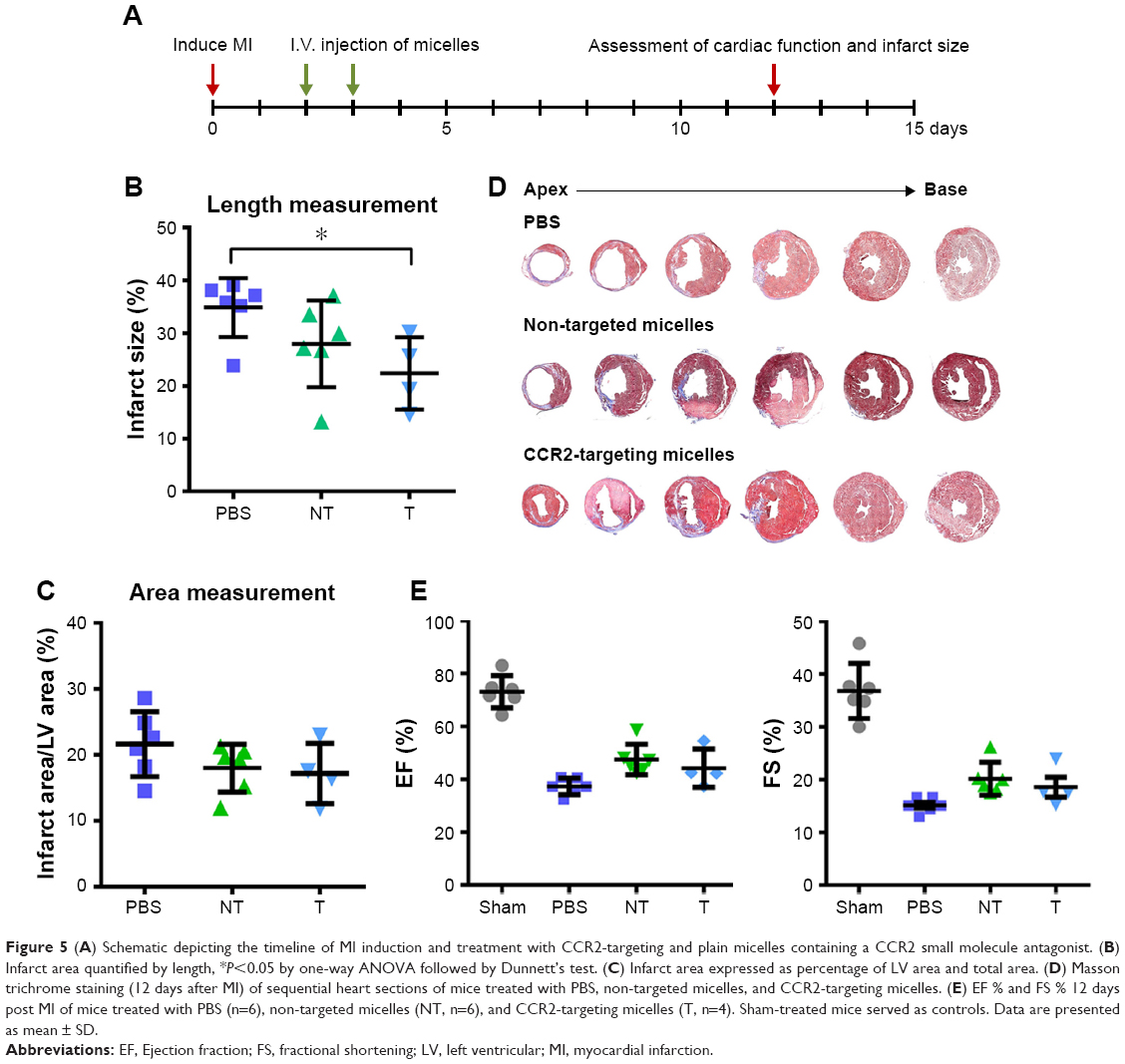 | Figure 5 (A) Schematic depicting the timeline of MI induction and treatment with CCR2-targeting and plain micelles containing a CCR2 small molecule antagonist. (B) Infarct area quantified by length, *P<0.05 by one-way ANOVA followed by Dunnett’s test. (C) Infarct area expressed as percentage of LV area and total area. (D) Masson trichrome staining (12 days after MI) of sequential heart sections of mice treated with PBS, non-targeted micelles, and CCR2-targeting micelles. (E) EF % and FS % 12 days post MI of mice treated with PBS (n=6), non-targeted micelles (NT, n=6), and CCR2-targeting micelles (T, n=4). Sham-treated mice served as controls. Data are presented as mean ± SD. |
Discussion
Inflammatory responses are an important factor in tissue repair and cardiac remodeling after MI. Inflammatory monocytes can negatively affect healing during MI, decrease cardiac function, and increase infarct size. For example, CCR2 knockout mice showed significantly improved healing after MI compared with wild-type mice,23 at least, in part, due to the lack of CCR2 expression preventing monocyte migration to the infarct and attenuation of LV remodeling after MI. CCR2 knockout mice showed a decrease in collagen deposition in the infarcted area, which could ultimately contribute to fibrosis. Similar effects were observed when lipid nanoparticles containing anti-CCR2 siRNA were administered to mice 1 hour after injury in a mouse model of reperfused MI.24 While early reperfusion is the optimal treatment for acute MI, many patients present after the critical 6 hours time frame to impact myocardial infarct size.4 In fact, less than half of all patients are treated within 12 hours after the onset of infarction.25 This underscores the need for adjunctive therapies that can ameliorate the decline in LV function when administered at later time points.
The overall objective of this study was to assess the effects of PEG-DSPE lipid micelles loaded with a CCR2 small molecule antagonist on inflammatory cell migration and cardiac function. To address the need for therapies that can be given independently of reperfusion and that are less time-sensitive, the CCR2-targeting and plain micelles were administered 2 days after MI. A secondary objective was to evaluate if decorating the PEG-DSPE micelles with a CCR2-targeting ligand could further improve therapeutic effects. For this purpose, we engineered PEG-DSPE lipid micelles to deliver a CCR2 small molecule antagonist to inflammatory monocytes infiltrating the heart after MI.
The CCR2-targeting PEG-DSPE lipid micelles loaded with a CCR2 small molecule antagonist showed significantly greater binding to CCR2-positive macrophages than plain CCR2-loaded micelles (Figure 3). The CCR2-targeting PEG-DSPE lipid micelles also effectively decreased inflammatory monocyte infiltration into the infarcted area of the heart and significantly reduced the number of splenic inflammatory monocytes (Figure 4). Smaller, non-significant reductions in inflammatory monocytes in the spleen and heart were also observed with plain, non-targeted micelles. The ability of the CCR2-targeting PEG-DSPE lipid micelles to more efficiently reduce the number of inflammatory monocytes in both the heart and the spleen is likely due to their ability to specifically bind to CCR2-expressing cells.
There are several limitations of our study. First, our in vivo pilot studies were focused on identifying the acute effects of modulating the inflammatory response with delayed administration of anti-inflammatory micelle therapy administered 48 hours after infarction. While we demonstrated very effective in vitro and in vivo inhibition of inflammatory monocytes by CCR2-targeting PEG-DSPE lipid micelles, this translated into only modest improvements in cardiac function and reductions in infarct size in vivo (Figure 5). This was likely a reflection of the short-time interval after treatment to assessing the pathology and functional parameters where LV remodeling is not yet fully established. In addition to the timing of initial therapy and the duration of the therapy could also impact the physiological effects in vivo. In light of the effective in vivo reductions in pro-inflammatory monocytes in the heart and spleen, more chronic studies with longer term and repeated administration of targeted micelles will be required to assess the impact on postinfarction remodeling.
Conclusion
PEG-DSPE micelles present a promising platform for cardiac therapeutics. CCR2-targeting PEG-DSPE micelles can significantly inhibit the number of inflammatory monocytes in the heart when administered intravenously. Although quantitative measurements did not reach statistical significance, the improvements in cardiac function and infarct size are encouraging and provide the basis for further improving CCR2-based small molecule therapeutics. No deleterious effects of our initial in vivo studies were seen but additional in vivo studies are needed to optimize the timing and duration of CCR2 dosing to achieve optimal improvement in cardiac function under in vivo conditions.
Acknowledgments
We would like to thank Dr Joseph Spernyak (Roswell Park Cancer Institute) for technical assistance in the use of the IVIS Spectrum in vivo imager and funding by the NIH (S10 OD 016450). We acknowledge support by the NIH through awards HL-126082 (JN&JMC), EB-021454 (JN), EB-023262 (JN), HL-61610 (JMC), the National Center for Advancing Translational Sciences UL1-TR-001412 and the Department of Veterans Affairs 1IO1BX002659 (JMC).
Disclosure
The authors report no conflicts of interest in this work.
References
Finegold JA, Asaria P, Francis DP. Mortality from ischaemic heart disease by country, region, and age: statistics from World Health Organisation and United Nations. Int J Cardiol. 2013;168(2):934–945. | ||
Naghavi M, Abajobir AA, Abbafati C. GBD 2016 Causes of Death Collaborators. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1151–1210. | ||
Reddy K, Khaliq A, Henning RJ. Recent advances in the diagnosis and treatment of acute myocardial infarction. World J Cardiol. 2015;7(5):243–276. | ||
Sheifer SE, Rathore SS, Gersh BJ, et al. Time to presentation with acute myocardial infarction in the elderly: associations with race, sex, and socioeconomic characteristics. Circulation. 2000;102(14):1651–1656. | ||
Gerczuk PZ, Kloner RA. An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Coll Cardiol. 2012;59(11):969–978. | ||
Ferguson SW, Wang J, Lee CJ, et al. The microRNA regulatory landscape of MSC-derived exosomes: a systems view. Sci Rep. 2018;8(1):1419. | ||
Lo CY, Weil BR, Palka BA, Momeni A, Canty JM, Neelamegham S. Cell surface glycoengineering improves selectin-mediated adhesion of mesenchymal stem cells (MSCs) and cardiosphere-derived cells (CDCs): Pilot validation in porcine ischemia-reperfusion model. Biomaterials. 2016;74:19–30. | ||
Weil BR, Canty JM. Stem cell stimulation of endogenous myocyte regeneration. Clin Sci. 2013;125(3):109–119. | ||
Weil BR, Suzuki G, Leiker MM, Fallavollita JA, Canty JM. Comparative efficacy of intracoronary allogeneic mesenchymal stem cells and cardiosphere-derived cells in Swine with hibernating myocardium. Circ Res. 2015;117(7):634–644. | ||
Ruparelia N, Godec J, Lee R, et al. Acute myocardial infarction activates distinct inflammation and proliferation pathways in circulating monocytes, prior to recruitment, and identified through conserved transcriptional responses in mice and humans. Eur Heart J. 2015;36(29):1923–1934. | ||
Sager HB, Kessler T, Schunkert H. Monocytes and macrophages in cardiac injury and repair. J Thorac Dis. 2017;9(Suppl 1):S30–S35. | ||
França CN, Izar MCO, Hortêncio MNS, et al. Monocyte subtypes and the CCR2 chemokine receptor in cardiovascular disease. Clin Sci. 2017;131(12):1215–1224. | ||
Gill KK, Kaddoumi A, Nazzal S. PEG-lipid micelles as drug carriers: physiochemical attributes, formulation principles and biological implication. J Drug Target. 2015;23(3):222–231. | ||
Nguyen J, Sievers R, Motion JP, Kivimäe S, Fang Q, Lee RJ. Delivery of lipid micelles into infarcted myocardium using a lipid-linked matrix metalloproteinase targeting peptide. Mol Pharm. 2015;12(4):1150–1157. | ||
Paulis LE, Geelen T, Kuhlmann MT, et al. Distribution of lipid-based nanoparticles to infarcted myocardium with potential application for MRI-monitored drug delivery. J Control Release. 2012;162(2):276–285. | ||
Ma P, Mumper RJ. Paclitaxel Nano-Delivery Systems: A Comprehensive Review. J Nanomed Nanotechnol. 2013;4(2):1000164. | ||
Deci MB, Liu M, Dinh QT, Nguyen J. Precision engineering of targeted nanocarriers. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2018:e1511. | ||
Nguyen J, Cooke JRN, Ellis JA, et al. Cationizable lipid micelles as vehicles for intraarterial glioma treatment. J Neurooncol. 2016;128(1):21–28. | ||
Nguyen J, Hossain SS, Cooke JRN, et al. Flow arrest intra-arterial delivery of small TAT-decorated and neutral micelles to gliomas. J Neurooncol. 2017;133(1):77–85. | ||
Takagawa J, Zhang Y, Wong ML, et al. Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area- and length-based approaches. J Appl Physiol. 2007;102(6):2104–2111. | ||
Deci MB, Ferguson SW, Scatigno SL, Nguyen J. Modulating Macrophage Polarization through CCR2 Inhibition and Multivalent Engagement. Mol Pharm. 2018;15(7):2721–2731. | ||
Swirski FK, Nahrendorf M, Etzrodt M, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325(5940):612–616. | ||
Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am J Pathol. 2004;165(2):439–447. | ||
Leuschner F, Dutta P, Gorbatov R, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29(11):1005–1010. | ||
Schömig A, Ndrepepa G, Kastrati A. Late myocardial salvage: time to recognize its reality in the reperfusion therapy of acute myocardial infarction. Eur Heart J. 2006;27(16):1900–1907. |
Supplementary materials
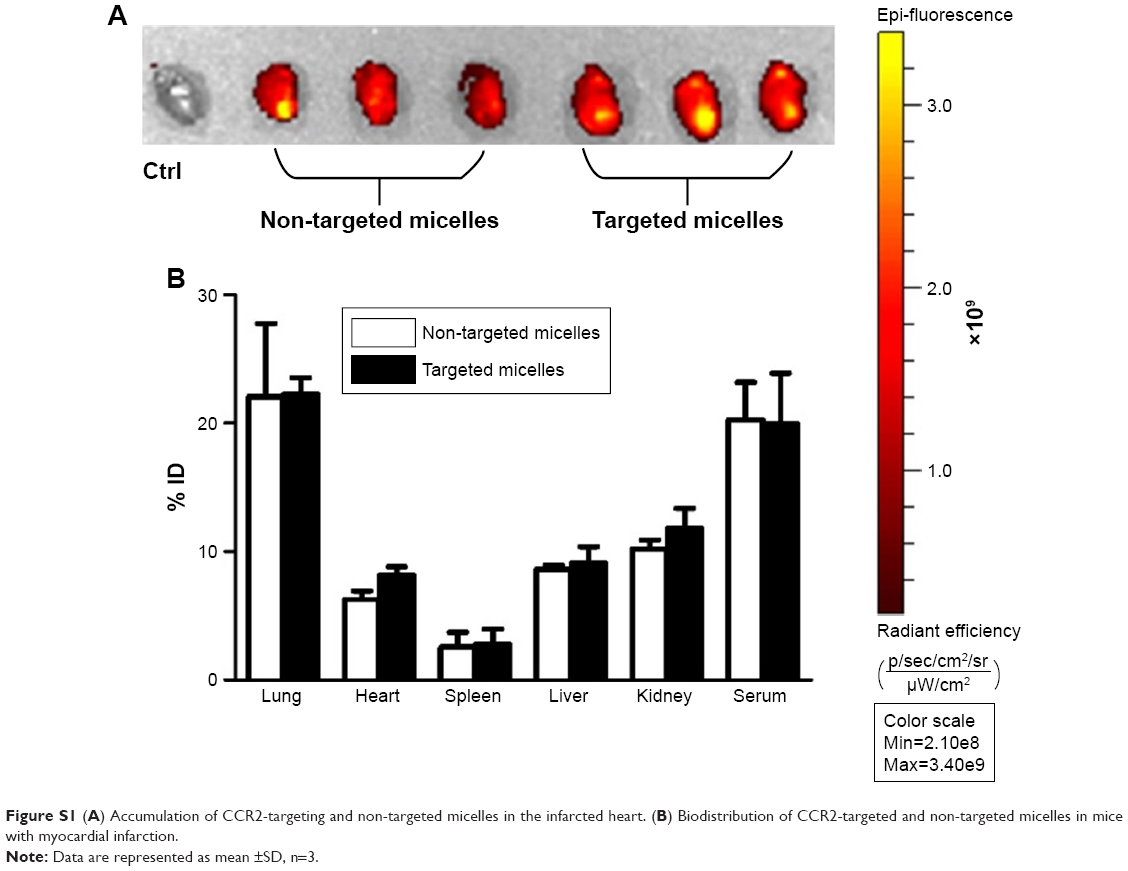 | Figure S1 (A) Accumulation of CCR2-targeting and non-targeted micelles in the infarcted heart. (B) Biodistribution of CCR2-targeted and non-targeted micelles in mice with myocardial infarction. |
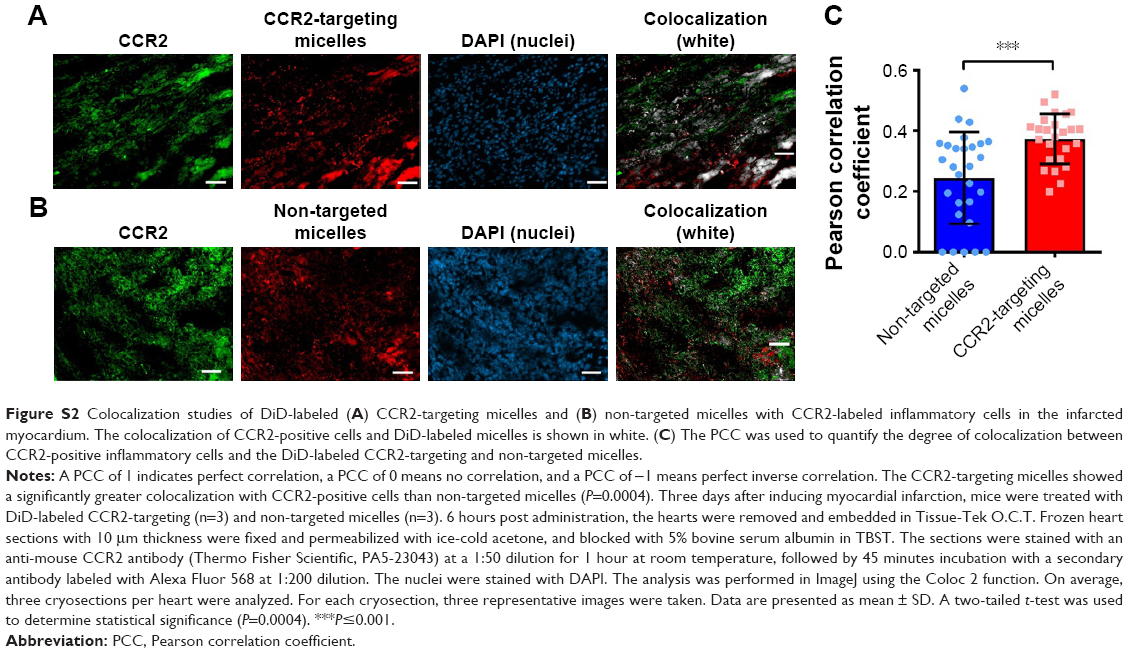 | Figure S2 Colocalization studies of DiD-labeled (A) CCR2-targeting micelles and (B) non-targeted micelles with CCR2-labeled inflammatory cells in the infarcted myocardium. The colocalization of CCR2-positive cells and DiD-labeled micelles is shown in white. (C) The PCC was used to quantify the degree of colocalization between CCR2-positive inflammatory cells and the DiD-labeled CCR2-targeting and non-targeted micelles. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.