")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Dose-dependent effects of atorvastatin on myocardial infarction
Authors Barbarash O, Gruzdeva O, Uchasova E , Belik E , Dyleva Y , Karetnikova V
Received 9 April 2015
Accepted for publication 12 May 2015
Published 29 June 2015 Volume 2015:9 Pages 3361—3368
DOI https://doi.org/10.2147/DDDT.S86344
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Olga Barbarash, Olga Gruzdeva, Evgenya Uchasova, Ekaterina Belik, Yulia Dyleva, Victoria Karetnikova
Federal State Budgetary Institution, Research Institute for Complex Issues of Cardiovascular Diseases, Kemerovo, the Russian Federation
Background: Dyslipidemia is a key factor determining the development of both myocardial infarction (MI) and its subsequent complications. Dyslipidemia is associated with endothelial dysfunction, activation of inflammation, thrombogenesis, and formation of insulin resistance. Statin therapy is thought to be effective for primary and secondary prevention of complications associated with atherosclerosis.
Methods: This study examined 210 patients with Segment elevated MI (ST elevated MI) who were treated with atorvastatin from the first 24 hours after MI. Group 1 (n=110) were given atorvastatin 20 mg/day. Group 2 (n=100) were given atorvastatin 40 mg/day. At days 1 and 12 after MI onset, insulin resistance levels determined by the homeostasis model assessment of insulin resistance index, lipid profiles, and serum glucose, insulin, adipokine, and ghrelin levels were measured.
Results: Free fatty acid levels showed a sharp increase during the acute phase of MI. Treatment with atorvastatin 20 mg/day, and especially with 40 mg/day, resulted in a decrease in free fatty acid levels. The positive effect of low-dose atorvastatin (20 mg/day) is normalization of the adipokine status. Administration of atorvastatin 20 mg/day was accompanied with a statistically significant reduction in glucose levels (by 14%) and C-peptide levels (by 38%), and a decrease in the homeostasis model assessment of insulin resistance index on day 12.
Conclusion: Determination of atorvastatin dose and its use during the in-hospital period and subsequent periods should take into account changes in biochemical markers of insulin resistance and adipokine status in patients with MI.
Keywords: myocardial infarction, statin, insulin resistance, adipokines, atorvastatin
Introduction
A key factor determining the development of both myocardial infarction (MI) and its consequent complications is dyslipidemia, which is associated with endothelial dysfunction, activation of inflammation, thrombogenesis, and formation of insulin resistance.1 In cardiology practice, administration of statins, which inhibit 3-hydroxy-3-methylglutaryl-CoA reductase (HMG-CoA reductase), the main effect of which is hypolipidemic, is generally recognized for primary and secondary prevention of atherosclerosis and its manifestations.2,3 Pleiotropic effects are observed through both lipid-dependent mechanisms and the absence of the mechanisms with statin administration. More than 40 pleiotropic effects of statins have been identified, among which are proven beneficial effects on migration and the functional state of macrophages, smooth muscle cells in vessel walls, improvement in the functional state of endothelium, and normalization of rheological and reduction in thrombogenic blood properties.4 Statins can modulate the expression of some genes involved in atherogenesis, which may significantly increase their range of application.
The long-term effects of statins are the most commonly studied.5 In recent years, however, studies have proved the ability of statins to have a short-term effect also, particularly in the treatment of patients with acute MI.5,6 Previous research has demonstrated the adverse effects of high doses of statins on carbohydrate metabolism indicators.7
This study was a comparative analysis of the effects of atorvastatin at doses of 20 mg/day and 40 mg/day on lipid parameters, adipokine status, insulin resistance markers, prothrombotic status, and inflammatory parameters in patients with MI in the dynamics of the in-hospital setting.
Materials and methods
Ethical considerations
The study was conducted in accordance with the principles of the Declaration of Helsinki, and the study protocol was approved by the Ethical Committee of the Research Institute for Complex Issues of Cardiovascular Diseases under the Siberian Branch of the Russian Academy of Medical Sciences. All patients gave written informed consent.
Study design
The study was an open, prospective, randomized, comparative, and controlled study. Inclusion criteria were those confirmed and approved by the Russian Society of Cardiology in 2007 and specified in “Acute ST-elevated myocardial infarction in patients hospitalized in the first 24 hours after the symptoms onset”. Exclusion criteria were the following: diabetes mellitus, previously identified or newly diagnosed at the time of admission; statin therapy for the month prior to MI; serious diseases affecting prognosis (anemia, renal failure, hepatic failure, oncological diseases); infectious inflammatory diseases during exacerbation; autoimmune diseases; and long-term treatment with corticosteroids.
Two-hundred ten patients were enrolled who had ST-elevated MI and who were treated with atorvastatin, which inhibits HMG-CoA reductase, from the first 24 hours of MI. Group 1 (n=110) were given atorvastatin at a dose of 20 mg/day. Group 2 (n=100) were given atorvastatin at a dose of 40 mg/day. The dose was not increased in either group. The study presents the results of observations during the period of 12 days from initial administration.
The clinical characteristics of the patients are presented in Table 1. In Group 1, drug administration was stopped in one patient because of the development of dyspeptic events; the patient experienced nausea within a week of being given atorvastatin. In Group 2, drug administration was stopped in one patient because of the development of dyspeptic events; the patient experienced epigastric pain and nausea within 5 days of being given atorvastatin. The control group consisted of 33 people aged 58 years (57.2; 59.2) without cardiovascular and endocrine system diseases.
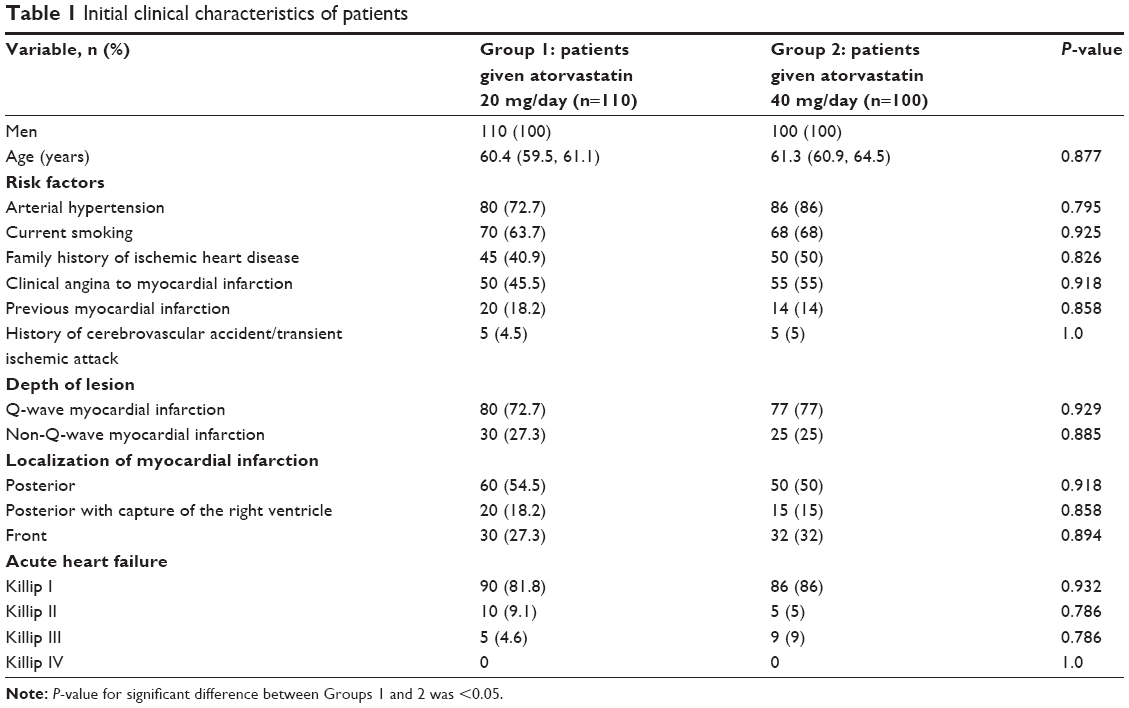 | Table 1 Initial clinical characteristics of patients |
The groups were comparable by age, sex, and the presence of coronary artery disease (CAD) risk factors, such as arterial hypertension, smoking, and obesity. Body mass index (BMI) was calculated using Quetelet’s equation as a ratio of body weight (kg) to height squared (m). BMI in Group 1 was 26.8 (26.12; 27.12) kg/m2 and in Group 2 was 25.3 (24.88; 25.67) kg/m2 (P=0.117). More than 40% of patients in both groups had a family history of CAD.
Percutaneous coronary intervention of the infarct-dependent artery was performed as reperfusion therapy in all patients. The groups were comparable by the presence of coronary atherosclerosis, which was based on results of a coronary angiography on admission, which showed multi-vessel disease (more than three vessels) in 66.7%.
MI patients received medical therapy according to the 2007 National Cardiology Society Guidelines. Throughout the observation and treatment period in hospital (12 days on average), β-adrenergic blocking agents, angiotensin-converting enzyme inhibitors, Ca2+-channel antagonists, diuretics, nitrates, aspirin, heparin, and clopidogrel were given in equal frequency to the two groups (Table 2).
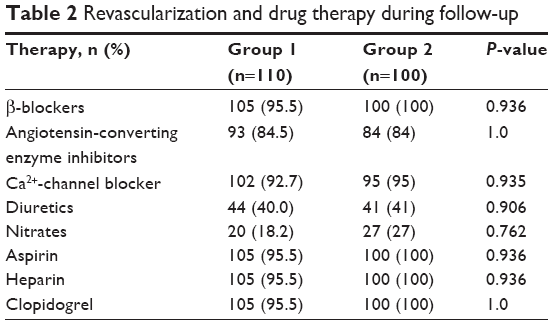 | Table 2 Revascularization and drug therapy during follow-up |
Assays
Blood serum and plasma were tested. Serum was separated from venous blood by centrifugation at 3,000× g for 20 minutes and stored at −70°C. At days 1 and 12 after MI onset, serum glucose, insulin, and C-peptide levels were measured with diagnostics laboratory kits (Diagnostic Systems Laboratories, Inc., Webster, TX, USA). Free fatty acid (FFA), total cholesterol (TC), triacylglycerol (TAG), low-density lipoprotein cholesterol (LDL), very-low-density lipoprotein cholesterol (VLDL), apolipoprotein B (apo-B), high-density lipoprotein cholesterol (HDL), and apolipoprotein A1 (apo-A1) levels were measured at the same study time points using standard Thermo Fisher Scientific test systems (Thermo Fisher Scientific Oy, Vantaa, Finland) in a Konelab 30i biochemistry analyzer (Thermo Fisher Scientific Oy). C-peptide and insulin concentrations were measured by enzyme-linked immunosorbent assay (ELISA) with BioMedica (Sydney, Australia) and Diagnostic Systems Laboratories, Inc. laboratory kits, respectively. The intra-assay coefficients of variation (CVs) for insulin and C-peptide ELISA were 3.8% and 4.2%, respectively, and the inter-assay CVs were 6.9% and 7.9%, respectively. Adipokine (leptin, adiponectin, resistin, and ghrelin) levels were measured using BioVendor assay kits (Brno, Czech Republic), and intra-assay CVs were 5.9% and 6.8%. Prothrombotic potential was assessed by determining plasminogen activator inhibitor-1 (PAI-1) levels, and was measured using Technoclone GmbH assay kits (Vienna, Austria), and intra-assay CVs were 4.8% and 5.7%.
The following formula was used to calculate the atherogenic index: atherogenic index (total relative value units) = (TC – HDL)/HDL.
C-reactive protein (CRP) and interleukin-6 (IL-6) levels were examined by ELISA. Serum glucose, insulin, and C-peptide levels were measured to assess carbohydrate metabolism and to diagnose insulin resistance. The homeostasis model assessment of insulin resistance (HOMA-IR) index was calculated on days 1 and 12 after MI onset. A HOMA-IR value >2.77 corresponds to insulin resistance.
Statistical analysis
Statistical analyses were performed using the nonparametric Mann–Whitney test for unpaired samples and the Wilcoxon test for paired samples. Results are presented as median values, unless otherwise specified, and 25% and 75% quartiles. Spearman’s correlation coefficient was calculated to analyze correlations between variables. Statistical analyses were performed using Statistica 6.0 (StatSoft, Tulsa, OK, USA).
Results
The main clinic–anamnestic data are shown in Table 1. The majority of patients in both groups were characterized by Q-wave MI. Back localization of MI prevailed in both groups. Activity of creatine phosphokinase-MB in both groups did not differ significantly. The maximum activity of creatine phosphokinase-MB was 129.6 U/L (25% and 75% quartiles: 112.32, 152.32) in Group 1 and 111.7 U/L (25% and 75% quartiles: 98.31, 125.98) in Group 2 (P=0.870).
More than 80% of patients in both groups did not show clinical signs of acute heart failure, which was estimated by Killip classifications (Table 1).7 More than 40% of patients had arrhythmias and cardiac conduction disorders, assessed using commonly accepted criteria. The parameters of the structural and functional states of the left ventricle did not differ significantly between groups. Median values of left ventricular ejection fraction, estimated at the first 24 hours of disease onset, were 53.5% (25% and 75% quartiles: 52.4, 54.5) in Group 1 and 49.7% (25% and 75% quartiles: 49.1, 50.5) in Group 2 (P=0.376). Mean values of left ventricular end-diastolic volume were 153 mL (25% and 75% quartiles: 148.2, 156.4) in Group 1 and 144.9 mL (25% and 75% quartiles: 139.2, 149.5) in Group 2 (P=0.068). Mean values of left ventricular end-systolic volume were 67 mL (25% and 75% quartiles: 62.3, 68.9) in Group 1 and 68.6 mL (25% and 75% quartiles: 66.5, 69.8) in Group 2 (P=0.402).
Changes in lipid-transport function parameters in the blood were observed for both groups on day 1 after MI onset. Both groups showed higher levels of TC, LDL, TAG, and apo-B, higher apo-B/apo-A1 ratio, and decreased levels of HDL and apo-A1 compared with controls. A two- to threefold increase in the atherogenic index was observed for both groups compared with the control group (Table 3). Common patterns of the effects of atorvastatin administration on the studied parameters were observed during the early in-hospital period, as well as dose-dependent effects, in particular, the pronounced hypocholesterolemic effect of atorvastatin (dose: 40 mg/day) (Table 3). A significant decrease in the initially high levels of cholesterol and its atherogenic fractions on day 12 was observed, with the atherogenic index decreasing by 36% compared with day 1. A less pronounced hypocholesterolemic effect was observed with the lower dose of atorvastatin. Atorvastatin administration (20 mg/day) resulted in TC reduction by 10% and the atherogenic index reduction by 26%, as well as a 1.2-fold increase in the concentration of apo-A1 on day 12 compared with day 1.
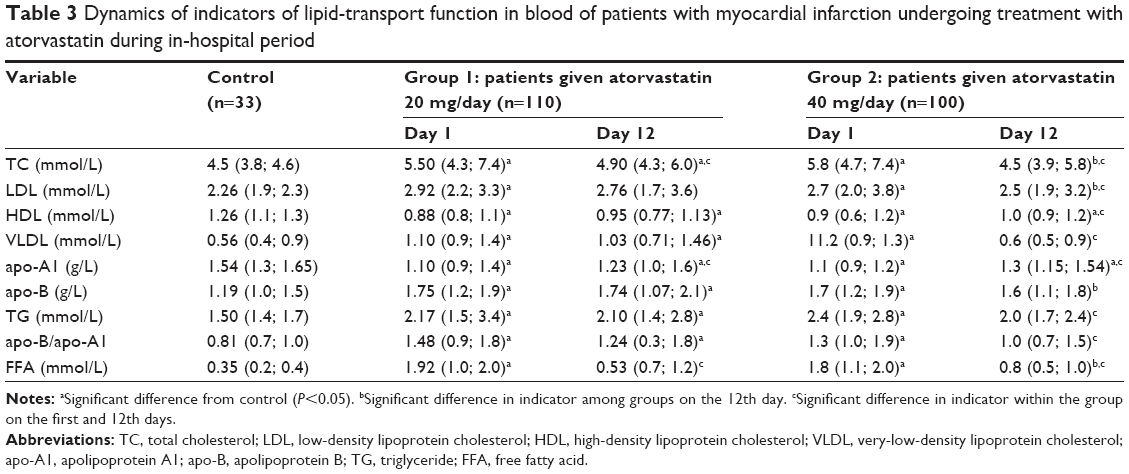 | Table 3 Dynamics of indicators of lipid-transport function in blood of patients with myocardial infarction undergoing treatment with atorvastatin during in-hospital period |
Pronounced changes in FFA levels were observed (Table 3). On day 1 after MI, FFA levels in both groups showed a ninefold increase compared with the control group. During the in-hospital period, there was a decrease in FFA levels in both groups, with the decrease being more pronounced in patients receiving atorvastatin 40 mg/day. In this group, FFA levels decreased by 83.3% compared with baseline values, and were 1.6-fold greater than the baseline levels of the control group. Atorvastatin 20 mg/day resulted in a decrease in FFA levels by 72.4% compared with day 1, but the FFA levels were still 2.6-fold greater than the baseline levels of the control group.
Changes in leptin, resistin, and adiponectin concentrations were compared among groups at days 1 and 12 after MI (Table 4). On admission, patients had a twofold increase in leptin levels compared with the control group. Atorvastatin 20 mg/day resulted in a decrease in leptin levels by 32.8% on day 12 compared with day 1. Patients who received 40 mg/day showed a 16.8% decrease compared with day 1. Compared with the control group, leptin levels were higher by 36% in patients receiving atorvastatin 40 mg/day.
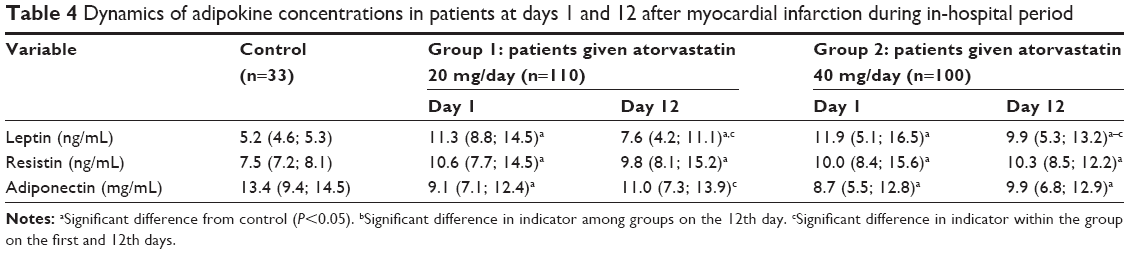 | Table 4 Dynamics of adipokine concentrations in patients at days 1 and 12 after myocardial infarction during in-hospital period |
The statistically significant increase in adiponectin levels was only observed for the atorvastatin 20 mg/day group (P<0.05). Atorvastatin 40 mg daily did not produce a similar effect. Resistin concentrations during the acute phase and treatment did not change significantly (P>0.05).
Patients with MI on day 1 showed a 1.4- to 1.5-fold increase in glucose concentration, a 1.3- to 1.5-fold increase in C-peptide levels, and a 1.2- to 1.3-fold increase in HOMA-IR index compared with the control group (Table 5). Administration of atorvastatin 20 mg/day resulted in a statistically significant reduction in glucose levels (by 14%) and C-peptide levels (by 38%), and a decrease in HOMA-IR index to baseline values on day 12. A stronger effect was seen with atorvastatin 40 mg/day. In addition to glucose reduction on day 12, C-peptide decreased by 44%, insulin decreased by 44%, and HOMA-IR index decreased by 43% compared with the acute phase of the disease. It should be noted that atorvastatin 40 mg/day significantly decreased the concentrations of insulin and C-peptide, and decreased the HOMA-IR index compared with baseline values, which are considered adverse effects of atorvastatin on carbohydrate metabolism.
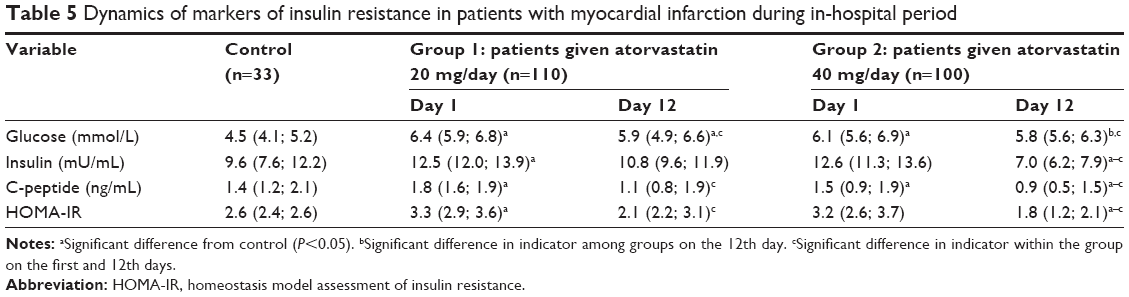 | Table 5 Dynamics of markers of insulin resistance in patients with myocardial infarction during in-hospital period |
Nonspecific inflammation is a common pathogenic link in the development of MI, with CRP and IL-6 used as key markers. In the current study, CRP increased 20-fold on day 1 after MI, and IL-6 showed a six- to sevenfold increase compared with the control group (Table 6). Atorvastatin 40 mg/day reduced CRP by 63.4%, and atorvastatin 20 mg/day reduced CRP by 23%. There were no statistically significant dose-dependent effects of atorvastatin on IL-6. IL-6 decreased by 49.2% in Group 1 and 44.3% in Group 2. In both groups, after the initiation of treatment, the concentrations of the inflammatory markers remained significantly higher compared with the control group.
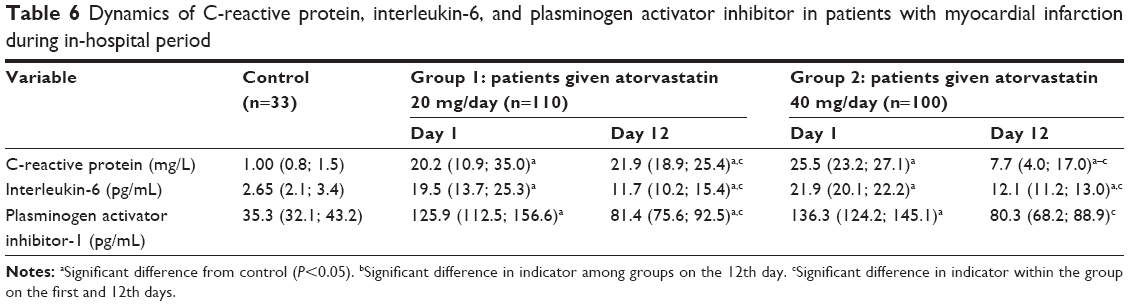 | Table 6 Dynamics of C-reactive protein, interleukin-6, and plasminogen activator inhibitor in patients with myocardial infarction during in-hospital period |
Evaluation of PAI-1, a key marker of thrombogenesis, showed that the level of PAI-1 increased fourfold compared with the control group on day 1 after MI. Atorvastatin administration produced favorable effects during the in-hospital period, allowing a reduction in the prothrombotic potential, but a dose-dependent effect was not pronounced. On day 12, atorvastatin reduced the levels of PAI-1 by 35.3% and 41.1% in Groups 1 and 2, respectively.
Discussion
Statins are widely used for secondary prevention of cardiovascular events in patients with MI. They can reduce mortality and improve prognosis in patients with CAD and acute coronary syndrome.2,8 Statins work by inhibiting the activity of a key enzyme in cholesterol synthesis, HMG-CoA reductase, by normalizing the levels of its lipid-transport forms. This can reduce the progression of atherosclerosis and the development of cardiovascular complications.8
Long-term use of statins has proved its dose-dependent effects on lipid profiles. For example, a meta-analysis of clinical studies demonstrated that the lipid-lowering efficiency of statins for LDL was directly proportional to the dose over 1 year of follow-up.9 Early intensive statin therapy can significantly reduce the risk of mortality and cardiovascular events over 2 years or more of treatment. The degree of reduction of LDL has no significant effect on these results, suggesting that statins have pleiotropic effects and/or the activity is dependent on the dose used.9,10
The results of the current study suggest the early (within 12 days of receiving atorvastatin), dose-dependent, lipid-lowering effect of atorvastatin. The most pronounced effects were seen with a dose of 40 mg/day, which is consistent with previous research. For example, Plenge et al reported that administration of atorvastatin in patients with MI in the early stages of the disease was associated with decreased levels of TC and LDL. In that study, a dose of 40 mg/day resulted in a significant decrease compared with 10 mg/day (P<0.05). The authors consider the fast action to be a positive factor in reducing ischemic attack recurrence.11 In the current study, hypolipidemic activity manifested at the low dose of atorvastatin (20 mg/day), and may be characterized by a decrease in both cholesterol levels and the atherogenic index, which reflects recovery of lipid status in patients with MI during the in-hospital period.
Pronounced effects of atorvastatin on FFA levels were observed. FFA levels showed a sharp increase during the acute phase of the disease, but treatment with atorvastatin 20 mg/day, and especially with 40 mg/day, resulted in a decrease in FFA levels. One possible mechanism for this effect may be improvement in the peroxisome proliferator-activated receptor, which restores metabolic processes in myocardium.2 The decline in FFA levels with statin administration is also probably associated with a decrease in FFA and TAG synthesis in the liver and their release into the blood.2 An experimental mouse study reported that administration of atorvastatin to fructose-fed mice reduced FFA and TAG concentrations.2 The authors suggest that atorvastatin blocks the expression of the nuclear protein carbohydrate response element-binding protein, which is responsible for the synthesis of FFA from carbohydrates.
It is thought that FFA plays a major role in reducing the sensitivity of peripheral tissues to the actions of insulin, contributing to the development of insulin resistance, and may be a factor provoking the development of diabetes.1,11 Reducing FFA levels can therefore be considered to give a protective effect against the development of insulin resistance in patients with MI. There is, however, no consensus in the literature on the impact of statins, including the effects of atorvastatin on the development of insulin resistance. Some authors have suggested that atorvastatin therapy produces a dose-dependent increase in insulin resistance.14 Other researchers do not share this view, and suggest that atorvastatin is safe in terms of the drug-related development of insulin resistance. Chu et al reported that atorvastatin at doses of 10 mg/day and 80 mg/day did not affect insulin sensitivity or leptin and adiponectin levels in diabetic patients with hyperlipidemia during 12 weeks of treatment.14 Inconsistencies in the data found in the literature may be the result of different doses and different follow-up periods.
The results of the current study suggest that the beneficial effect of atorvastatin on insulin resistance is provided at a dose of 20 mg/day. This dose was found to normalize the HOMA-IR index, and provide a moderate decrease in glucose and C-peptide levels to baseline values. Such effects may reflect a reduction in insulin resistance, restoring the sensitivity of peripheral tissues to the actions of insulin. These findings are consistent with previous studies that confirm the positive effect of statins on peripheral tissue sensitivity to insulin and carbohydrate metabolism.12
The administration of atorvastatin 40 mg/day showed more pronounced decreases in the parameters reflecting pancreatic function (insulin and C-peptide levels), and through these parameters, reflected the status of carbohydrate metabolism, not only compared with patients treated with the lower dose but also compared with healthy controls. This suggests the possible suppression of insulin synthesis via a decrease in C-peptide secreted by the pancreas in patients with MI receiving atorvastatin 40 mg/day.
Experimental data suggest that atorvastatin blocks the synthesis of ubiquinone (also known as coenzyme Q10), an essential factor providing mitochondrial electron transport. This may lead to a decrease in the intensity of tissue respiration and ATP synthesis in pancreatic cells, and may delay the secretion of C-peptide and insulin.12 Such an effect cannot be considered positive, especially over the long term.
Adipokines are known to play important roles in the regulation of insulin sensitivity, glucose homeostasis, and lipid metabolism.14,15 Data on the effects of atorvastatin on adipokine levels are controversial: atorvastatin is reported to not produce any effects, and is also reported to result in an increase or decrease in adipokine levels.13,15 In the current study, atorvastatin 20 mg/day significantly modulated adipokine levels. We suggest that restoration of the adipokine status does not require high doses of statins; the positive effect is already achieved at a dose of 20 mg/day. With a dose of 40 mg/day, leptin levels were observed to remain elevated. This elevation, in combination with a reduction in insulin concentration, can lead to hyperleptinemia and metabolic dysregulation, and worsen myocardial ischemia, and may be regarded as a poor prognostic factor.
In recent years, studies have examined the importance of inflammation in the development of MI and its associated complications. Statins, which activate peroxisome proliferator-activated receptor alpha, are assumed to reduce the activity of nuclear factor kappa B, which regulate the expression of many genes whose products (cytokines, chemokines, cell adhesion molecules) are involved in the inflammatory and immune responses.16
Several differences in the influence of atorvastatin at the specific doses on proinflammatory markers, which are significantly increased in the acute phase of MI, were observed. Plasma levels of CRP decreased to a greater extent in patients treated with atorvastatin 40 mg/day compared with 20 mg/day. There was no dose-dependent effect on IL-6. Both groups during treatment showed a reduction in cytokine concentrations, but the concentrations did not reach the same level seen in the control group. This finding, taken with the reduction in insulin secretion, may suggest a contribution to the development of latent forms of insulin resistance in patients taking atorvastatin 40 mg/day.
IL-6 is known to be able to induce insulin resistance and enhance production of mediators of endothelial dysfunction in arterioles and capillaries, with the involvement of proinflammatory serine/threonine kinase suppressor proteins, and cytokine signaling and protein tyrosine phosphatases acting as negative regulators of physiological insulin.17,18
Patients with MI are known to have an elevated prothrombotic status. Statins have an anticoagulant action, the mechanism of which is complex. Statins decrease plasma fibrinogen levels and normalize the lipid composition of blood cell membranes, inhibit ADP-dependent platelet aggregation, inhibit thromboxane production, and diminish concentrations of tissue PAI-1 in patients with CAD.19 The results of the current study indicate the absence of a dose-dependent effect of atorvastatin on PAI-1 levels. Despite atorvastatin reducing concentrations of PAI-1, high prothrombotic activity was still present in the early in-hospital period because of the remaining high PAI-1 levels. High PAI-1 levels may be involved in the development of insulin resistance in the post-hospital follow-up period in patients receiving atorvastatin, especially at high doses.20
Conclusion
Dose-related pleiotropic effects of atorvastatin were observed in the early in-hospital period in the current study. Atorvastatin 40 mg/day showed a more pronounced effect on the recovery of lipid status in patients with MI compared with atorvastatin 20 mg/day. Correction of insulin resistance was more effective at 20 mg/day compared with 40 mg/day, and to a lesser extent was dependent on the effects of atorvastatin on inflammatory markers and thrombogenesis. The positive effect of low-dose atorvastatin (20 mg/day) is the normalization of adipokine status, which is a favorable prognostic sign. Determination of the dose of atorvastatin and its use in the in-hospital period and subsequent periods should take into account the changes in biochemical markers of insulin resistance and adipokine status in patients with MI.
Limitations
Our study has strengths and weaknesses. Overall, the trial was designed to assess the effect of atorvastatin on lipid parameters, adipokine status, insulin resistance markers, prothrombotic status, and inflammatory parameters in patients with MI. A limitation in our study was the short observation period – 12 days (in-hospital period).
Acknowledgment
The authors wish to thank Olga Akbasheva for assistance in writing this article.
Disclosure
The authors declare that they have no competing interests. This research received no grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
Cefalu WT. Insulin resistance: cellular and clinical concepts. Exp Biol Med. 2001;226:13–26. | ||
Rodríguez-Calvo R, Barroso E, Serrano L, et al. Atorvastatin prevents carbohydrate response element binding protein activation in the fructose-fed rat by activating protein kinase A. Hepatology. 2009;49:106–115. | ||
Ichihara K, Satoh K. Disparity between angiographic regression and clinical event rates with hydrophobic statins. Lancet. 2002;359:2195–2198. | ||
Besler C, Doerries C, Giannotti G, Lüscher TF, Landmesser U. Pharmacological approaches to improve endothelial repair mechanisms. Expert Rev Cardiovasc Ther. 2008;6:1071–1082. | ||
Muhlestein JB, Anderson JL, Horne BD, et al; Intermountain Heart Collaborative Study Group. Early effects of statins in patients with coronary artery disease and high C-reactive protein. Am J Cardiol. 2004;94:1107–1112. | ||
Hulten E, Jackson JL, Douglas K, George S, Villines TC. The effect of early, intensive statin therapy on acute coronary syndrome. Arch Intern Med. 2006;166:1814–1821. | ||
Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther. 2011;5:325–380. | ||
Killip T, Kimball JT. Treatment of myocardial infarction in a coronary care unit. A two year experience with 250 patients. Am J Cardiol. 1967;20:457–464. | ||
Rizzo M, Berneis K. Low-density lipoprotein size and cardiovascular risk assessment. QJM. 2006;99:1–14. | ||
Jones PH, Davidson MH, Stein EA, et al; STELLAR Study Group. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol. 2003;92:152–160. | ||
Plenge JK, Hernandez TL, Weil KM, et al. Simvastatin lowers C-reactive protein within 14 days: an effect independent of low-density lipoprotein cholesterol reduction. Circulation. 2002;106:1447–1452. | ||
Dresner A, Laurent D, Marcucci M, et al. Effects of free fatty acids on glucose transport and IRS-1–associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259. | ||
Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol. 2010;55:1209–1216. | ||
Chu CH, Lee JK, Lam HC, et al. Atorvastatin does not affect insulin sensitivity and the adiponectin or leptin levels in hyperlipidemic type 2 diabetes. J Endocrinol Invest. 2008;31:42–47. | ||
Mantzoros CS, Magkos F, Brinkoetter M, et al. Leptin in human physiology and pathophysiology. Am J Physiol Endocrinol Metab. 2011;301:567–584. | ||
Li S, Shin HJ, Ding EL, van Dam RM. Adiponectin levels and risk of type 2 diabetes: a systematic reviewand meta-analysis. JAMA. 2009;302:179–188. | ||
Ortego M, Bustos C, Hernández-Presa MA, et al. Atorvastatin reduces NF-kB activation and chemokine expression in vascular smooth muscle cells and mononuclear cells. Atherosclerosis. 1999;147:253–261. | ||
Gabay C, Kushner I. Acute phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. | ||
Son JW, Kim DJ, Lee CB, et al. Effects of patient-tailored atorvastatin therapy on ameliorating the levels of atherogenic lipids and inflammation beyond lowering low-density lipoprotein cholesterol in patients with type 2 diabetes. J Diabetes Investig. 2013;4:466–474. | ||
Ni XQ, Zhu JH, Yao NH, Qian J, Yang XJ. Statins suppress glucose-induced plasminogen activator inhibitor-1 expression by regulating RhoA and nuclear factor-κB activities in cardiac microvascular endothelial cells. Exp Biol Med (Maywood). 2013;238:37–46. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.