")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Dexmedetomidine protects H9c2 cardiomyocytes against oxygen-glucose deprivation/reoxygenation-induced intracellular calcium overload and apoptosis through regulating FKBP12.6/RyR2 signaling
Authors Yuan M, Meng XW , Ma J, Liu H , Song SY, Chen QC, Liu HY, Zhang J , Song N, Ji FH , Peng K
Received 22 June 2019
Accepted for publication 23 August 2019
Published 2 September 2019 Volume 2019:13 Pages 3137—3149
DOI https://doi.org/10.2147/DDDT.S219533
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Mei Yuan,1,2,* Xiao-Wen Meng,1,* Jiao Ma,1,* Hong Liu,3 Shao-Yong Song,1 Qing-Cai Chen,1 Hua-Yue Liu,1 Juan Zhang,1 Nan Song,1 Fu-Hai Ji,1 Ke Peng1
1Department of Anesthesiology, First Affiliated Hospital of Soochow University, Suzhou, Jiangsu 215006, People’s Republic of China; 2Department of Anesthesiology, Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou, Jiangsu 215008, People’s Republic of China; 3Department of Anesthesiology and Pain Medicine, University of California Davis Health System, Sacramento, CA 95817, USA
Correspondence: Fu-Hai Ji; Ke Peng
Department of Anesthesiology, First Affiliated Hospital of Soochow University, 188 Shizi Street, Suzhou, Jiangsu 215006, People’s Republic of China
Tel +86 5 126 778 0055; +86 5 126 778 0159
Email [email protected]; [email protected]
*These authors contributed equally to this work
Purpose: Intracellular calcium ([Ca2+,]i) overload is a major cause of cell injury during myocardial ischemia/reperfusion (I/R). Dexmedetomidine (DEX) has been shown to exert anti-inflammatory and organ protective effects. This study aimed to investigate whether pretreatment with DEX could protect H9c2 cardiomyocytes against oxygen-glucose deprivation/reoxygenation (OGD/R) injury through regulating the Ca2+, signaling.
Methods: H9c2 cardiomyocytes were subjected to OGD for 12 h, followed by 3 h of reoxygenation. DEX was administered 1 h prior to OGD/R. Cell viability, lactate dehydrogenase (LDH) release, level of [Ca2+,]i, cell apoptosis, and the expression of 12.6-kd FK506-binding protein/ryanodine receptor 2 (FKBP12.6/RyR2) and caspase-3 were assessed.
Results: Cells exposed to OGD/R had decreased cell viability, increased LDH release, elevated [Ca2+,]i level and apoptosis rate, down-regulated expression of FKBP12.6, and up-regulated expression of phosphorylated-Ser2814-RyR2 and cleaved caspase-3. Pretreatment with DEX significantly blocked the above-mentioned changes, alleviating the OGD/R-induced injury in H9c2 cells. Moreover, knockdown of FKBP12.6 by small interfering RNA abolished the protective effects of DEX.
Conclusion: This study indicates that DEX pretreatment protects the cardiomyocytes against OGD/R-induced injury by inhibiting [Ca2+,]i overload and cell apoptosis via regulating the FKBP12.6/RyR2 signaling. DEX may be used for preventing cardiac I/R injury in the clinical settings.
Keywords: dexmedetomidine, H9c2 cardiomyocytes, oxygen-glucose deprivation/reoxygenation, apoptosis, intracellular calcium overload, FKBP12.6/RyR2
Introduction
Myocardial ischemia/reperfusion (I/R) injury is a major contributing factor of morbidity and mortality in patients with ischemic heart disease or after cardiac surgery.1,2 Studies identified various molecular and cellular mechanisms involved in the process of myocardial I/R.3–7 Of these, intracellular calcium [Ca2+]i overload is a critical one that leads to cardiomyocyte apoptosis and cell death.8–10
Dexmedetomidine (DEX) is a highly selective α2-adrenoceptor agonist with sedative, anxiolytic, analgesic, and sympatholytic properties.11,12 Our previous studies showed that DEX use was associated with improved outcomes and reduced mortality in patients undergoing cardiac surgery.13,14 In a rat myocardial I/R model, DEX administration prior to ischemia (pretreatment) reduced the myocardial infarct size by inhibiting inflammation via the α2-adrenergic receptor activation.15 DEX was also shown to inhibit muscarine-induced [Ca2+]i elevation in the cultured dorsal root ganglion (DRG) cells via the muscarinic type 3 receptors.16 However, whether DEX exerts cardiac protection through regulating the Ca2+ signaling is unclear.
The 12.6-kd FK506-binding protein (FKBP12.6, also known as calstabin-2), a member of the FKBPs family, regulates Ca2+ release from cardiac sarcoplasmic reticulum (SR) via the interaction with cardiac ryanodine receptor 2 (RyR2).17,18 In pathological conditions, such as heart failure, binding of FKBP12.6 to RyR2 was evidently reduced.4 Besides, He et al found a remarkably decreased ratio of FKBP12.6/RyR2 and an increased level of [Ca2+]i in myocardial infarction rats.19
Of note, RyR2 phosphorylation is a critical regulator of the channel function.20 Phosphorylation of serine 2814 (Ser2814) on cardiac RyR2 was significantly increased in a time-dependent manner following reperfusion in wild-type mice, while knock-in mice with an inactivated RyR2 Ser2814 phosphorylation site had improved outcomes after reperfusion.21,22 Studies also revealed that the regulation of Ca2+ in SR and cytoplasm was associated with the apoptotic signaling pathway.12,23,24 In the apoptotic process, caspase-3 is known as an executioner that functions in DNA fragmentation and degradation of cytoskeletal proteins.25
In this study, the effect of DEX pretreatment on Ca2+ signaling and apoptosis in H9c2 cardiomyocytes during oxygen-glucose deprivation/reoxygenation (OGD/R) was investigated. We hypothesized that DEX could protect the cells against OGD/R-induced injury through alleviating [Ca2+]i overload and inhibiting caspase-3 dependent apoptosis via the FKBP12.6/RyR2 signaling pathway.
Materials and methods
Cell culture
H9c2 cardiomyocytes, a rat embryonic heart-derived cell line (Shanghai Institute of Life Sciences, Chinese Academy of Sciences, Shanghai, China), were cultured in Dulbecco’s modified Eagle’s medium (DMEM, HyClone, USA) supplemented with 10% fetal bovine serum (FBS, Biological Industries, Israel), 100 U/mL penicillin, and 100 mg/mL streptomycin. Cells were grown in an incubator containing 5% CO2 at 37 °C for at least 24 h. Cells of 70–80% confluence were used for the experiments.
OGD/R model
H9c2 cardiomyocytes were treated with glucose-free DMEM and incubated at 37 °C in a hypoxia chamber (95% N2 and 5% CO2), as previously described.26 Cells were subjected to OGD for 12 h, and then cultured in the medium replaced by normal DMEM for 3 h of reoxygenation.
Experimental protocols
To determine the optimal concentration of DEX (Jiangsu Hengrui Medicine Co., Ltd, Jiangsu, China) against OGD/R, cells were randomly divided into 5 groups: the control group (cells were cultured in DMEM with 10% FBS), the OGD/R group (cells underwent OGD/R), and 3 DEX groups (cells were treated with 0.1, 1, and 10 μM DEX for 1 h prior to OGD/R). The concentrations of DEX were based on our previous study.27
To investigate the effects of FKBP12.6 knock-down by small interfering RNA (siRNA) (si-FKBP12.6; GenePharma, Shanghai, China) on H9c2 cardiomyocytes under OGD/R, cells were divided into 6 groups: the control group, the OGD/R group, the D1+OGD/R group (1 μM DEX prior to OGD/R), the si-FKBP12.6+D1+OGD/R group, the vehicle+D1+OGD/R group, and the mismatch+D1+OGD/R group. In the si-FKBP12.6+D1+OGD/R group, cells were transfected with FKBP12.6 siRNA using the RNAifectinTM (20 nM siRNA in 1 μL RNAifectinTM; Applied Biological Materials, Richmond, BC, Canada) for 24 or 48 h before DEX treatment. In the vehicle+D1+OGD/R group, only 1 μL RNAifectinTM was applied. In the mismatch+D1+OGD/R group, cells were transfected with negative control sequences using RNAifectinTM. The siRNA oligo sequences are shown in Table 1.
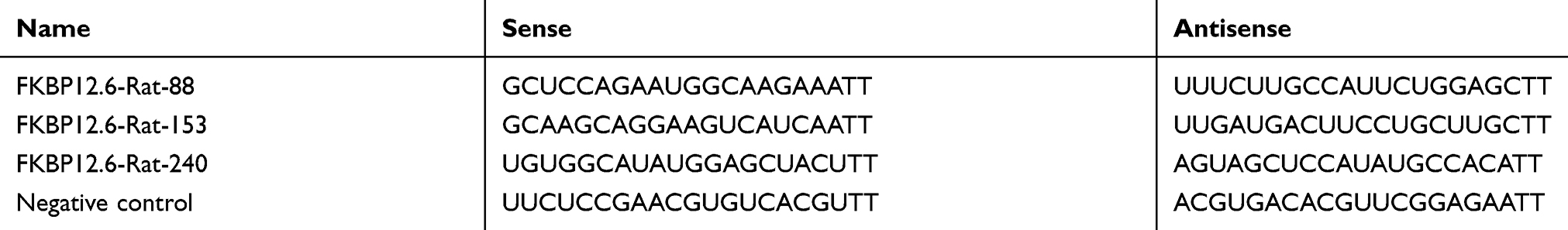 |
Table 1 siRNA oligo sequences |
Cell viability assay
Cell viability was measured by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (M2128, Sigma Chemical Co. St. Louis, MO, USA) assay, as previously described.28 Briefly, cells were cultured in 96-well plates at a density of 1×104 cells per well. At the end of oxygenation, 20 μL MTT solution (5 mg/mL) was added to each well for an additional incubation of 4 h at 37 °C. Then, 150 μL dimethyl sulfoxide (DMSO) (D4540, Sigma, St. Louis, MO, USA) was added and formazan crystals were dissolved in a shaker for 10 min. The absorbance value at 570 nm was measured with a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Lactate dehydrogenase release assay
Cytotoxicity was measured by using the lactate dehydrogenase (LDH) Assay kit (ab65393, Abcam, Cambridge, UK). At the end of oxygenation, cell cultures were centrifuged at 600×g for 10 min. The supernatant (10 μL/well) were extracted into another transparent 96-well plate and incubated with 100 μL LDH reaction agent at room temperature for 30 min. The absorbance value at 490 nm was measured with the microplate reader.
Quantitative real-time PCR
Total RNA was extracted with Trizol reagent (Invitrogen, Carlsbad, CA, USA). The reverse transcription was performed using Thermo Scientific RevertAid First Strand cDNA synthesis kit (Thermo Fisher scientific, Waltham, MA, USA). Quantitative real-time PCR (RT-qPCR) was conducted with EvaGreen qPCR MasterMix (Applied Biological Materials, Richmond, BC, Canada) in a 20 μL reaction volume using an ABI Prism 7500 Sequence Detection System (Applied Biosystems, Bedford, MA, USA). Amplification process included a pre-incubation at 95 °C for 5 min, followed by amplification of the target gene for 40 cycles (for each cycle: denaturation at 94 °C for 30 s, annealing at 62 °C for 30 s, and elongation at 72 °C for 32 s). The expression of GAPDH was used for normalization. Three replicates were tested for each sample. The primer oligo sequences are shown in Table 2.
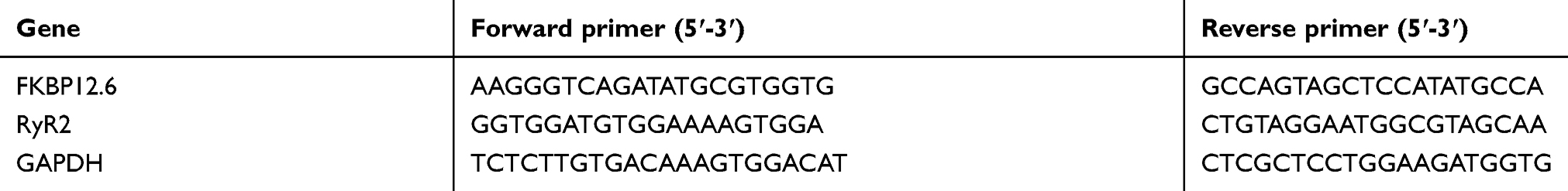 |
Table 2 Primer oligo sequences |
Western blots
Total protein was extracted and the protein concentration was determined using a bicinchoninic acid reagent kit (Beyotime, Shanghai, China), as previously described.27 After electrophoresis on polyacrylamide gels (Bio-Rad, California, USA), proteins were transferred to polyvinylidene difluoride (PVDF) membranes at 200 mA at 4 °C for 2 h. Then, the membranes were blocked for 2 h at room temperature, and incubated with the following primary antibodies at 4 °C overnight: mouse anti-FKBP12.6 (1:1000, Santa Cruz Biotechnology, CA, USA), rabbit anti-RyR2 (1:1000, Signalway Antibody LLC, MD, USA), rabbit anti-pSer2814-RyR2 (1:5000, Badrilla, Leeds, Yorkshire, UK), rabbit anti-Caspase-3 (1:1000, Cell Signaling Technology, Beverly, MA, USA), and mouse anti-β-actin (1:1000, Cell Signaling Technology, Beverly, MA, USA). Then, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:5000, Santa Cruz Biotechnology, CA, USA) for 2 h at room temperature. Bands were visualized by a ChemiDocTM XRS+ System (Bio-Rad, Richmond, CA) with an enhanced chemiluminescence kit (ECL, Beyotime, Shanghai, China). The protein expression was normalized to β-actin as a control.
Intracellular calcium analysis
Level of [Ca2+]i was measured using the Fluo-3/AM staining (39294-1ML, Sigma, St. Louis, MO, USA) and Pluronic F127 (Sigma). To obtain the working solution, Pluronic F127 was added to Fluo-3/AM, which was diluted with Hanks’ solution (Ca2+-free). The final concentration was 10 μM for Fluo-3/AM and 0.05% (w/v) for Pluronic F127. At the end of oxygenation, cells were incubated with working solution in dark environment at 37 °C for 30 min. Cells were then washed with Ca2+-free Hanks’ solution for three times. After incubation at 37 °C for 30 min again, fluorescence intensity was determined by a microscope (Live Cell Imaging CellR, Olympus, Tokyo, Japan).
Hoechst staining
Cells were fixed with 4% formaldehyde for 10 min at room temperature, washed with phosphate-buffered saline (PBS) for three times, and incubated with 20 mmol/L Hoechst 33258 (Sigma) for 10 min at a dark room. The apoptotic cells with concentrated and fragmented bright blue nuclei were determined using a fluorescence microscopy (Olympus, Tokyo, Japan). Images were captured from random fields and data were presented as percentage of apoptotic cells to total cells.
Statistical analyses
Statistical analyses were performed using the GraphPad Prism software (version 7.0, GraphPad, San Diego, CA, USA). Due to the small samples, the Shapiro–Wilk test was used for normality check.29 Normally distributed data were expressed as mean ± standard error of the mean and compared using one-way or two-way analysis of variance followed by Bonferroni or Dunnet post-test as appropriate, whereas skewed data were expressed as median (interquartile range) and compared with non-parametric Mann–Whitney U test. A two-tailed value of P<0.05 indicates a statistically significant difference.
Results
DEX attenuated H9c2 cell injury induced by OGD/R
In the OGD/R group, H9c2 cells lost the normal shape. Specifically, cells became small and round, with decreased cell refraction. Pretreatment with 0.1, 1, and 10 μM DEX improved cell morphology (Figure 1A). MTT and LDH release assays were then used to measure the cell viability and cytotoxicity. OGD/R exposure significantly reduced cell viability (mean =52.9% of control, P<0.01) and increased LDH release (mean =205.6% of control, P<0.01), which was alleviate by 1 and 10 μM DEX (cell viability: mean =76.2% and 75.7% of control, P<0.01; LDH: mean =138.0% and 138.8% of control, P<0.01) (Figure 1B and C). The use of 0.1 μM DEX was less effective, and the effect of 1 μM DEX was comparable to that of 10 μM. Based on these results, 1 μM DEX was used in the following experiments.
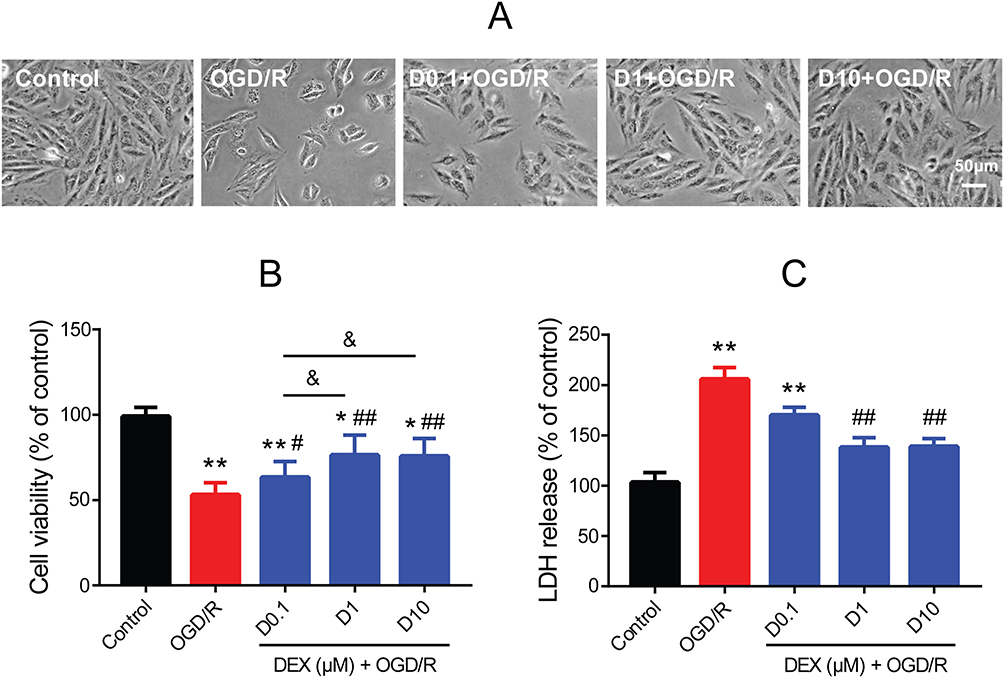 |
Figure 1 Effect of DEX pretreatment on cell viability and cytotoxicity in H9c2 cells after OGD/R. (A) Morphological changes after OGD/R with or without DEX. Scale bar =50 μm. (B) Cell viability by MTT assay (n=8). (C) Cytotoxicity by LDH release assay (n=8). *P<0.05, **P<0.01 vs the control group; #P<0.05, ##P<0.01 vs the OGD/R group; &P<0.05 for the comparisons shown. |
DEX inhibited [Ca2+]i overload in H9c2 cells during OGD/R
By using the Fluo-3/AM, a membrane permeable fluorescent Ca2+ indicator, a significantly elevated [Ca2+]i level was observed in the OGD/R group compared to the control group (mean fold change =1.76, P<0.01). Both 1 and 10 μM DEX pretreatment effectively blocked the increase in [Ca2+]i (mean fold change =1.25 and 1.21, P<0.01), but 0.1 μM DEX did not (Figure 2A and B). There was no significant difference between 1 and 10 μM DEX. Besides, DEX had no impact on [Ca2+]i levels in normal conditions (Figure 2C).
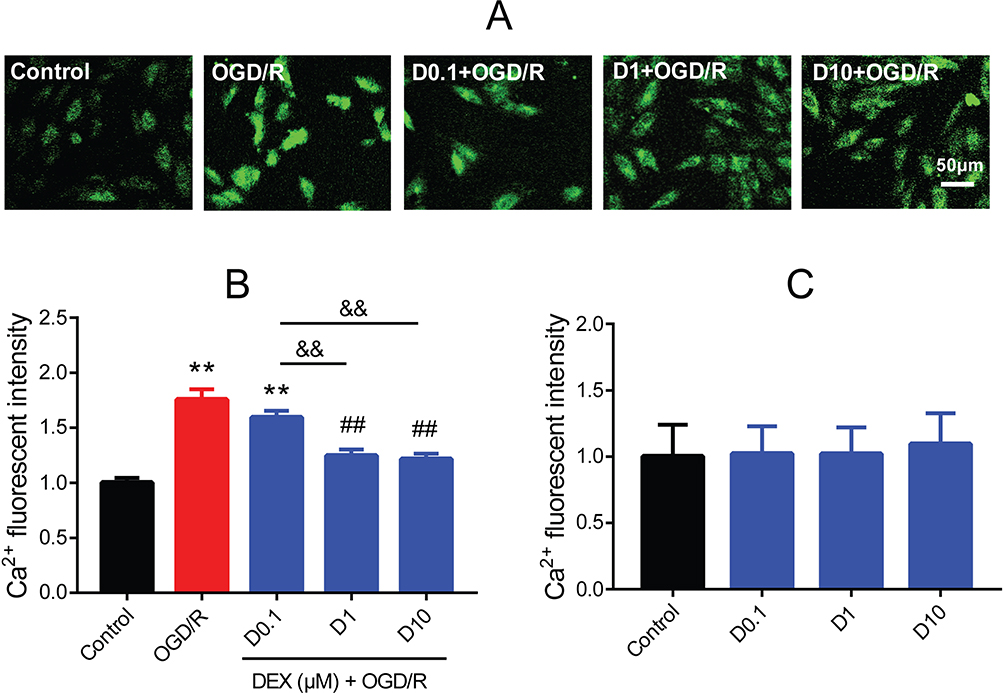 |
Figure 2 Effect of DEX pretreatment on [Ca2+]i of H9c2 cells after OGD/R. (A) Fluo-3/AM (green) showing the [Ca2+]i. Scale bar =50 μm. (B) Fluorescent intensity of [Ca2+]i in the control, OGD/R, and DEX groups (n=5). (C) Fluorescent intensity of [Ca2+]i in normal cells (n=5). **P<0.01 vs the control group; ##P<0.01 vs the OGD/R group; &&P<0.01 for the comparisons shown. |
DEX regulated FKBP12.6/RyR2 expression in H9c2 cells during OGD/R
To investigate whether FKBP12.6 and RyR2 were involved in the DEX-elicited protection against OGD/R in H9c2 cells, the mRNA and protein expression were measured. The RT-qPCR and Western blot results showed significantly down-regulated expression of FKBP12.6 after OGD/R, both in the mRNA and protein levels (mean fold change =0.51 and 0.59, P<0.01), which was reversed by pretreatment with 1 μM DEX (mean fold change =0.98 and 0.83, P<0.01) (Figure 3A–C). The expression of total RyR2 was not significantly altered by OGD/R or DEX (Figure 3D–F). OGD/R exposure significantly increased the expression of phosphorylated-Ser2814-RyR2 (p-S2814-RyR2) and the ratio of p-S2814-RyR2 to total RyR2 (P<0.01), which was blocked by DEX (P<0.05) (Figure 3G and H).
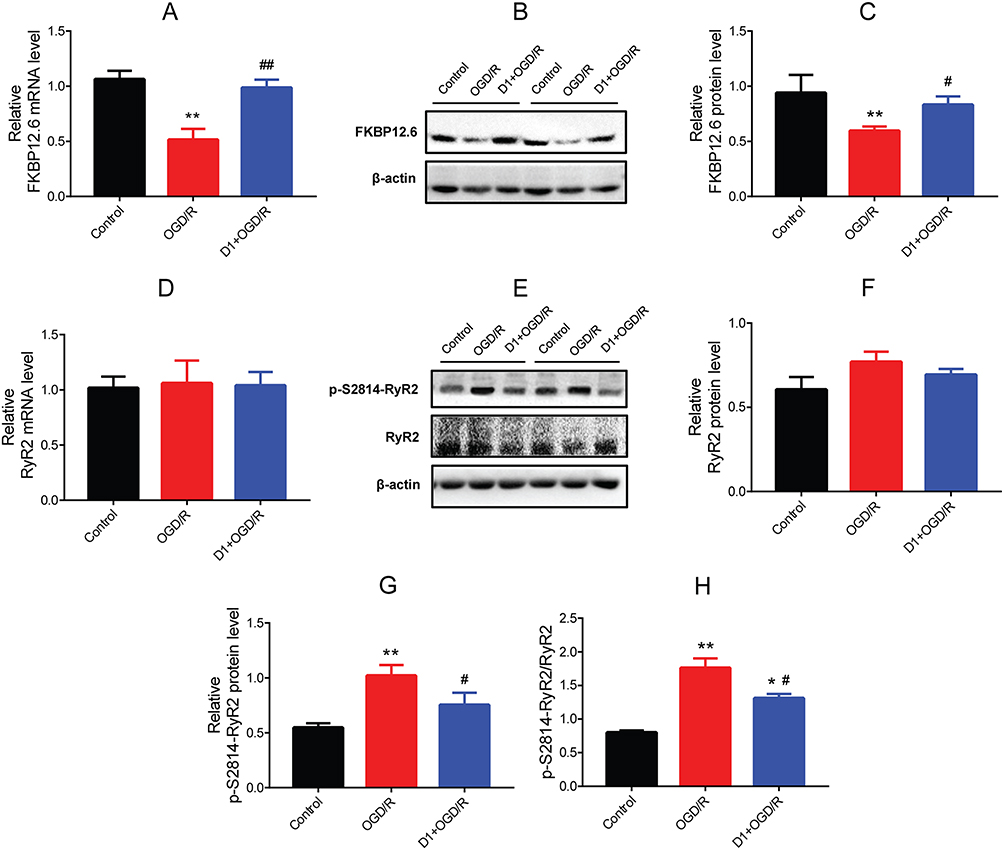 |
Figure 3 Effect of DEX pretreatment on the expression of FKBP12.6/RyR2 in H9c2 cells after OGD/R. (A) RT-qPCR showing FKBP12.6 mRNA level (n=5). (B, C) Western blot showing FKBP12.6 protein expression (n=5). (D) RT-qPCR showing RyR2 mRNA level (n=5). (E–H) Western blot showing p-S2814-RyR2 and total RyR2 protein expression and the ratio of p-S2814-RyR2 to RyR2 (n=5). *P<0.05, **P<0.01 vs the control group; #P<0.05, ##P<0.01 vs the OGD/R group. |
DEX reduced caspase-3 dependent apoptosis in H9c2 cells during OGD/R
The effect of DEX on cell apoptosis during OGD/R was investigated. The Hoechst staining results showed morphological changes typical of apoptosis (nuclear condensation and fragmentation) and elevated apoptosis rate (mean =54.0%, P<0.01) in OGD/R cells. The cell apoptosis was alleviated by pretreatment with 1 and 10 μM DEX (mean =28.3% and 28.6%, P<0.01), but not 0.1 μM DEX (Figure 4A and B). The protein expression of cleaved caspase-3 was up-regulated during OGD/R (P<0.01), while DEX reduced the expression of cleaved caspase-3 and the ratio of cleaved caspase-3 to caspase-3 (P<0.05) (Figure 4C–E). The expression of caspase-3 remained unchanged during the process (Figure 4F).
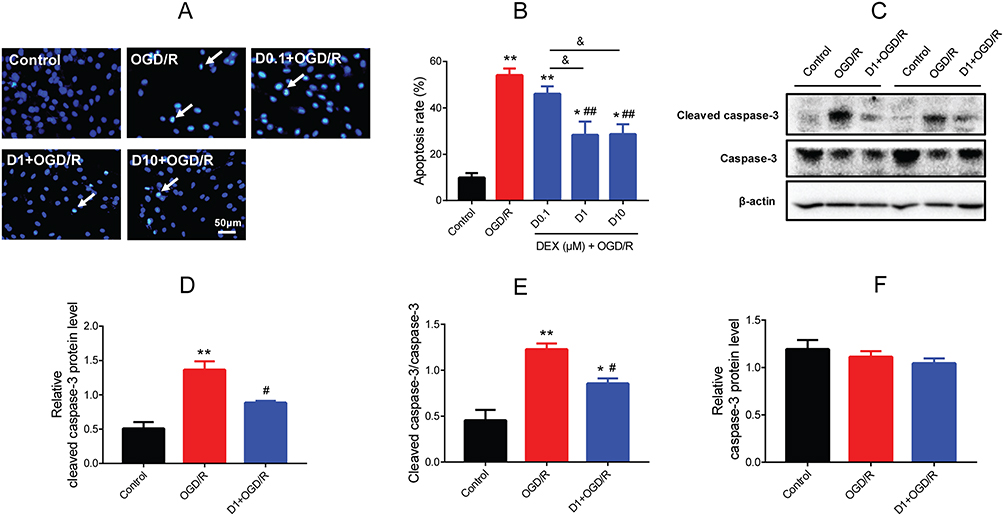 |
Figure 4 Effect of DEX pretreatment on apoptosis in H9c2 cells after OGD/R. (A) Hoechst 33,342 staining showing the apoptotic cells (white arrows). Scale bar =50 μm. (B) Apoptosis rate in the control, OGD/R, and DEX groups (n=5). (C–F) Western blot showing cleaved caspase-3, caspase-3, and the ratio of cleaved caspase-3 to caspase-3 (n=5). *P<0.05, **P<0.01 vs the control group; #P<0.05, ##P<0.01 vs the OGD/R group; &P<0.05 for the comparisons shown. |
Knockdown of FKBP12.6 abolished DEX-elicited protection against OGD/R-induced injury
To investigate the role of FKBP12.6 in the protective effects of DEX, cells were transfected with three different FKBP12.6 siRNAs (si-88, si-153, and si-240) for 24 and 48 h. The siRNAs significantly reduced FKBP12.6 mRNA levels compared to the control, vehicle, or mismatch groups, with better knockdown efficacy for 48 h (mean fold change =0.07 for si-88, 0.15 for si-153, and 0.12 for si-240, P<0.01) (Figure 5A). Next, Western blot results showed that si-88 was the most efficient one to inhibit the protein expression of FKBP12.6 (mean fold change =0.37, P<0.01) (Figure 5B and C). Therefore, transfection with si-88 for 48 h was selected in the subsequent experiments. The use of si-FKBP12.6 reversed the protective effect of DEX pretreatment against OGD/R-induced injury in H9c2 cells, according to the cell viability and LDH release assays (P<0.01) (Figure 5D and E).
 |
Figure 5 Knockdown efficacy of FKBP12.6 siRNAs and their effect on cell viability and cytotoxicity of H9c2 cells after OGD/R. (A) RT-qPCR showing FKBP12.6 mRNA expression after transfection with three siRNAs for 24 and 48 h (n=5). (B, C) Western blot showing FKBP12.6 protein levels after 48 h of transfection (n=5). (D) Cell viability by MTT assay (n=8). (E) Cytotoxicity by LDH release assay (n=8). *P<0.05, **P<0.01 vs the control group of 24 h (A); ##P<0.01 vs the control group of 48 h (A); *P<0.05, **P<0.01 vs the control group (C–E); ##P<0.01 vs the OGD/R group (D, E); &P<0.05 for the comparisons shown, &&P<0.01 for the comparisons shown. |
Knockdown of FKBP12.6 abolished DEX-elicited regulation of Ca2+ signaling during OGD/R
Next, whether pretreatment with DEX inhibited [Ca2+]i overload in H9c2 cells during OGD/R through a direct action on FKBP12.6/RyR2 signaling was assessed. The Fluo-3/AM staining results showed that si-FKBP12.6 significantly blocked DEX-induced inhibition of [Ca2+]i levels (P<0.01) (Figure 6A and B). In addition, DEX pretreatment significantly inhibited the activation of p-S2814-RyR2 and reduced the ratio of p-S2814-RyR2 to total RyR2 (P<0.05), which was reversed by si-FKBP12.6 (P<0.01 for p-S2814-RyR2, P<0.05 for p-S2814-RyR2/RyR2) (Figure 6C–E). The protein expression of total RyR2 showed no significant change (Figure 6F).
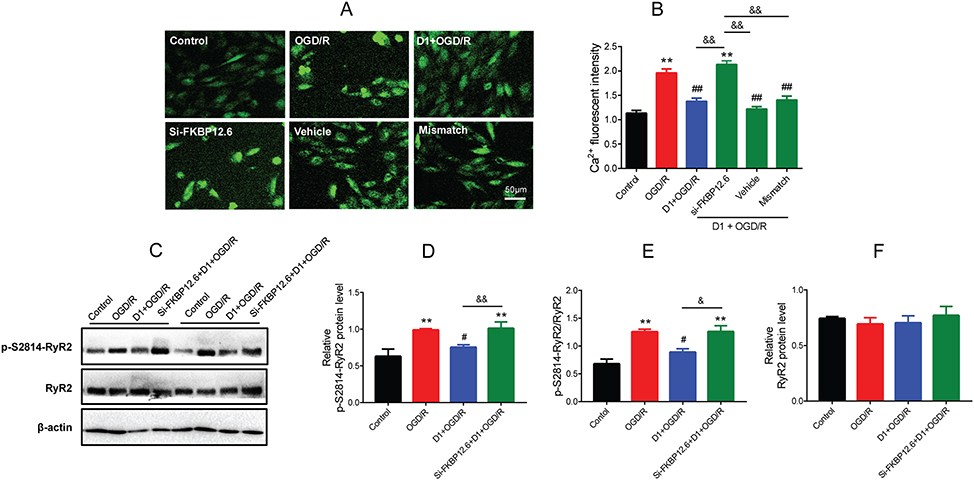 |
Figure 6 Effect of FKBP12.6 knockdown by siRNA on [Ca2+]i and RyR2 expression of H9c2 cells after OGD/R. (A) Fluo-3/AM showing [Ca2+]i levels in cells with si-FKBP12.6 transfection. (B) Fluorescent intensity of [Ca2+]i showing a high level of [Ca2+]i after si-FKBP12.6 transfection (n=5). (C–F) Western blot showing p-S2814-RyR2 and total RyR2 protein expression in cells with si-FKBP12.6 transfection (n=5). **P<0.01 vs the control group; #P<0.05, ##P<0.01 vs the OGD/R group; &P<0.05 for the comparisons shown, &&P<0.01 for the comparisons shown. |
Knockdown of FKBP12.6 abolished DEX-elicited inhibition of cell apoptosis during OGD/R
Furthermore, whether pretreatment with DEX inhibited cell apoptosis via the FKBP12.6/RyR2 signaling pathway was determined. The Hoechst 33258 staining results showed that si-FKBP12.6 eliminated the inhibition of apoptosis by DEX in H9c2 cells (P<0.01) (Figure 7A and B). Next, the effect of si-FKBP12.6 on the caspase-3 signaling was also assessed. DEX significantly reduced the expression of cleaved caspase-3 during OGD/R and the ratio of cleaved caspase-3 to caspase-3 (P<0.05), and these effects were blocked by si-FKBP12.6 (P<0.05) (Figure 7C–E). The expression of caspase-3 did not show significant change throughout the process (Figure 7F).
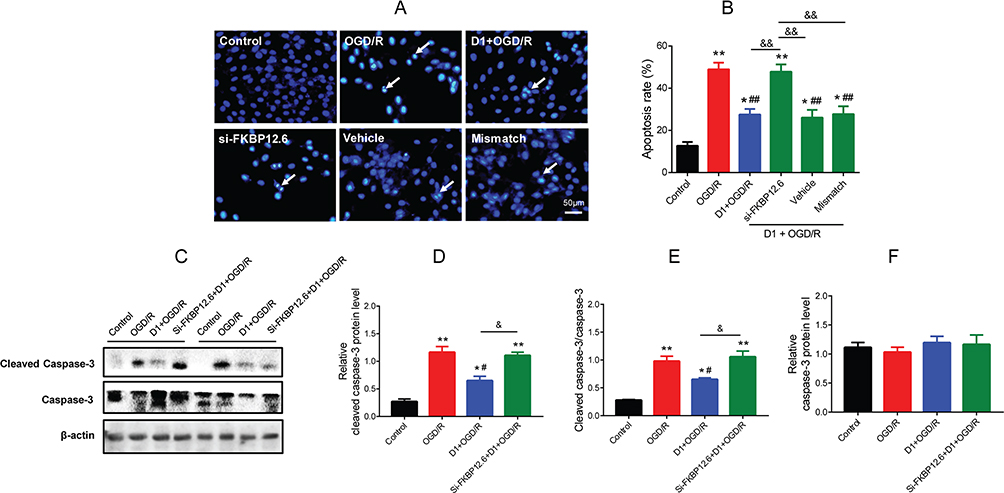 |
Figure 7 Effect of FKBP12.6 knockdown by siRNA on cell apoptosis in H9c2 cells after OGD/R. (A) Hoechst 33,342 staining showing the apoptotic cells (white arrows). (B) Apoptosis rate after si-FKBP12.6 transfection (n=5). (C–F) Western blot showing cleaved caspase-3 and caspase-3 protein expression in cells with si-FKBP12.6 transfection (n=5). *P<0.05, **P<0.01 vs the control group; #P<0.05, ##P<0.01 vs the OGD/R group; &P<0.05 for the comparisons shown, &&P<0.01 for the comparisons shown. |
Discussion
Some possible mechanisms may underlie the DEX-induced protective effects against myocardial I/R injury, including inhibition of inflammation, activation of the eNOS/NO signaling, and maintaining a direct action.15,30,31 It is also highlighted that disordered Ca2+ homeostasis is a leading cause of myocardial I/R injury.8–10,32 DEX has been shown to influence Ca2+ signaling in several types of cells. One study found that DEX alleviated [Ca2+]i overload in rat hippocampal and DRG neurons during cerebral ischemia.33 Another study reported that DEX modulated histamine-induced Ca2+ signaling in cancer cell lines.34 Based on our current findings, this is the first study to demonstrate that pretreatment with DEX protects H9c2 cardiomyocytes against OGD/R-induced injury by inhibiting [Ca2+]i overload and caspase-3 dependent apoptosis through a direct regulation of the FKBP12.6/RyR2 signaling (Figure 8).
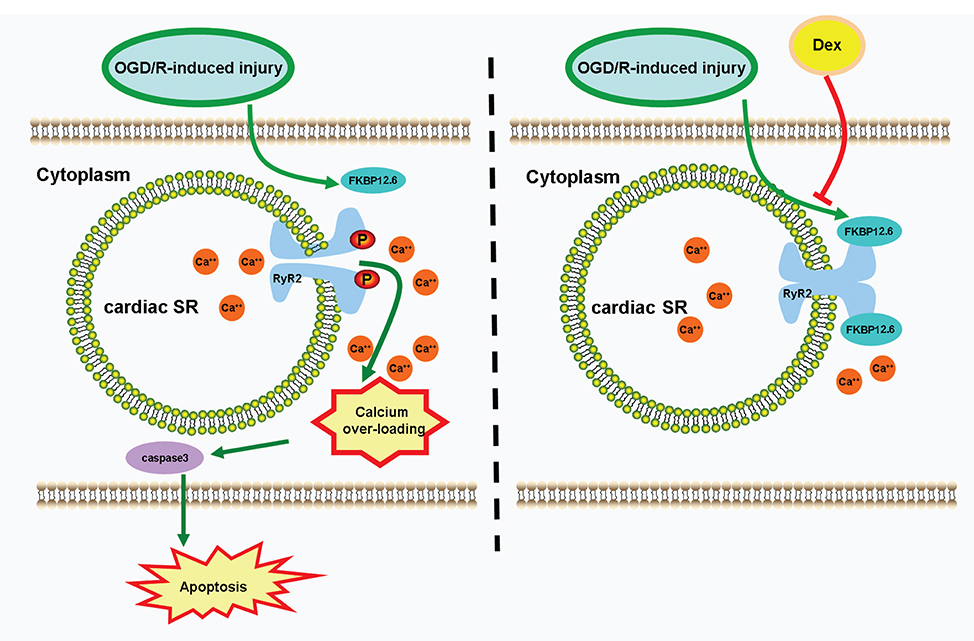 |
Figure 8 Schematic depicting DEX protects H9c2 cells against OGD/R-induced [Ca2+]i overload and apoptosis. During OGD/R, the down-regulation of FKBP12.6 leads to decreased combination of FKBP12.6 and RyR2, releasing Ca2+ from cardiac SR through phosphorylated RyR2 channels. The increased cytosolic free Ca2+ leads to [Ca2+]i overload, triggering caspase-3 dependent cell apoptosis. DEX pretreatment attenuates [Ca2+]i overload and apoptosis in H9c2 cells during OGD/R through a direct regulation of FKBP12.6/RyR2 signaling. Abbreviations: DEX, dexmedetomidine; SR, sarcoplasmic reticulum; FKBP12.6, 12.6-kd FK506-binding protein; RyR2, ryanodine receptor 2. |
Studies showed that RyR2 dysfunction resulted in [Ca2+]i overload during myocardial I/R injury.35,36 The suppression of RyR2-Ser2814 phosphorylation attenuated myocardial I/R injury by inhibiting cell apoptosis.9 In our study, the increased RyR2-Ser2814 phosphorylation led to [Ca2+]i overload in H9c2 cells exposed to OGD/R, which was inhibited by pretreatment with DEX. [Ca2+]i overload eventually initiates the apoptotic process where caspase-3 protein acts as a critical executioner.12,24 A study found that DEX protected spinal cord against I/R-induced injury through inhibiting apoptosis and caspase-3 activation,12 which was in line with the current results in H9c2 cells.
As a regulatory protein, FKBP12.6 stabilizes the RyR channels, preventing Ca2+ leakage from the SR.37,38 The decreased FKBP12.6 expression has been noted during rat myocardial I/R.39 Our results showed that FKBP12.6 expression was down-regulated during OGD/R in H9c2 cells. Specifically, the disassociation of FKBP12.6 from RyR2 increased the spontaneous Ca2+ spark frequency and Ca2+ transient amplitude.40,41 FKBP12.6-deficient mice developed abnormal cardiac function, leading to exercise-induced cardiac arrhythmia and sudden death.42 On the other hand, FKBP12.6 over-expression improved cardiac function in mice after myocardial infarction.43 Another study showed that FK506 induced disassociation of FKBP12.6 from RyR2, promoting SR stress-mediated apoptosis.44 Our study suggests that the protective effect of DEX against OGD/R-induced injury in H9c2 cells was blocked by si-FKBP12.6, revealing the important role of FKBP12.6 in the OGD/R-induced [Ca2+]i overload and DEX-mediated protection. In this regard, FKBP12.6 may be a promising therapeutic target for alleviating cardiac I/R injury.
Our previous study indicated that DEX pretreatment alleviated myocardial I/R injury by inhibiting inflammation and down-regulating the expression of toll-like receptors 4 (TLR4)-myeloid differentiation primary response 88-nuclear factor-kappa B via the α2-adrenergic receptor.15 A recent study showed that TLR4 activation induced Ca2+ leakage via regulating FKBP12.6 dissociation from RyR2 in septic cardiomyocytes.45 Thus, we hypothesized that DEX may attenuate myocardial I/R injury by inhibiting [Ca2+]i overload via TLR4-mediated FKBP12.6/RyR2 signaling pathway. In addition to the action on α2 adrenergic receptors, DEX may act on ion channels and membrane potential.46 The diverse effects of DEX may be associated with its organ protective effects.47 Using the whole cell patch clamp technique, one study showed that DEX attenuated L-type calcium channel and reduced Ca2+ inward flow in rat ventricular myocytes.48 Another study found that DEX inhibited Ca2+ inward flow through voltage gated calcium channels and glutamate receptors in rat DRG and hippocampus.33 Taken together, DEX may protect the cardiomyocytes through a direct effect on calcium-related ion channels.
The concentrations of DEX (0.1, 1, and 10 µM) in this study are higher than those clinically used for surgery and sedation. For humans, the maximum tolerated concentration of DEX is 15 ng/mL (equivalent to 75 nM).49 For cultured cardiomyocytes, however, cell activity did not decrease when DEX concentrations <30 µM.50 In our previous study, pretreatment with 1 µM DEX offered the best protection against hypoxia/reoxygenation-induced injury in H9c2 and primary neonatal rat cardiomyocytes.27 In a recent study, 150 μM was the maximum safe concentration for DEX in PC12 cells, and 50 μM was eventually selected for the subsequent experiments.51 Other studies used DEX 50 ng/mL (equivalent to 250 nM) on pacemaker cells in sinoatrial nodes of rabbits and DEX 200 ng/mL (equivalent to 1 µM) on rat ventricular myocytes.48,52 Therefore, it is possible that the safe concentration of DEX varies in different species and cell types.
This study has several limitations. First, as the interaction of FKBP12.6 and RyR2 has been well identified,18,37,38 the FKBP-RyR2 complex was not assessed by using a coimmunoprecipitation technique. Second, the present study focused on the regulation mechanism of DEX on the FKBP12.6/RyR2 signaling during OGD/R injury. However, Ca2+ reuptake mediated by sarcoplasmic reticulum Ca2+-ATPase (SERCA) may have been another important mechanism underlying the effects of DEX, as many studies showed that SERCA played an important role on the regulation of Ca2+ homeostasis in myocardial I/R injury.53–55 Third, the current results in H9c2 cardiomyocytes may not be fully extrapolated to animal models or even humans. Last, whether DEX-induced protection against I/R-induced injury is associated with the above-mentioned ion channels needs to be clarified in our future studies.
In conclusion, this is the first study showing that pretreatment with DEX alleviated OGD/R-induced [Ca2+]i overload and cell apoptosis in H9c2 cardiomyocytes through regulating the FKBP12.6/RyR2 signaling pathway. DEX may be used as a therapeutic agent for patients at high risk of myocardial I/R injury.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81873925 and 81671880 to FHJ, 81701098 to XWM, 81601659 to KP, and 81601666 to JZ), Jiangsu Provincial Medical Youth Talents Program (QNRC2016741 to KP), Jiangsu Provincial Medical Innovation Team (CXTDA2017043 to FHJ), and Suzhou Key Disease Program (LCZX201603 to FHJ).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Benjamin EJ, Muntner P, Alonso A, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56–e528.
2. Aghaei M, Motallebnezhad M, Ghorghanlu S, et al. Targeting autophagy in cardiac ischemia/reperfusion injury: a novel therapeutic strategy. J Cell Physiol. 2019. doi:10.1002/jcp.28345
3. Li D, Wang X, Huang Q, Li S, Zhou Y, Li Z. Cardioprotection of CAPE-oNO2 against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF-kappaB pathway in vivo and in vitro. Redox Biol. 2017;15:62–73. doi:10.1016/j.redox.2017.11.023
4. Scoote M, Williams AJ. The cardiac ryanodine receptor (calcium release channel): emerging role in heart failure and arrhythmia pathogenesis. Cardiovasc Res. 2002;56(3):359–372. doi:10.1016/s0008-6363(02)00574-6
5. Li J, Xiang X, Gong X, Shi Y, Yang J, Xu Z. Cilostazol protects mice against myocardium ischemic/reperfusion injury by activating a PPARgamma/JAK2/STAT3 pathway. Biomed Pharmacother. 2017;94:995–1001. doi:10.1016/j.biopha.2017.07.143
6. Xie XJ, Fan DM, Xi K, et al. Suppression of microRNA-135b-5p protects against myocardial ischemia/reperfusion injury by activating JAK2/STAT3 signaling pathway in mice during sevoflurane anesthesia. Biosci Rep. 2017;37(3). doi:10.1042/BSR20170186
7. Toldo S, Marchetti C, Mauro AG, et al. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int J Cardiol. 2016;209:215–220. doi:10.1016/j.ijcard.2016.02.043
8. Mattiazzi A, Argenziano M, Aguilar-Sanchez Y, Mazzocchi G, Escobar AL. Ca2+ Sparks and Ca2+ waves are the subcellular events underlying Ca2+ overload during ischemia and reperfusion in perfused intact hearts. J Mol Cell Cardiol. 2015;79:69–78. doi:10.1016/j.yjmcc.2014.10.011
9. Di Carlo MN, Said M, Ling H, et al. CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J Mol Cell Cardiol. 2014;74:274–283. doi:10.1016/j.yjmcc.2014.06.004
10. Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res. 2012;94(2):168–180. doi:10.1093/cvr/cvs116
11. Gerlach AT, Murphy CV, Dasta JF. An updated focused review of dexmedetomidine in adults. Ann Pharmacother. 2009;43(12):2064–2074. doi:10.1345/aph.1M310
12. Sun Z, Zhao T, Lv S, Gao Y, Masters J, Weng H. Dexmedetomidine attenuates spinal cord ischemia-reperfusion injury through both anti-inflammation and anti-apoptosis mechanisms in rabbits. J Transl Med. 2018;16(1):209. doi:10.1186/s12967-018-1583-7
13. Yang Y, Duan W, Jin Z, et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J Pineal Res. 2013;55(3):275–286. doi:10.1111/jpi.12070
14. Ji F, Li Z, Young N, Moore P, Liu H. Perioperative dexmedetomidine improves mortality in patients undergoing coronary artery bypass surgery. J Cardiothorac Vasc Anesth. 2014;28(2):267–273. doi:10.1053/j.jvca.2013.06.022
15. Zhang JJ, Peng K, Zhang J, Meng XW, Ji FH. Dexmedetomidine preconditioning may attenuate myocardial ischemia/reperfusion injury by down-regulating the HMGB1-TLR4-MyD88-NF-small ka, CyrillicB signaling pathway. PLoS One. 2017;12(2):e0172006. doi:10.1371/journal.pone.0172006
16. Takizuka A, Minami K, Uezono Y, et al. Dexmedetomidine inhibits muscarinic type 3 receptors expressed in Xenopus oocytes and muscarine-induced intracellular Ca2+ elevation in cultured rat dorsal root ganglia cells. Naunyn Schmiedebergs Arch Pharmacol. 2007;375(5):293–301. doi:10.1007/s00210-007-0168-4
17. Xiao YF, Zeng ZX, Guan XH, et al. FKBP12.6 protects heart from AngII-induced hypertrophy through inhibiting Ca(2+)/calmodulin-mediated signalling pathways in vivo and in vitro. J Cell Mol Med. 2018;22(7):3638–3651. doi:10.1111/jcmm.13645
18. Zhang X, Tallini YN, Chen Z, et al. Dissociation of FKBP12.6 from ryanodine receptor type 2 is regulated by cyclic ADP-ribose but not beta-adrenergic stimulation in mouse cardiomyocytes. Cardiovasc Res. 2009;84(2):253–262. doi:10.1093/cvr/cvp212
19. He H, Shi M, Zeng X, et al. Decreased FKBP12.6 expression and enhanced endothelin receptor signaling associated with arrhymogenesis in myocardial infarction rats. Phytother Res. 2008;22(8):1115–1124. doi:10.1002/ptr.2470
20. Fauconnier J, Roberge S, Saint N, Lacampagne A. Type 2 ryanodine receptor: a novel therapeutic target in myocardial ischemia/reperfusion. Pharmacol Ther. 2013;138(3):323–332. doi:10.1016/j.pharmthera.2013.01.015
21. Ling H, Gray CB, Zambon AC, et al. Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ Res. 2013;112(6):935–944. doi:10.1161/CIRCRESAHA.112.276915
22. Chelu MG, Sarma S, Sood S, et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119(7):1940–1951. doi:10.1172/jci37059
23. Yin B, Hou XW, Lu ML. Astragaloside IV attenuates myocardial ischemia/reperfusion injury in rats via inhibition of calcium-sensing receptor-mediated apoptotic signaling pathways. Acta Pharmacol Sin. 2018;40(5):599–607. doi:10.1038/s41401-018-0082-y
24. Meng Y, Li WZ, Shi YW, Zhou BF, Ma R, Li WP. Danshensu protects against ischemia/reperfusion injury and inhibits the apoptosis of H9c2 cells by reducing the calcium overload through the p-JNK-NF-kappaB-TRPC6 pathway. Int J Mol Med. 2016;37(1):258–266. doi:10.3892/ijmm.2015.2419
25. Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6(2):99–104. doi:10.1038/sj.cdd.4400476
26. Jiang WB, Zhao W, Chen H, et al. Baicalin protects H9c2 cardiomyocytes against hypoxia/reoxygenation-induced apoptosis and oxidative stress through activation of mitochondrial aldehyde dehydrogenase 2. Clin Exp Pharmacol Physiol. 2017;45:3003–311.
27. Gao JM, Meng XW, Zhang J, et al. Dexmedetomidine protects cardiomyocytes against hypoxia/reoxygenation injury by suppressing TLR4-MyD88-NF-kappaB signaling. Biomed Res Int. 2017;2017:1674613. doi:10.1155/2017/1674613
28. Ma LQ, Yu Y, Chen H, et al. Sweroside alleviated aconitine-induced cardiac toxicity in H9c2 cardiomyoblast cell line. Front Pharmacol. 2018;9:1138. doi:10.3389/fphar.2018.01138
29. Ghasemi A, Zahediasl S. Normality tests for statistical analysis: a guide for non-statisticians. Int J Endocrinol Metab. 2012;10(2):486–489. doi:10.5812/ijem.3505
30. Riquelme JA, Westermeier F, Hall AR, et al. Dexmedetomidine protects the heart against ischemia-reperfusion injury by an endothelial eNOS/NO dependent mechanism. Pharmacol Res. 2016;103:318–327. doi:10.1016/j.phrs.2015.11.004
31. Yoshikawa Y, Hirata N, Kawaguchi R, Tokinaga Y, Yamakage M. Dexmedetomidine maintains its direct cardioprotective effect against ischemia/reperfusion injury in hypertensive hypertrophied myocardium. Anesth Analg. 2018;126(2):443–452. doi:10.1213/ANE.0000000000002452
32. Kristiansen SB, Haanes KA, Sheykhzade M, Edvinsson L. Endothelin receptor mediated Ca(2+) signaling in coronary arteries after experimentally induced ischemia/reperfusion injury in rat. J Mol Cell Cardiol. 2017;111:1–9. doi:10.1016/j.yjmcc.2017.07.013
33. Akpinar H, Naziroglu M, Ovey IS, Cig B, Akpinar O. The neuroprotective action of dexmedetomidine on apoptosis, calcium entry and oxidative stress in cerebral ischemia-induced rats: contribution of TRPM2 and TRPV1 channels. Sci Rep. 2016;6:37196. doi:10.1038/srep37196
34. Yang D, Hong JH. Dexmedetomidine modulates histamine-induced Ca(2+) signaling and pro-inflammatory cytokine expression. Korean J Physiol Pharmacol. 2015;19(5):413–420. doi:10.4196/kjpp.2015.19.5.413
35. Zucchi R, Ronca F, Ronca-Testoni S. Modulation of sarcoplasmic reticulum function: a new strategy in cardioprotection? Pharmacol Ther. 2001;89(1):47–65. doi:10.1016/S0163-7258(00)00103-0
36. Fauconnier J, Meli AC, Thireau J, et al. Ryanodine receptor leak mediated by caspase-8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc Natl Acad Sci U S A. 2011;108(32):13258–13263. doi:10.1073/pnas.1100286108
37. Liao B, Zheng YM, Yadav VR, Korde AS, Wang YX. Hypoxia induces intracellular Ca2+ release by causing reactive oxygen species-mediated dissociation of FK506-binding protein 12.6 from ryanodine receptor 2 in pulmonary artery myocytes. Antioxid Redox Signal. 2011;14(1):37–47. doi:10.1089/ars.2009.3047
38. Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi:10.1146/annurev.physiol.67.040403.114521
39. Na T, Huang ZJ, Dai DZ, Zhang Y, Dai Y. Abrupt changes in FKBP12.6 and SERCA2a expression contribute to sudden occurrence of ventricular fibrillation on reperfusion and are prevented by CPU86017. Acta Pharmacol Sin. 2007;28(6):773–782. doi:10.1111/j.1745-7254.2007.00580.x
40. McCall E, Li L, Satoh H, Shannon TR, Blatter LA, Bers DM. Effects of FK-506 on contraction and Ca2+ transients in rat cardiac myocytes. Circ Res. 1996;79(6):1110–1121. doi:10.1161/01.res.79.6.1110
41. Xin HB, Senbonmatsu T, Cheng DS, et al. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature. 2002;416(6878):334–338. doi:10.1038/416334a
42. Wehrens XH, Lehnart SE, Huang F, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113(7):829–840. doi:10.1016/s0092-8674(03)00434-3
43. Huang F, Shan J, Reiken S, Wehrens XH, Marks AR. Analysis of calstabin2 (FKBP12.6)-ryanodine receptor interactions: rescue of heart failure by calstabin2 in mice. Proc Natl Acad Sci U S A. 2006;103(9):3456–3461. doi:10.1073/pnas.0511282103
44. Tang J, Ge Y, Yang L, et al. ER stress via CHOP pathway is involved in FK506-induced apoptosis in rat fibroblasts. Cell Physiol Biochem. 2016;39(5):1965–1976. doi:10.1159/000447894
45. Yang J, Zhang R, Jiang X, et al. Toll-like receptor 4-induced ryanodine receptor 2 oxidation and sarcoplasmic reticulum Ca(2+) leakage promote cardiac contractile dysfunction in sepsis. J Biol Chem. 2018;293(3):794–807. doi:10.1074/jbc.M117.812289
46. Chen BS, Peng H, Wu SN. Dexmedetomidine, an alpha2-adrenergic agonist, inhibits neuronal delayed-rectifier potassium current and sodium current. Br J Anaesth. 2009;103(2):244–254. doi:10.1093/bja/aep107
47. Cai Y, Xu H, Yan J, Zhang L, Lu Y. Molecular targets and mechanism of action of dexmedetomidine in treatment of ischemia/reperfusion injury. Mol Med Rep. 2014;9(5):1542–1550. doi:10.3892/mmr.2014.2034
48. Zhao J, Zhou CL, Xia ZY, Wang L. Effects of dexmedetomidine on L-type calcium current in rat ventricular myocytes. Acta Cardiologica Sinica. 2013;29(2):175–180.
49. Ebert TJ, Hall JE, Barney JA, Uhrich TD, Colinco MD. The effects of increasing plasma concentrations of dexmedetomidine in humans. Anesthesiology. 2000;93(2):382–394. doi:10.1097/00000542-200008000-00016
50. Peng K, Qiu Y, Li J, Zhang ZC, Ji FH. Dexmedetomidine attenuates hypoxia/reoxygenation injury in primary neonatal rat cardiomyocytes. Exp Ther Med. 2017;14(1):689–695. doi:10.3892/etm.2017.4537
51. Tan Y, Bi X, Wang Q, et al. Dexmedetomidine protects PC12 cells from lidocaine-induced cytotoxicity via downregulation of Stathmin 1. Drug Des Devel Ther. 2019;13:2067–2079. doi:10.2147/DDDT.S199572
52. Pan X, Zhang Z, Huang YY, Zhao J, Wang L. Electrophysiological effects of dexmedetomidine on sinoatrial nodes of rabbits. Acta Cardiologica Sinica. 2015;31(6):543–549.
53. Kang SM, Lim S, Song H, et al. Allopurinol modulates reactive oxygen species generation and Ca2+ overload in ischemia-reperfused heart and hypoxia-reoxygenated cardiomyocytes. Eur J Pharmacol. 2006;535(1–3):212–219. doi:10.1016/j.ejphar.2006.01.013
54. Wang R, Yang M, Wang M, et al. Total saponins of Aralia elata (Miq) seem alleviate calcium homeostasis imbalance and endoplasmic reticulum stress-related apoptosis induced by myocardial ischemia/reperfusion injury. Cell Physiol Biochem. 2018;50(1):28–40. doi:10.1159/000493954
55. Zhang CM, Gao L, Zheng YJ, Yang HT. Berbamine protects the heart from ischemia/reperfusion injury by maintaining cytosolic Ca(2+) homeostasis and preventing calpain activation. Circ J. 2012;76(8):1993–2002. doi:10.1253/circj.cj-11-1431
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.