")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Different effects of statins on induction of diabetes mellitus: an experimental study
Received 5 May 2015
Accepted for publication 6 September 2015
Published 24 November 2015 Volume 2015:9 Pages 6211—6223
DOI https://doi.org/10.2147/DDDT.S87979
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Wang Zhao, Shui-Ping Zhao
Department of Cardiology, The Second Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China
Background: To determine the effect of different statins on the induction of diabetes mellitus.
Materials and methods: Four statins (atorvastatin, pravastatin, rosuvastatin, and pitavastatin) were used. Cytotoxicity, insulin secretion, glucose-stimulated insulin secretion, and G0/G1 phase cell cycle arrest were investigated in human pancreas islet β cells, and glucose uptake and signaling were studied in human skeletal muscle cells (HSkMCs).
Results: Human pancreas islet β cells treated with 100 nM atorvastatin, pravastatin, rosuvastatin, and pitavastatin had reduced cell viability (32.12%, 41.09%, 33.96%, and 29.19%, respectively) compared to controls. Such cytotoxic effect was significantly attenuated by decreasing the dose to 10 and 1 nM, ranged from 1.46% to 17.28%. Cells treated with 100 nM atorvastatin, pravastatin, rosuvastatin, and pitavastatin had a reduction in the rate of insulin secretion rate by 34.07%, 30.06%, 26.78%, and 19.22%, respectively. The inhibitory effect was slightly attenuated by decreasing the dose to 10 and 1 nM, ranging from 10.84% to 29.60%. Insulin secretion stimulated by a high concentration of glucose (28 mmol/L) was significantly higher than a physiologic concentration of glucose (5.6 mmol/L) in all treatment groups. The glucose uptake rates at a concentration of 100 nM were as follows: atorvastatin (58.76%) < pravastatin (60.21%) < rosuvastatin (72.54%) < pitavastatin (89.96%). We also found that atorvastatin and pravastatin decreased glucose transporter (GLUT)-2 expression and induced p-p38 MAPK levels in human pancreas islet β cells. Atorvastatin, pravastatin, and rosuvastatin inhibited GLUT-4, p-AKT, p-GSK-3β, and p-p38 MAPK levels in HSkMCs.
Conclusion: Statins similar but different degree of effects on pancreas islet β cells damage and induce insulin resistance in HSkMC.
Keywords: statins, insulin, glucose, human pancreas islet β cell, human skeletal muscle cells
Introduction
Statins are used to reduce low-density lipoprotein cholesterol (LDL-C) levels and are associated with reduced cardiovascular events in a wide range of patient populations. However, recent studies suggested that statins are associated with an increased risk of developing type 2 diabetes mellitus (T2DM).1 The Justification for the Use of Statins in Primary Prevention (JUPITER) study (N=17,802) showed that treatment with rosuvastatin (20 mg/day for 1.9 years) was associated with a 25% increase in relative risk for T2DM among middle-aged men and women versus placebo,2 which was accompanied by a small, but significant increase in glycated hemoglobin (HbA1c). A meta-analysis of 13 statin clinical trials (n=91,140 patients without T2DM) indicated that standard doses of statins (atorvastatin [10 mg], pravastatin [40 mg], simvastatin [40 mg], or rosuvastatin [20 mg]) were associated with a 9% increased risk for T2DM over 4 years (odds ratio [OR] =1.09, 95% confidence interval [CI] =1.02–1.17), with little heterogeneity (I2=11%) between trials.3 Based on these studies, the US Food and Drug Administration changed the labeling requirements for manufactures to include a warning about the possibility of increased blood glucose and HbA1c levels. The European Medicines Agency also issued a statement on the small increased risk for T2DM with the use of statins.1
Currently, seven statins are available for the treatment of dyslipidemia (atorvastatin, simvastatin, rosuvastatin, pitavastatin, lovastatin, pravastatin, and fluvastatin). The risk for the T2DM development of these statins differs.4 More recently, a meta-analysis of 17 randomized controlled trials (RCTs), including 113,394 patients without preexisting T2DM, showed that several statins was associated with the lowest risk for developing T2DM (OR=0.90, 95% CI=0.71–1.35 for 10–20 mg pravastatin/day; OR=1.04, 95% CI=0.75–1.46 for 10 mg atorvastatin/day; and OR=1.11, 95% CI=0.81–1.52 for 10 mg rosuvastatin/day).5 When comparing the effects of moderate- and high-dose of statins (lovastatin [20–40 mg] < pravastatin [40 mg] < atorvastatin [80 mg] < simvastatin [40 mg] < rosuvastatin [20 mg]), the order of risk was similar, and the results remained the same in several other ranking analyses.6
T2DM is a complex disorder characterized by pancreatic β cell dysfunction and insulin resistance of skeletal muscle, adipose tissue, and the liver.4 The precise reasons for the increased incidence of T2DM with statins have not been well demonstrated. However, possible explanations included residual confounding factors, such as improved survival with statin treatment and a greater chance of identifying incident T2DM in statin-treated patients.7–9 Several biological mechanisms have also been proposed for the diabetogenic effects of statins.7–9 Under normal conditions, glucose enters pancreatic beta cells,7 skeletal muscle cells,10 and adipocytes11 via insulin-responsive transmembrane glucose transporter (GLUT)-2 and -4. A number of animal- and cell-based studies have reported statin-associated alterations in glucose metabolism that could result in the decreased insulin secretion and/or insulin resistance,7–9 some of which also reported the differential effect among different statins. Thus, further research into the mechanisms underlying statin-mediated T2DM is urgently needed.12–14
In the present study, four statins (atorvastatin, pravastatin, rosuvastatin, and pitavastatin) were studied to determine their effect on human pancreas islet β cells and human skeletal muscle cells (HSkMCs). To investigate the potential mechanisms for the diabetogenic effects of statins, cell viability and insulin secretion were analyzed in human pancreas islet β cells, and glucose uptake was studied in HSkMCs.
Materials and methods
Cell lines, chemicals, and reagents
This work was approved by the Second Xiangya Hospital of Central South University. The human pancreas islet β cells were obtained from PriCells (Wuhan Pricells Biotechnology & Medicine Co., Ltd., Wuhan, People’s Republic of China). The HSkMCs were obtained from ScienCell (Carlsbad, CA, USA). Dulbecco’s Modified Eagle’s Medium (DMEM) medium and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Human insulin enzyme-linked immunosorbent assay kits were purchased from Shanghai Chuanxiang Biotechnology (Shanghai, People’s Republic of China). PKC immunoblot kits were obtained from Boster (Wuhan Boster Biological Engineering Co., Ltd.,). Glucose (hexokinase method) reagent was purchased from Shanghai Fenghui Medical Technology Co., Ltd. (Shanghai, People’s Republic of China). All the antibodies used in this study were purchased from Cell Signaling Technology (Beverly, MA, USA). Pitavastatin (livalo®) was a gift from Kowa Company, Ltd. (Nagoya, Japan). Atorvastatin, pravastatin, and rosuvastatin were purchased from other commercial pharmaceutical companies. No ethics statement was required from the institutional review board for the use of these human cell lines.
Cell culture and treatment
Human pancreas islet β cells and HSkMC were grown at 37°C in DMEM medium supplemented with 10% FBS, 100 IU/mL of penicillin, and 100 mg/mL of streptomycin in a 5% CO2/95% air-humidified atmosphere incubator according to standard protocols. When the adherent cells reached sub-confluence, the cells were passaged after trypsinization (0.25% trypsin and 0.03% EDTA [ethylenediaminetetraacetic acid]). The culture cells harvested by trypsinization were then used for experiments.
Cell viability assay
The effects of the drugs on cell proliferation were evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Cell Titer 96 Aqueous; Promega, Beijing, People’s Republic of China) for five groups (control, dimethyl sulfoxide [DMSO]; atorvastatin: 1, 10, and 100 nM; pravastatin: 1, 10, and 100 nM; rosuvastatin: 1, 10, and 100 nM; pitavastatin: 1, 10, and 100 nM). Briefly, 5×104 cells per well were seeded in triplicate in 90 μL of culture medium without FBS containing 5 g/L of bovine serum albumin (BSA) in 96-well flat-bottom microculture plates for 24 hours. The cellular morphology was observed at 12, 24, and 48 hours using an inverted microscope. Then, 100 μL of MTT solution (1 mg/mL in phosphate buffer saline [PBS]) was added and incubated for 4 hours. The medium was aspirated and replaced with 150 μL/well of DMSO to dissolve the formazan salt. The color intensity of the formazan solution, which reflects the cell growth conditions, was measured at a wavelength of 550 nm using a microplate spectrophotometer (Thermomax; Molecular Devices LLC, Sunnyvale, CA, USA).
Cell cycle analysis
The cells were harvested at 48 hours after different statin treatments and washed twice using PBS, which was precooled at 4°C. Cells were resuspended with 1% BSA-PBS before staining with 10 μL of propidium iodide (20 mg/L; Sigma–Aldrich, St Louis, MO, USA) and 100 mg/L of RNase A (Sigma–Aldrich). The cells were incubated at 37°C for 30 minutes before being analyzed for DNA content using a Calibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Insulin secretion
The level of insulin secretion was measured 48 hours after disposition using an enzyme-linked immunosorbent assay kit following centrifugation (3,000× g for 20 minutes) of the cultural supernatants. The insulin concentrations were calculated using the optical density at 450 nm. Every test was repeated in triplicate.
GSIS measured by radio immunoassay
To evaluate glucose-stimulated insulin secretion (GSIS) after the different treatments, the cells were washed and placed in Krebs-Ringer bicarbonate buffer (KRBB, [115 mM NaCl, 24 mM NaHCO3, 5 mM KCl, 1 mM MgCl2·6H2O, and 1 mM CaCl2·2H2O)] supplemented with 5.6 mmol/L glucose for a quiescent period of 1 hour. Next, cells were incubated for 1 hour in KRBB containing 5.6 or 28 mmol/L of glucose, respectively. The supernatants were collected and the insulin concentrations were determined using a radio immunoassay method. Every sample was repeated in triplicate.
Glucose uptake assay
Glucose uptake was measured in parallel plates using a previously described protocol.15 Briefly, cells were washed twice in PBS 48 hours after disposition and serum-free media was added for a 30-minute preincubation at 37°C in a 5% CO2 incubator before incubation for 2 hours at 37°C in glucose-free DMEM containing 1.3 μCi/mL of 2-deoxy-D-[1-3H]glucose (Amersham Biosciences, Buckinghamshire, UK). The cells were then placed on ice and immediately washed three times with ice-cold DMEM to terminate the reactions. The cells were then inspected for monolayer detachment and lysed in 0.5 mL (200 g/L) of KOH with scraping. Cell lysate (100 μL) was used for protein content measurement with Coomassie light blue. After washing twice with cold absolute ethyl alcohol, the lysates were centrifuged at 3,000× g for 5 minutes. The sediment (glycogen) was repeatedly washed with cold absolute ethyl alcohol over 3 hours on filter paper. The lysates were dried and transferred to 10 mL of Ready-Gel scintillation fluid (Beckman Coulter Inc., Fullerton, CA, USA), and the incorporated radioactivity was assessed in a liquid scintillation counter (1219 RackBeta; PerkinElmer, Waltham, MA, USA). Three independent experiments were conducted for each of the selected test compounds after a treatment for 48 hours.
Western blotting
The cells were collected in PBS and lysed 48 hours after disposition. The cell lysate was centrifuged at 12,000× g for 10 minutes, and the supernatant was used for Western blotting. Each sample (40 μg of cytoplasmic or nuclear protein) was separated by 10% SDS-PAGE and the proteins were transferred to nitrocellulose membranes (Amersham Biosciences). The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T [pH 7.4]) for 1 hour at room temperature. The membranes were washed with 0.1% TBS-T, then incubated with primary antibodies against GLUT-2, GLUT-4, PKA, PKC, p38 MAPK, phosphorylated p38 MAPK (Thr 180/Tyr182), Akt, phosphorylated Akt, GSK-3α, phosphorylated GSK-3α, GSK-3β, and phosphorylated GSK-3β in 0.1% TBS-T overnight at 4°C. The membrane was washed three times with 0.1% TBS-T. The membranes were then incubated with secondary antibodies including anti-rabbit and anti-mouse HRP antibodies for 1 hour at room temperature followed by three washings with 0.1% TBS-T. The immune complexes were visualized using chemiluminescence ECL Plus™ detection reagents following the manufacturer-suggested procedure. The reacted membranes were exposed to a chemiluminescence detector (CHEMI-SMART-3126 WL/26MX; Vilberlourmat, Marne la Vallee, France). Bands of interest were analyzed using ImageJ software.
Results
Statin-induced cytotoxicity in human pancreas islet β cells
We first examined the cytotoxic effects of statins on human pancreas islet β cells using the MTT reduction assay. Statins decreased cell viability of human pancreas islet β cells in a dose-dependent manner (Figure 1). Cells treated with 100 nM atorvastatin, pravastatin, rosuvastatin, and pitavastatin for 24 hours had reduced cell viability (32.12%, 41.09%, 33.96%, and 29.19% reduction, all P<0.001 vs controls) relative to control cells, respectively. This cytotoxic effect was significantly attenuated by decreasing the dose to 10 and 1 nM (reduction rate ranged from 1.46% to 17.28%, all P<0.05 for 10 nM and P>0.05 for 1 nM vs controls), suggesting that statin-induced cytotoxic effect was dose-dependent (Figure 1).
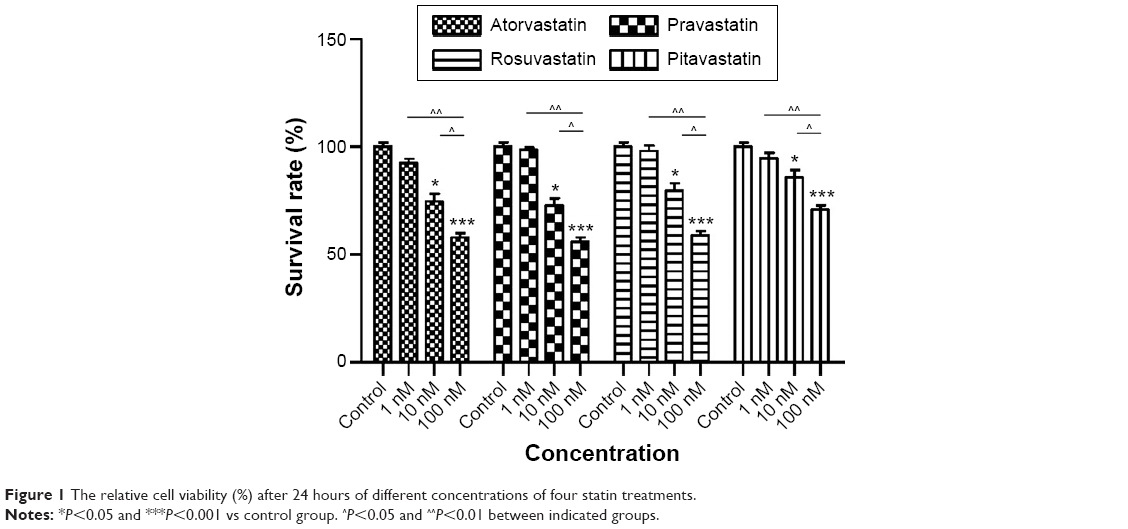 | Figure 1 The relative cell viability (%) after 24 hours of different concentrations of four statin treatments. |
Statins decrease insulin secretion
Cells treated with 100 nM atorvastatin, pravastatin, rosuvastatin, and pitavastatin for 24 hours had reduced insulin secretion rate (34.07%, 30.06%, 26.78%, and 19.22%, all P<0.001) relative to control cells, respectively. This inhibitory effect was slightly attenuated with decreased doses of 10 and 1 nM with a range of 10.84% to 29.60% (all P<0.05 10 vs 100 nM and all P<0.01 1 vs 100 nM), suggesting that statins suppressed insulin secretion in a dose-dependent manner. However, there was no significant correlation between the decreased cell survival rate and insulin secretion rate in all four statin treatments (all P>0.05; data not shown).
GSIS
The insulin secretion stimulated by a high glucose concentration (28 mmol/L) was significantly higher than a physiologic concentration (5.6 mmol/L) in all statin treatment groups, ranging from 53.44 to 78.32 ng/mL and from 35.78 to 54.22 ng/mL, respectively (data not shown). Upon physiologic glucose level (5.6 mmol/L) stimulation, the insulin secretion by pancreas islet β cells was the highest in pitavastatin group (range 46.29–51.05 ng/mL) and the lowest in pravastatin group (range 35.77–40.99 ng/mL) at the three concentration levels (data not shown in figure). The findings were similar under high glucose concentration (28 mmol/L) stimulation condition, ranging from 71.94 to 78.32 ng/mL in the pitavastatin group and from 53.44 to 65.86 ng/mL in pravastatin group at the three concentration levels (data not shown in figure). Therefore, we obtained a similar finding in a further analysis using the high glucose stimulation/low glucose stimulation index. The glucose stimulation index significantly decreased between control and 100 nM statin treatment of all four statins (Figure 2). Besides, pravastatin showed a significant decreased glucose stimulation index even at a relatively low concentration (10 nM; Figure 2).
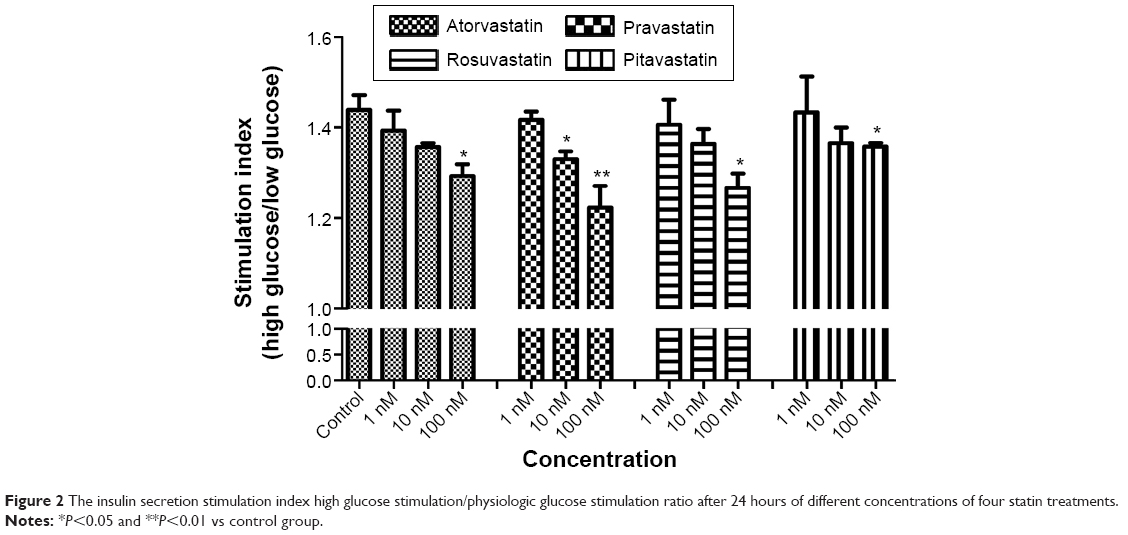 | Figure 2 The insulin secretion stimulation index high glucose stimulation/physiologic glucose stimulation ratio after 24 hours of different concentrations of four statin treatments. |
Statins-induced G0/G1 phase cell cycle arrest in human pancreas islet β cells
Statin-induced G0/G1 phase cell cycle arrest in human pancreas islet β cells also showed a dose-dependent pattern. The four statins induced G0/G1 cell cycle arrest at a low concentration (1 nM), where the G0/G1 DNA content was higher in statin-treated cells compared with control treatment in both cell lines. At a moderate concentration (10 nM), the G0/G1 DNA content of the four statin-treated groups was approximately the same as the low concentration (1 nM). At a high concentration (100 nM), the G0/G1 DNA content of the four drug-treatment groups was much higher than the low and moderate concentration groups, rising to 80.36%–86.28%. The proliferation of human pancreas islet β cells was significantly inhibited by statins at a high concentration (Figures 3 and 4).
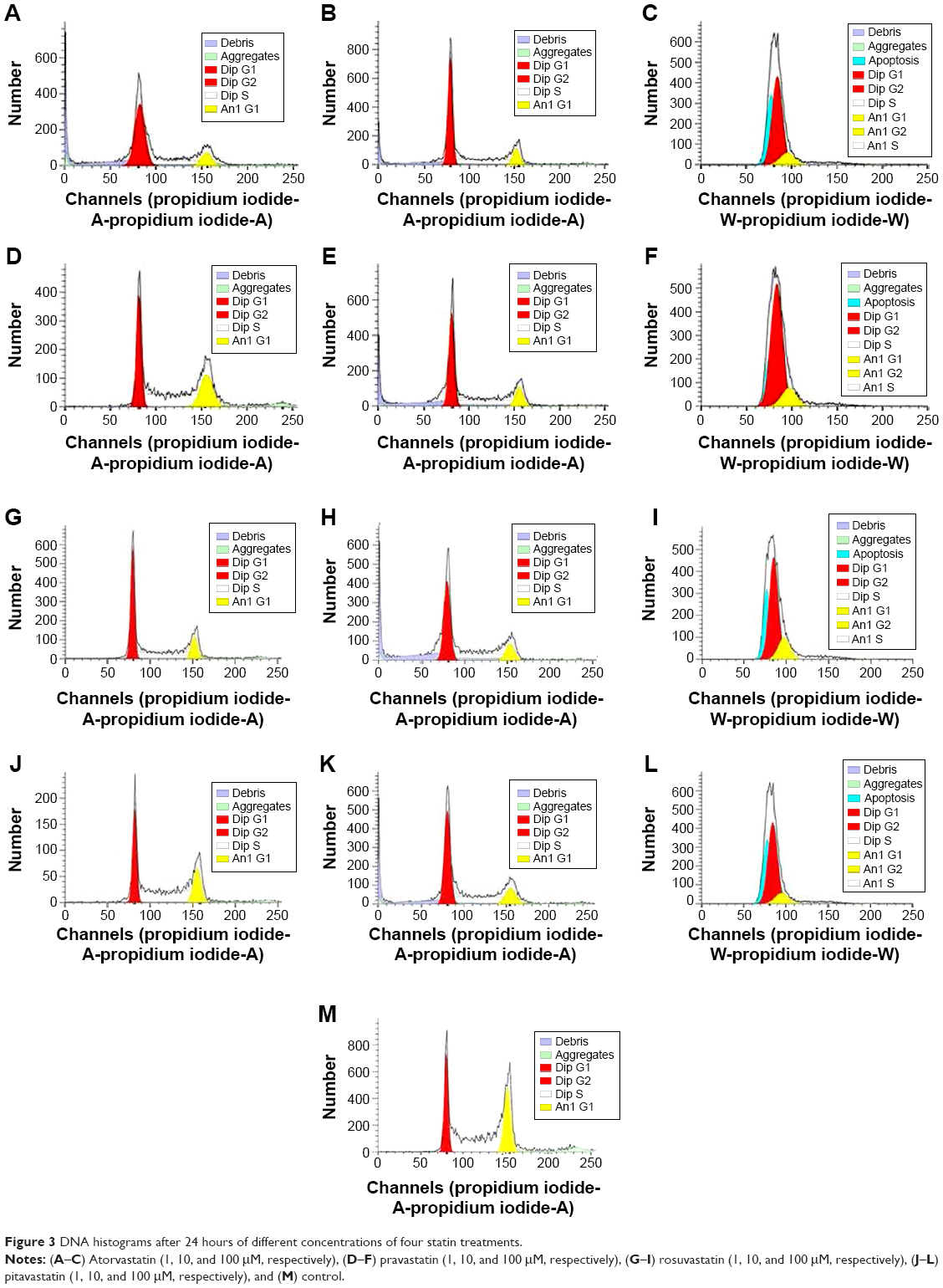 | Figure 3 DNA histograms after 24 hours of different concentrations of four statin treatments. |
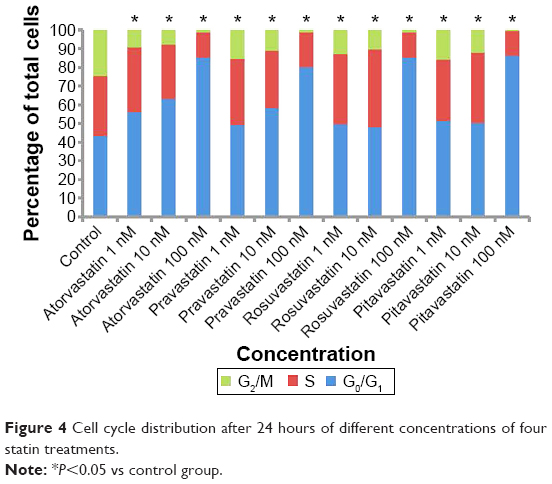 | Figure 4 Cell cycle distribution after 24 hours of different concentrations of four statin treatments. |
Decreased glucose uptake in HSkMCs
Exposure of HSkMCs to statins for 48 hours decreased [3H]-2-deoxy-glucose uptake compared to basal values. The glucose uptake rates at a concentration of 100 nM were as follows: atorvastatin (58.76%) < pravastatin (60.21%) < rosuvastatin (72.54%) < pitavastatin (89.96%). The decrease in glucose uptake was dose-dependent at all three concentrations (1, 10, and 100 nM) for atorvastatin and pravastatin. Pitavastatin had a less pronounced influence on the glucose uptake in HSkMCs at all three concentrations (Figure 5).
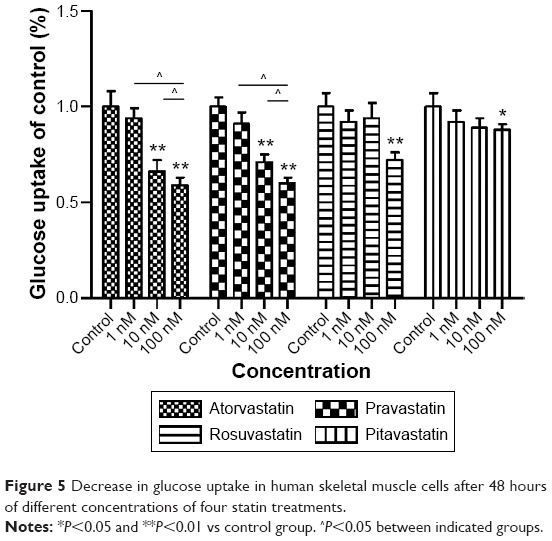 | Figure 5 Decrease in glucose uptake in human skeletal muscle cells after 48 hours of different concentrations of four statin treatments. |
Statin-induced GLUT-2 suppression and p-p38 MAPK overexpression in human pancreas islet β cells
As shown in Figure 6, atorvastatin and pravastatin elicited a concentration-dependent inhibition of GLUT-2 expression in human pancreas islet β cells, while rosuvastatin and pitavastatin showed a slight increase in GLUT-2 expression. However, the four statins showed a concentration-dependent increase in p38 MAPK phosphorylation with a maximum of sevenfold to13-fold increase over control values at a concentration of 100 nM statin. The expression of total p38 MAPK was approximately the same among the four statins-treated groups and the control group. There was no significant difference of PKA levels in statin-treated groups at all three concentrations compared to the control group.
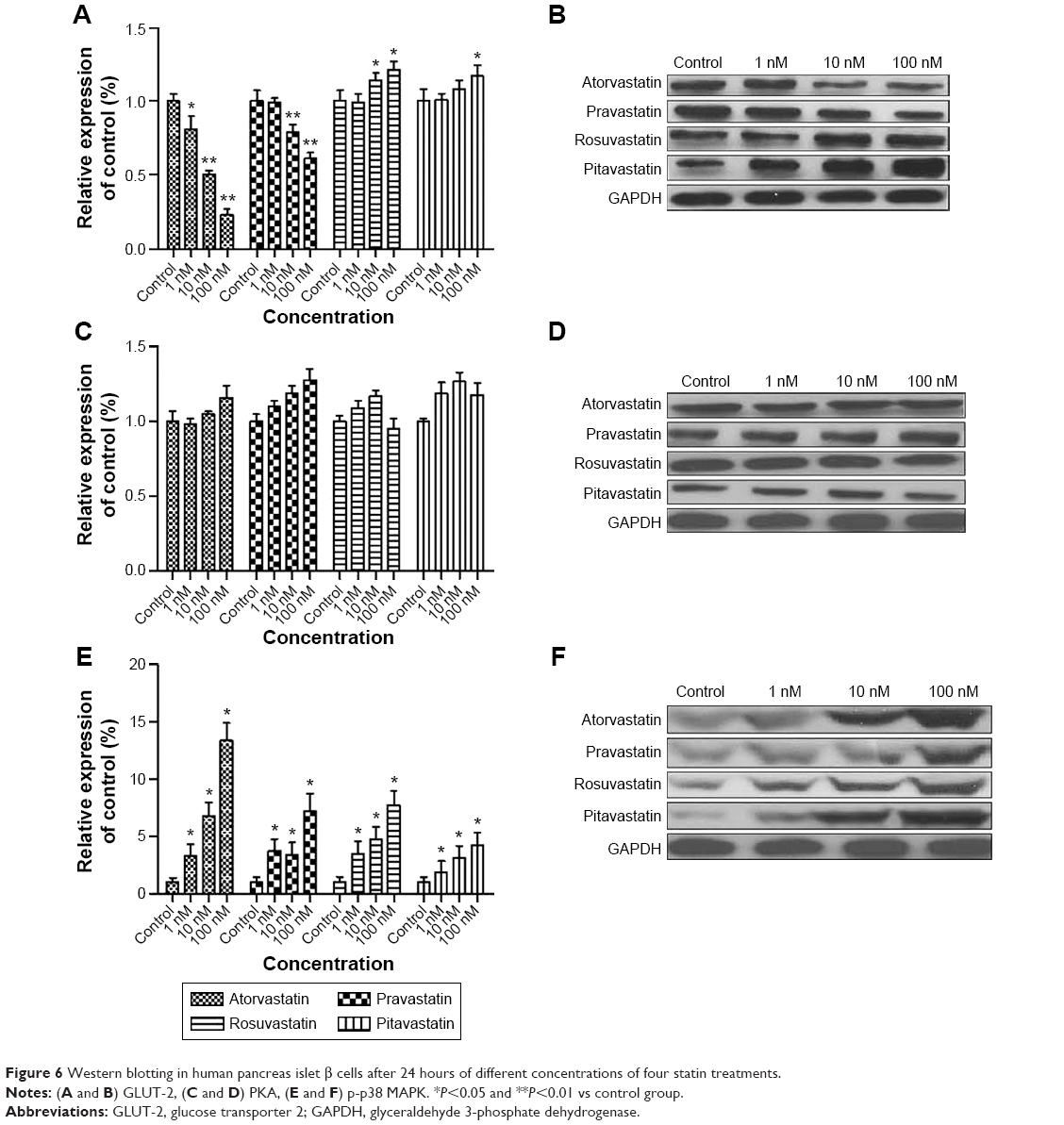 | Figure 6 Western blotting in human pancreas islet β cells after 24 hours of different concentrations of four statin treatments. |
Western blotting in HSkMCs
After statin treatment (Figure 7), GLUT-4 expression in HSkMCs was suppressed in the atorvastatin, pravastatin, and rosuvastatin groups and over expressed in the pitavastatin group compared to the control group in a concentration-dependent manner. The p-AKT was suppressed in all four statin-treated groups compared to the control group. The maximum suppression effect occurred in the atorvastatin group (100 nM). The expressions of p-GSK-3b and p-p38 MAPK were suppressed in all four statin-treated groups compared to the control group in a concentration-dependent manner.
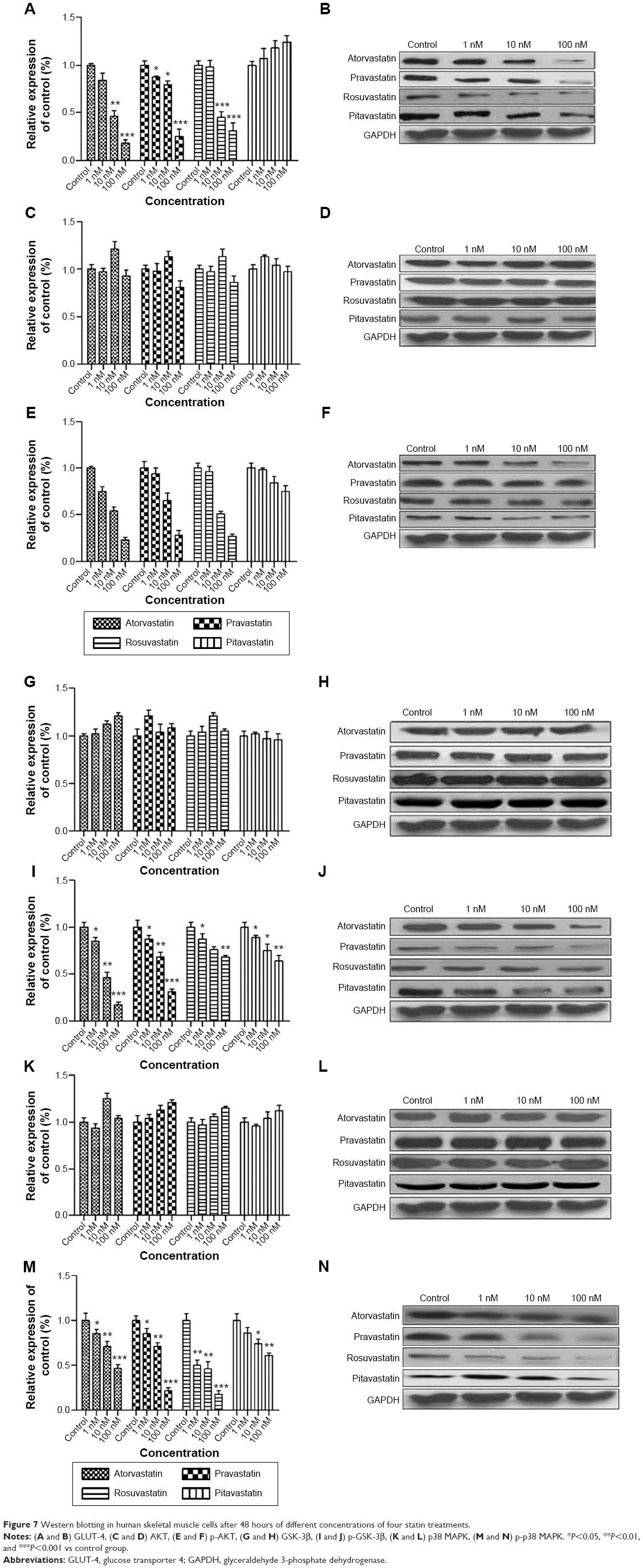 | Figure 7 Western blotting in human skeletal muscle cells after 48 hours of different concentrations of four statin treatments. |
Altered human pancreas islet β cells and HSkMCs phenotype normalized after MAPK and AKT inhibitor treatment
To further mechanistically prove that the altered human pancreas islet β cells and HSkMCs phenotype were due to induced MAPK signaling and suppressed MAPK/AKT signaling pathways, respectively, we treated the two types of cells with MAPK and AKT inhibitors. Inhibiting MAPK signaling in human pancreas islet β cells pretreated with atorvastatin led to the normalized cell survival rate (Figure 8A), and so as the glucose secretion upon stimulation reflected by glucose stimulation index (Figure 8B). These data suggested a causational relation between the induced MAPK signaling and statin treatment in human pancreas islet β cells. On the other hand, treating HSkMCs with MAPK and AKT signaling inhibitors (SB203580 and MK2206, respectively) led to decreased glucose uptake of, which recapitulate the glucose uptake change in HSkMCs treated with statin (Figure 8C and D).
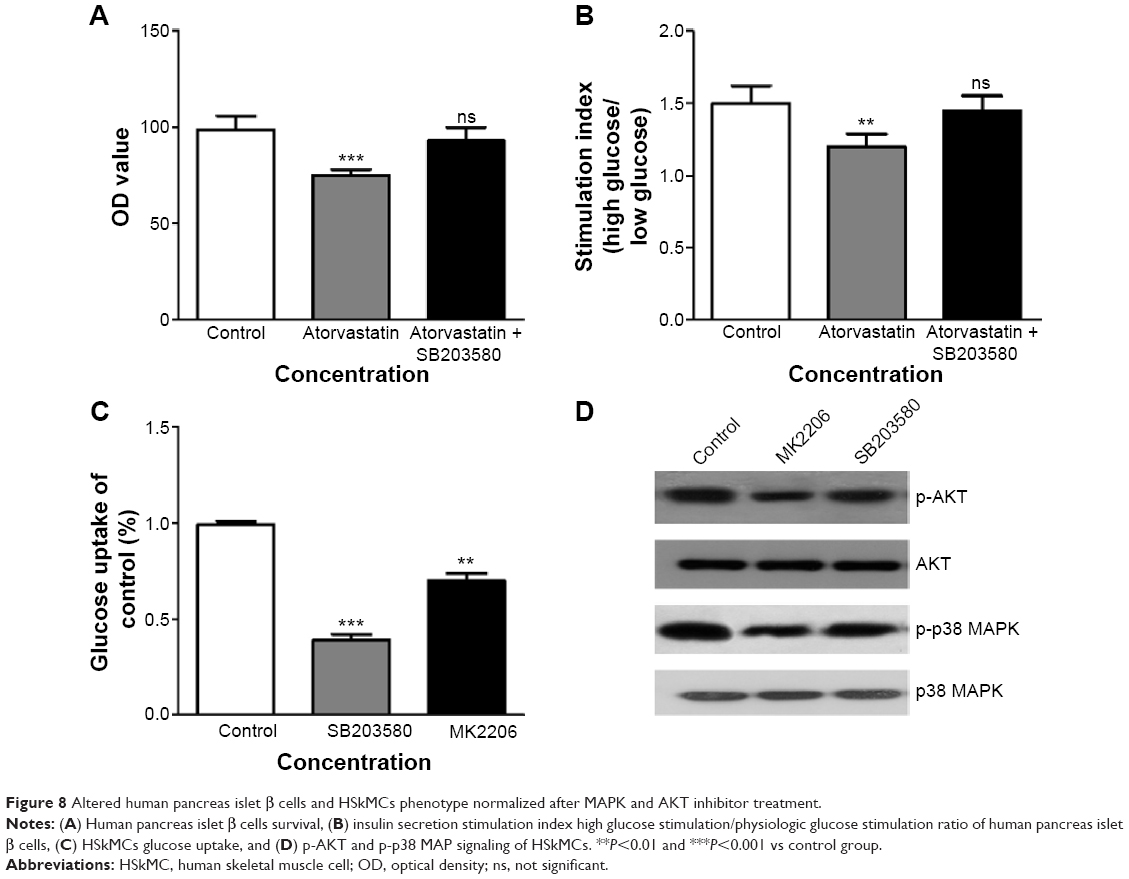 | Figure 8 Altered human pancreas islet β cells and HSkMCs phenotype normalized after MAPK and AKT inhibitor treatment. |
Discussion
Lipophilic statins have pleiotropic actions that might cause unfavorable metabolic effects, such as reduction of insulin secretion and exacerbation of insulin resistance.16–18 Recent large-scale RCTs have raised the possibility that lipophilic statins might increase the rate of new onset diabetes.19–21 Specifically, in the Heart Protection Study, 335 subjects developed diabetes in the simvastatin group, whereas 293 subjects developed diabetes in the placebo group (hazard ratio [HR] =1.15, 95% CI=0.98–1.35, P=0.10).7 In the Anglo-Scandinavian Cardiac Outcomes Trial, the atorvastatin group developed diabetes with an HR of 1.15 (95% CI=0.91–1.44). In the JUPITER trial, rosuvastatin (20 mg) significantly increased the rate of onset of new diabetes (3.0% vs 2.4%, P=0.01) with a significant increase in HbA1c (5.9% vs 5.8%, P=0.001).21 Another meta-analysis of RCTs suggested potential differences between individual statins, with pravastatin showing a trend toward a reduction in risk (risk ratio [RR] =0.84; 95% CI=0.86–1.49) and atorvastatin, rosuvastatin, and simvastatin demonstrated a significant increase in risk (RR=1.14; 95% CI=1.02–1.28) versus placebo.22
A randomized, single-blind, placebo-controlled parallel study showed that despite beneficial reductions in LDL-C and Apo B levels after a 2-month treatment of atorvastatin, atorvastatin resulted in significant increased fasting insulin and HbA1c levels, consistent with insulin resistance and increased ambient glycemia in hypercholesterolemic patients.23 In the present study, we found 100 nM atorvastatin, pravastatin, and rosuvastatin treatment caused more significant statin-induced cytotoxicity in human pancreas islet β cells and statin-reduced insulin secretion compared with pitavastatin (Figures 1 and 9). Statin treatment (all four statins) reduced GSIS under high glucose (28 mmol/L) and physiologic conditions (5.6 mmol/L), with a greater decrease under high glucose conditions. Inhibition of GSIS was greatest in the pravastatin treatment group and lowest in the pitavastatin treatment group under high glucose and physiologic conditions.
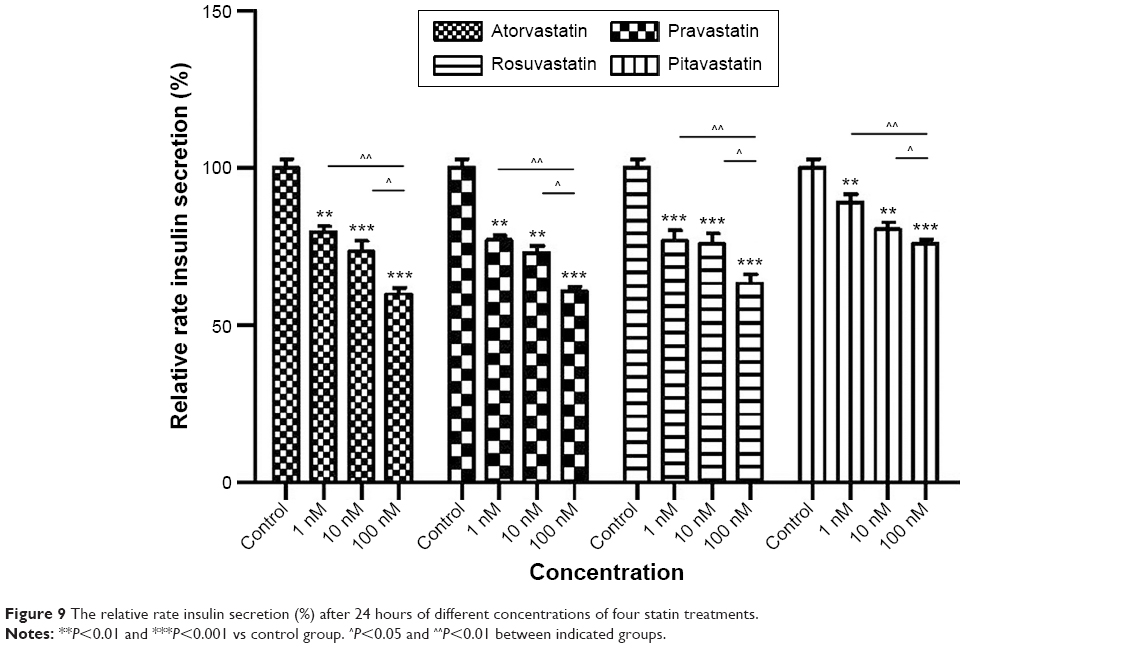 | Figure 9 The relative rate insulin secretion (%) after 24 hours of different concentrations of four statin treatments. |
One issue has to be discussed is whether there was a potential association between the pancreatic cell toxicity and decreased insulin secretion. First, we did not find a significant correlation between the decreased pancreatic cell survival rate and the decrease insulin secretion in all the four statins at any dose (all P>0.05). Besides, another evidence that does not support the correlation between pancreas β cell death and decreased insulin secretion was that low-dose statin treatment (1 nM) did not show a significant decrease survival rate (Figure 9), while the insulin secretion rate was already significantly decreased (Figure 1, 1 nM columns). In other words, these data suggested a decreased insulin secretion prior to the cell viability decrease or possibly cell death. This indicates a dosage window of a potential decreased insulin secretion without the confounding effect of decreased cell viability. However, we admit that at a high dose of statin use (100 nM), it is hard to differentiate or distinguish the effect of decreased insulin secretion due to pancreatic cell dysfunction from the decreased pancreatic cell viability.
In this study, we found a more profound effect of statin on decreased pancreas β cell survival rate at higher dose than at low or median dose, suggesting a dose-dependent effect of statin on cell survival. Several studies and reviews have investigated and studied the adverse effect of statin.5,6 Most of the clinical studies or meta-analysis suggested that the side effect of statin including statin-induced diabetes takes place when patients take a relatively high dose of statin. Thus, the data from both ours and other studies have suggested the adverse effect of statins as a dose-dependent fashion.24,25
In glucose uptake experiment, we found little effect of pitavastatin on glucose uptake by HSkMCs at a low dose (1 nM) compared with other statins at this dose. A study carried out by Kawai et al could help to explain such finding. Kawai et al reported in their study that pitavastatin actually increased glucose uptake in KKAy mice.26 Although the exact mechanism has not been further unraveled in this human study, relatively increased glucose uptake compared with other statins may contribute to the effect. On the other hand, pharmacodynamics of different statin could be different due to the difference of ring structure and substituents although statins share similar molecular structure, granting some of them characteristics of hydrophobic and others hydrophilic. Therefore, the equivalent effective dose of different statins varies. It is possible that pitavastatin at the concentration of 1 nM is not sufficient enough or as effective as other statins in reducing the glucose uptake.
The influence on human pancreatic islet β cell proliferation was determined by cell cycle analysis using a Calibur flow cytometer. Induction of G0/G1 phase cell cycle arrest was dose-dependent and similar with all four statins. Atorvastatin induced G0/G1 phase cell cycle arrest at a low concentration (1 nM), whereas the G0/G1 DNA content was higher in the treated cells compared with control treatment in both cell lines. At a higher concentration (100 nM) for all four statins, sub-G0/G1 population cells appeared in approximately 80% of the DNA histograms, indicating the presence of apoptotic cells in both cell lines.
Thus, the beneficial effects of statins must have begun before the development of prediabetes and metabolic syndrome. Most observational studies in humans that reported an enhanced risk of diabetes were conducted in older subjects in whom statins were started very late in life.27,28 Even in the prospective intervention trial (JUPITER), the mean age of subjects at entry was 66 years, when statins were started.29 It would thus appear that for significant protection against T2DM, statins must be started early in life, and late introduction may not be associated with any beneficial effects.
We speculate on three important mechanisms that collectively account for statin-induced T2DM. First, there is a significant reduction in insulin secretion and GSIS on human pancreas islet β cells, suggesting that the statin-related effects on glucose tolerance are mediated by favorable changes in insulin sensitivity through GLUT-2 and p-p38 MAPK suppression. Second, statins reduce glucose uptake at different levels in HSkMCs through GLUT-4, p-GSK-3β, p-p38 MAPK, and p-AKT suppression. Third, high concentrations of statins induce human pancreas islet β cell apoptosis to further reduce insulin secretion, suggesting that a possible mechanism underlying the preventive role of statins in the development of T2DM may be an ability to preserve β cell function.
Another possible mechanism which may have contributed to a reduction in the risk for T2DM is the anti-inflammatory effects. Although we did not measure inflammatory markers in this study, there is an existing study, which clearly demonstrated that statins reduce inflammation.30 Inflammation has been associated with an increased risk of T2DM in animals31 and humans;32 however, further studies are required to elucidate the precise mechanism by which statins exert a protective effect in reducing the risk for T2DM.
Statin treatment in this study showed a dose-dependent decrease of pancreatic β cells survival rate, suggesting a possible toxic effect. One would raise the question whether long-term or high-dose statin therapy in clinical practice to lower the LDL level as much as possible could lead to the pancreatic cell toxicity, pancreas-related adverse effect, or even degeneration of the pancreas. There have been several studies reporting the association between low or high dose of statin and acute pancreatitis.33,34 However, a larger scale meta-analysis reported a contradictory result.34 Preiss et al reported that stain therapy was associated with a lower risk of pancreatitis in patients with normal or mildly elevated triglyceride levels.34 Similarly, contradictory results also exist regarding the association between statin usage and pancreatic cancer.35,36 Therefore, based on the clinical studies without mechanistic investigation, it is hard to conclude the effect of long-term high-dose statin treatment in pancreas pathology.
Last but not least, the finding of the potential risk of developing diabetes in patients receiving statin therapy, together with the data from our in vitro experiment, both raise the question whether the clinical practice guideline should be changed due to this potential risk. Several studies support the idea that statin should still be recommended in patients with high risk of having cardiovascular events despite of a potential risk of developing diabetes.8,20,22–24 First, although statins used at a certain dosage such as atorvastatin at 80 mg and simvastatin at 80 mg were associated with increased risks of diabetes mellitus, these statins still had a 5%–22% reduction in the cardiovascular events in a long run indicating a beneficial effect.34 Besides, there has been evidence suggesting that no matter with and without diabetes, people of increased cardiovascular risk have been substantial benefited from statin therapy with decreased major cardiovascular events and overall mortality.37 Since most clinical studies focusing on the risk of developing diabetes due to statin therapy has been only short-term follow-ups, the long-term possibility of statin-induced diabetes mellitus is still unknown.1,3 Therefore, it would be too harsh for physicians to stop recommending statin in reducing cardiovascular event for patients with high risk, based on the beneficial effects of statins. Further, the mechanism of stain-induced diabetes is still far from clear which need intense basis research and long-term clinical investigation. Therefore, before seeing more solid evidence supporting that the adverse effect of statin has overweight the beneficial effects, the retraction of statin therapy in patients with high risk should not be recommended, although patients of the potential risk should be informed of the potential risk of diabetes when using a high dose of statin.
Conclusion
The balance of evidence available suggests that statins are associated with an increased risk for T2DM and that there does appear to be a dose effect, with the risk of new-onset T2DM increasing with higher doses of statin therapy. Given the lifesaving benefits of statins in reducing cardiovascular disease events, the small absolute risk for the development of T2DM is clearly outweighed by the benefits for the majority of individuals for whom statin therapy is recommended (ie, people with existing cardiovascular disease or at moderate-to-high risk of such disorders) and clinical practice in these patients should not change. Although it is intuitive that higher doses of statins produce slightly higher adverse effects, a mechanism underlying the increased risk for T2DM has not been established. Nevertheless, the available data suggest a biological platform by which cholesterol may have both positive and negative effects on insulin secretion processes. Further research is required to determine the mechanisms by which statins impair glucose metabolism. It is possible that the risk for new-onset T2DM is associated with high-risk genotypes/phenotypes, thus there is a need to resolve this issue in adequately designed trials, and/or by utilizing existing databases with detailed data on insulin secretion and genetic information.
Disclosure
The authors report no conflicts of interest in this work.
References
Naveed AS, Henry G, Kausik R, et al. The use of statins in people at risk of developing diabetes mellitus: evidence and guidance for clinical practice. Atheroscler Suppl. 2014;15:1–15. | ||
Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. | ||
Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomized statin trials. Lancet. 2010;375:735–742. | ||
Sattar N, Taskinen MR. Statins are diabetogenic-myth or reality? Atheroscler Suppl. 2012;13:1–10. | ||
Navarese EP, Buffon A, Andreotti F, et al. Meta-analysis of impact of different types and doses of statins on new-onset diabetes mellitus. Am J Cardiol. 2013;111:1123–1130. | ||
Baker WL, Talati R, White CM, et al. Differing effect of statins on insulin sensitivity in non-diabetics: a systematic review and meta-analysis. Diabetes Res Clin Pract. 2010;87:98–107. | ||
Supale S, Li N, Brun T, et al. Mitochondrial dysfunction in pancreatic beta cells. Trends Endocrinol Metab. 2012;23:477–487. | ||
Sampson UK, Linton MF, Fazio S. Are statins diabetogenic? Curr Opin Cardiol. 2011;26:342–347. | ||
Koh KK, Sakuma I, Quon MJ, et al. Differential metabolic effects of distinct statins. Atherosclerosis. 2011;215:1–8. | ||
Koeks J, Schrauwen P. Muscle mitochondria and insulin resistance: a human perspective. Trends Endocrinol Metab. 2012;23:444–450. | ||
Kusminski CM, Scherer PE. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol Metab. 2012;23:435–443. | ||
Rosenbaum D, Dallongeville J, Sabouret P, et al. Discontinuation of statin therapy due to muscular side effects: a survey in real life. Nutr Metab Cardiovasc Dis. 2012;23:871–875. | ||
Draeger A, Monastyrskaya K, Mohaupt M, et al. Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. J Pathol. 2006;210:94–102. | ||
Golomb BA, Evans MA, Dimsdale JE, et al. Effects of statins on energy and fatigue with exertion: results from a randomized controlled trial. Arch Intern Med. 2012;172:1180–1182. | ||
Ciaraldi TP, Abrams L, Nikoulina S, Mudaliar S, Henry RR. Glucose transport in cultured human skeletal muscle cells. Regulation by insulin and glucose in nondiabetic and non-insulin-dependent diabetes mellitus subjects. J Clin Invest. 1995;96:2810–2827. | ||
Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol. 1999;126:1205–1213. | ||
Kanda M, Satoh K, Ichihara K. Effects of atorvastatin and pravastatin on glucose tolerance in diabetic rats mildly induced by streptozotocin. Biol Pharm Bull. 2003;26:1681–1684. | ||
Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycemic control. Diabetologia. 2006;49:1881192. | ||
Collins R, Armitage J, Parish S, Sleigh P, Peto R, Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5,963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361:2005–2016. | ||
Sever PS, Dahlof B, Poulter NR, et al. Prevention of coronary and stroke events with atrovastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361:1149–1158. | ||
Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. | ||
Coleman CI, Reinhart K, Kluger J, With CM. The effect of statins on the development of new-onset type 2 diabetes: a meta-analysis of randomized controlled trials. Curr Med Res Opin. 2008;24:1359–1362. | ||
Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol. 2010;55:735–1097. | ||
Silva MA, Swanson AC, Gandhi PJ, et al. Statin-related adverse events: a meta-analysis. Clin Ther. 2006;28(1):26–35. | ||
Silva MA, Mattews ML, Jarvis C, et al. Meta-analysis of drug-induced adverse events associated with intensive-dose statin therapy. Clin Ther. 2007;29(2):253–260. | ||
Kawai Y, Sato-Ishida R, Motoyama A, et al. Place of pitavastatin in the statin armamentarium: promising evidence for a role in diabetes mellitus. Drug Des Devel Ther. 2011;5:283–297. | ||
Waters DD, Ho JE, DeMicco DA, et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol. 2011;57:1535–1545. | ||
Sheperd J, Blauw CJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomized controlled trial. Lancet. 2002;360:1623–1630. | ||
Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. New Engl J Med. 2008;359:2195–2207. | ||
Furuya DT, Poletto AC, Favaro RR, Martins JO, Zorn TM, Machado UF. Anti-inflammatory effect of atorvastatin ameliorates insulin resistance in monosodium glutamate-treated obese mice. Metabolism. 2010;59:395–399. | ||
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–614. | ||
Spranger J, Kroke A, Mohlig M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–817. | ||
Singh S, Loke YK. Statins and pancreatitis: a systematic review of observational studies and spontaneous case reports. Drug Saf. 2006;29(12):1123–1132. | ||
Preiss D, Tikkanen MJ, Welsh P, et al. Lipid–modifying therapies and risk of pancreatitis: a meta-analysis. JAMA. 2012;308(8):804–811. | ||
Cui X, Xie Y, Chen M, et al. Statin use and risk of pancreatic cancer: a meta-analysis. Cancer Causes Control. 2012;23(7):1099–1111. | ||
Gbelcova H, Lenicek M, Zelenka J, et al. Differences in antitumor effects of various statins on human pancreatic cancer. Int J Cancer. 2008;122(6):1214–1221. | ||
Ballantyne CM, Olsson AG, Cook TJ, et al. Influence of low high-density lipoprotein cholesterol and elevated triglyceride on coronary heart disease events and response to simvastatin therapy in 4S. Circulation.2001;104:3046–3051. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.