")
Back to Journals » International Journal of Nanomedicine » Volume 14
Development And In Vitro Characterization Of Bladder Tumor Cell Targeted Lipid-Coated Polyplex For Dual Delivery Of Plasmids And Small Molecules
Authors Samaddar S , Mazur J, Boehm D, Thompson DH
Received 29 July 2019
Accepted for publication 22 October 2019
Published 4 December 2019 Volume 2019:14 Pages 9547—9561
DOI https://doi.org/10.2147/IJN.S225172
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Shayak Samaddar, Joshua Mazur, Devin Boehm, David H Thompson
Department of Chemistry, Purdue University, Bindley Bioscience Center, West Lafayette, Indiana 47906, USA
Correspondence: David H Thompson
Department of Chemistry, Purdue University, Bindley Bioscience Center, 1203 W. State St., West Lafayette, Indiana 47906, USA
Tel +1 765-494-0386
Email [email protected]
Background: Bladder cancer is the fourth most common cancer in men and eleventh most common in women. Combination therapy using a gene and chemotherapeutic drug is a potentially useful strategy for treating bladder cancer in cases where a synergistic benefit can be achieved successfully. This approach relies on developing drug combinations using carrier systems that can load both hydrophilic genes and hydrophobic drugs. Ideally, the formulation for carrier system should be free of traditional high shear techniques such as sonication and extrusion to reduce shear-induced nucleic acid strand breakage. Moreover, the system should be able to protect the nucleic acid from enzymatic attack and deliver it specifically to the tumor site.
Materials and methods: A dual payload carrier system that was formulated using a simple flow mixing technique to complex anionic plasmid (EGFP-NLS) using a cationic polymer (CD-PEI2.5kD) followed by coating of the polyplex using lipid membranes. The resulting lipid-coated polyplex (LCP) formulations are targeted to bladder cancer cells by employing a bacterial adhesive peptide sequence, RWFV, that targets the LCP to the tumor stroma for efficiently delivering reporter plasmid, EGFP-NLS and a model small molecule drug, pyrene, to the cancer cells.
Results: Encapsulation efficiency of the peptide targeted carrier for the plasmid was 50% ± 0.4% and for pyrene it was 16% ± 0.4%. The ability of the targeted LCP to transfect murine bladder cancer cells was 4-fold higher than LCP bearing a scrambled peptide sequence. Fluorescence of cells due to pyrene delivery was highest after 4 hrs using targeted LCP. Finally, we loaded the peptide targeted LCP with anti-cancer agent, curcumin. The targeted formulation of curcumin resulted in only 45% viable cancer cells at a concentration of 5 μg/mL, whereas the empty and non-targeted formulations did not result any significant cell death.
Conclusion: These results demonstrate the specificity of the targeting peptide sequence in engaging tumor cells and the utility of the developed carrier platform to deliver a dual payload to bladder tumor cells.
Keywords: dual delivery, bladder cancer, gene therapy
Introduction
Non-muscle invasive bladder carcinoma (NMIBC) has the highest recurrence rate among all types of cancer.1 This requires constant monitoring and frequent hospitalization, making NMIBC very expensive to manage on a cost-per-patient basis from diagnosis to death.2 Recently, there has been an increased appreciation regarding the role of multidrug resistance (MDR) in the poor prognosis of patients with recurring tumors after first-line chemotherapeutic therapy.3–7 Kunze et al reported that siRNA-mediated silencing of genes like B-cell lymphoma 2 (BCL2), BCL2-like 1 (BCLXL), X-linked inhibitor of apoptosis (XIAP), and survivin, led to sensitization of MDR expressing bladder carcinoma cells such as T24 and J82 cells towards mitomycin-C and cisplatin chemotherapy.8 In a similar study, it was shown that downregulation of the MDR-associated protein-1 (MRP1) by emodin, a natural anthraquinone analogue, increased cisplatin mediated apoptosis in cisplatin-resistant T24 and J82 cells.9 Thus, the co-delivery of chemotherapeutic agents has the potential to overcome MDR by manipulating the expression profile of the MDR gene or activating synergistic pathways.10,11 The biggest challenge in this approach, however, is the availability of a suitable carrier system that can load two therapeutic molecules with dramatically different physical properties and deliver them to cancer cell targets.10,12 Nucleic acid therapeutics are anionic and hydrophilic nature with high molecular weights, whereas conventional chemotherapeutics are typically hydrophobic molecules with much lower molecular weights.
Multiple drug delivery systems have been developed that are capable of delivering nucleic acid and small molecule payloads for the treatment of different diseases, especially cancer.10,11 A major problem for urothelial carcinoma patients, however, is that intravesical administration of small molecule therapeutics affords poor retention within bladder due to constant urine influx and bladder voiding, resulting in sub-optimal therapeutic concentrations and modest clinical outcome.13 Development of a bladder tumor-targeted dual delivery system which can “stick” to cancerous sites and release dual payload within the targeted tumor cell may have advantages in treating urothelial carcinoma. We report a tumor-targeted dual delivery system as a potential model for delivery of small molecules and nucleic acids to bladder tumor cells. For this proof-of-concept study, we used EGFP-NLS as our reporter gene and pyrene as a hydrophobic small molecule reporter that is able to partition efficiently into lipid bilayers. Additionally, the inherent fluorescent quality of pyrene makes it easily quantifiable using its absorbance or fluorescence properties.14,15 Finally, we loaded a well-known anti-cancer agent, curcumin, which is hydrophobic in nature and has recently been evaluated as a intravesical therapy in a preclinical orthotopic rat model of bladder cancer as a curcumin-cyclodextrin complex to enhance its solubility.16
High molecular weight (M.W.; e.g., 10k and 25k) polyethyleneimine (PEI) has been widely used as a gene transfection agent in vitro.17 Unfortunately, these cationic polymers are highly cytotoxic, thereby limiting their in vivo applications.18,19 On the contrary, low molecular weight PEI has a better toxicity profile, however, it lacks the high transfection efficiency of its high molecular weight counterparts.20 Conjugating β-CD to PEI has been shown to enhance luciferase gene transfection in HEK293 cells by 3.7 times relative to unmodified PEI and significantly decreases the toxicity of the polymer.21 Reduction in cationic charge density and/or a change in polymer structure was proposed as the basis for the in reduction of toxicity relative to unmodified PEI. Additionally, the membrane destabilizing effect of cyclodextrin has been suggested to aid in endosome membrane destabilization for enhancing transfection.22,23 Our formulation utilizes β-CD and linear PEI (M.W. 2.5kD) as a conjugate (CD2.5kD-PEI) to electrostatically condense the plasmid into a polyplex. The polyplex is then further coated with a layer of fusogenic lipid mixture to produce a lipid-coated polyplex (LCP) that is also capable of encapsulating hydrophobic small molecules such as pyrene (Figure 1, inset). The exterior of the LCP is decorated with a lipopeptide sequence (GNRQRWFVVWLGSTNDPV) that is derived from fibronectin attachment protein. The peptide mimics a fragment of the bacterial adhesin present on the surface of Mycobacterium bovis. This peptide sequence has been shown to be responsible for the functional activity of fibronectin attachment protein, with the core tetrapeptide RWFV being absolutely critical for fibronectin engagement.24 Our formulation approach uses a microfluidic mixing technique to form LCP with enhanced targeting to bladder tumor cells when the GNRQRWFVVWLGSTNDPV peptide is present on the particle surface.
Materials And Methods
Materials
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-mPEG2k), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and cholesterol hemisuccinate were purchased from Avanti Polar Lipids. Linear poly(ethyleneimine) (2.5kD) was purchased from Polysciences. β-Cyclodextrin, pyrene and curcumin were purchased from Sigma. DNAase-I was purchased from New England Biolabs. EGFP-NLS was obtained as a gift from Prof. Rob Parton (Addgene plasmid# 67652). Slide-a-Lyzer dialysis cassettes (MWCO 20K, 3 mL) and cell culture reagents like Dulbecco’s modified Eagle medium (DMEM), Opti-MEM® reduced serum medium, 1× phosphate buffered saline solution (PBS) and GibcoTM TrypLE express enzyme (1×) were purchased from Fisher Scientific. Fetal bovine serum (FBS), Penicillin-Streptomycin 10/10 (100X) and L-glutamine –200 mM (100×) were purchased from Atlanta Biologicals. MB49 cells were obtained as a gift from Prof. Timothy Ratliff (Purdue University, Department of Comparative Pathobiology) and have been authenticated by STR profile. Ultrapure water (18.2 MΩ) was used for preparation of 20 mM HEPES buffer (pH 7.4). All solvents were of analytical grade, purchased from commercial sources. Buffers and solvents were filtered through 0.22 µm CA syringe filters (Macherey-Nagel Inc.) before use.
Synthesis Of CD-PEI
Poly(ethylenimine) (2.5 kDa) was used to introduce a single modification onto the primary β-cyclodextrin rim as previously described.25–27
Synthesis Of Lipopolymer Conjugates
The FAP sequence (GNRQRWFVVWLGSTNDPV) with a propargylglycine modification at C termini was synthesized by solid-phase peptide synthesis. Similarly, a control peptide sequence (scrambled peptide, SCR) GNRQWVRFVWLGSTNDPV) was also prepared where the order of the critical RWFV tetrapeptide was scrambled to WVRF. The alkyne modification on the peptide was utilized for a copper-catalyzed azide−alkyne cycloaddition (CuAAC) conjugation to DSPE-PEG2k-Azide. A detailed description of the method has been reported previously.28
Formulation Of Polyplex And Lipid-Coated Polyplex (LCP)
The formulation set-up comprised two syringes attached to 1/16” I.D. tubing connected by a T-junction. The T junction was followed by 6ʹ length tubing of the same I.D. wrapped around a plastic support 10 cm in length, 2.5 cm in width and 0.5 mm in height (Supplementary Figure 1). Optimal conditions for polyplex formation were determined by mixing the HEPES solution (20 mM) of CD2.5kD-PEI and 60 µg/mL EGFP-NLS at different N/P ratios of 5, 10, 15 and 20. N/P was defined as the ratio between the number of nitrogen atoms present in the polyethyleneimine tail of CD2.5kD-PEI to the number of phosphorus atoms in the plasmid backbone. Flow rates were varied from 10 µL/min to 5 mL/min. The flow was allowed to stabilize for at least 6 s before starting sample collection.
The polyplex formed above were then mixed with an 80% ethanolic solution of 0.5 mM, 1 mM, 2 mM, 4 mM or 8 mM mixture of lipids (34:54:10:2 CHEMS:DOPE:DSPC:DSPE-mPEG-RWFV). The flow rate was kept constant in this step at 5mL/min for each syringe. The resulting LCP were immediately diluted in HEPES buffer to <16% ethanol after collection. The diluted particles were then dialyzed using 20K MWCO dialysis cassettes against 500 volumes of 20mM HEPES for 24 hrs. All collected formulations were stored at 4ºC prior to analysis. For small molecule loading, 5 mol% of pyrene or curcumin was mixed with the ethanolic lipid solution. Non-targeted and scrambled LCP were prepared as controls using the method described above except that the DSPE-mPEG-RWFV was replaced by DSPE-mPEG (non-targeted control) or DSPE-mPEG-SCR (scrambled control). LCP prepared by bulk phase mixing employed 1mL of 60 µg plasmid to 1 mL of CD2.5kD-PEI in 20mM HEPES buffer at a N/P ratio of 10 in a glass vial, followed by brief vortexing for 30 s and incubation at 25°C for 30 mins. Lipid coating was performed using the lipid film hydration method. The lipid mixture in chloroform (1 mL of 4 mM mixture) was evaporated in a round bottom flask under reduced pressure and stored overnight under vacuum to remove residual solvent. Polyplex solution (2mL) was added to the dry lipid film, followed by hydration for 30 mins at 37ºC and vortexing for 4 mins. A schematic representation of the formulation steps using flow mixing is shown in Figure 1.
Dynamic Light Scattering (DLS) Measurements
The hydrodynamic diameters (number mean), polydispersity index (PDI) and zeta potentials (ζ) of the materials were evaluated using a particle size analyzer (Zetasizer Nano ZS, Malvern Instruments Ltd.) at 25°C with a scattering angle of 173°. Particle solutions (30 µL) were diluted to 1 mL with sterile filtered NanoPure water before analysis.
Gel-Shift Assay
The pDNA complexation ability of polymers mixed within the flow channel was determined by 1% agarose gel electrophoresis. The agarose gels were precast in 1× TBE buffer with GelRed dye (Biotium Inc.) at 1:10,000 dilution. Polyplex samples containing 0.1 μg of pDNA at different N/P ratios were added to loading dye (5:1 dilution) and loaded onto the gel, prior to initializing electrophoresis at a constant voltage of 55 V for 1.5 hrs in 1× TBE buffer. The pDNA bands were then visualized under a UV transilluminator at 365 nm.
TEM Images
Transmission electron microscopy (TEM) images of LCP were collected using a Tecnai G2 20 microscope equipped with a 200 kV LaB6 filament and fitted with a bottom mount 2k × 2k Gatan US1000 CCD camera. Samples were deposited onto Formvar/carbon-coated Cu 400-mesh grids and stained with 2% uranyl acetate prior to visualization. Further image analysis was performed using ImageJ software (NIH).
Quantifying Plasmid Content
Encapsulation efficiencies and loading efficiencies of plasmid DNA in LCP were determined using the membrane impermeable dye PicoGreenTM (Molecular Probes, Eugene, OR, USA) that binds specifically to double-stranded DNA and fluoresces only when bound. PicogreenTM fluorescence was measured with a spectrofluorometer (Biotek Synergy) using excitation and emission wavelengths of 490 and 525 nm, respectively. Plasmid content was determined by measuring the PicoGreenTM fluorescence after treating the LCP for 30 mins at 25ºC with 1% Triton X-100 for disrupting the lipid coat, followed by another 30 mins treatment with 0.15 mg/mL of heparin sulfate solution for decomplexing the polyplex.29,30 Plasmid concentrations were calculated by comparing PicoGreen fluorescence values to the calibration curve prepared using plasmid standards. The encapsulation efficiency (EE) and loading efficiencies (LE) were calculated using the following formula:

Quantifying Pyrene Content
HPLC analysis was used to measure the amount of pyrene that was encapsulated within the LCP. Briefly, 50 µL of the LCP was dried using a Vacufuge TM (Eppendorf) at 40ºC for 2 hrs. NanoPure water (30 µL) was added to the dried pellet, followed by addition of 70 µL of acetonitrile to dissolve the LCP. An Agilent 1200 HPLC system equipped with a XBridgeTM C18 column (2.1 mm × 100 mm, pore size 3.5 μm) and employing mobile phase A (water with 10 mM NH4OAc) and mobile phase B (75% acetonitrile:25% methanol with 10 mM NH4OAc) was used for analysis. A gradient elution was used from 95% A to 95% B over 4 mins at a flow rate of 250 µL/min, with pyrene absorbance monitored at 338 ± 4 nm using a diode array detector. Pyrene concentrations were calculated by comparing the pyrene absorbance for LCP to a calibration curve prepared using absorbance values obtained from standard pyrene concentration. The EE and LE were calculated using the following formula:

Quantifying Curcumin Content
Curcumin absorbance at 425 nm was used to quantify the amount of drug encapsulated within the LCP. A calibration curve prepared by spiking known amounts of curcumin into a solution of empty LCP. The following formula was used to determine the EE and LE.
DNAase Protection Assay
The ability of the LCP to protect the complexed DNA against nucleases was evaluated by incubation with DNase-I.31 In brief, LCP formed at N/P = 10 were incubated at 37°C in 20 mM HEPES buffer containing 2U DNase I. The reaction was quenched by addition of 10 mM EDTA solution to a final EDTA concentration of 2.5 mM and heating at 65ºC for 10 mins using a heat block. The particle solutions were then treated with 1% Triton X-100 for 30 mins at 25ºC to strip the lipid coating, followed by another 30 mins treatment at 25ºC with 0.15 mg/mL heparin to decomplex the polyplex and release the pDNA. Next, CHCl3 was added at a 1:1 ratio, the mixture vortexed, and then incubated for 15 mins to effect phase separation. The aqueous layer containing pDNA was collected and concentrated using an Eppendorf Vacufuge under reduced pressure at 25ºC. The extracted pDNA was analyzed using 1% agarose gel electrophoresis.
Quantifying Transfection Efficiency
MB49 cells were seeded in 96-well plates at a density of 15,000 cells per well in 100 μL of complete medium (DMEM containing 10% FBS, supplemented with 1% penicillin-streptomycin and 1% L-glutamine) and incubated for 24 hrs prior to transfection. The medium was replaced, and naked plasmid, non-targeted LCP, scrambled LCP, or targeted LCP were added at 0.1 μg DNA per well in 100 μL in Opti-MEM® media for 4 hrs. Then, the cells were rinsed with PBS and incubated at 37°C for an additional 4 hrs, 12 hrs or 24 hrs in complete media. Next, the cells were trypsinized and analyzed using a FC500 (Beckman Coulter) flow cytometer equipped with a 488 nm laser and 525 ± 20 nm detector. As positive control, Lipofectamine™ LTX was used according to the manufacturer’s protocol. All transfection experiments were performed in triplicate.
Quantifying Pyrene Delivery
MB49 cells were seeded in 24-well plates at a density of 2 × 105 cells per well in 500 μL of complete medium and incubated for 24 hrs prior to transfection. Media was replaced with either non-targeted LCP, scrambled LCP or targeted LCP at an equivalent dose of 0.05 μg/mL pyrene in 500 μL Opti-MEM® media for 4 hrs. Opti-MEM® media without LCP was used as a negative control. Next, the cells were rinsed with PBS and incubated at 37°C for an additional 4 hrs or 24 hrs in complete media. Cells were trypsinized and analyzed using a BD Fortessa flow cytometer equipped with a 355 nm UV laser and 379 ± 14 nm detector.
Cytotoxicity Evaluation
Cell viability was measured using CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega). MB49 cells were seeded at 15,000 cells per well in 96-well plates in growth medium and allowed to adhere overnight at 37°C. Cells were washed with PBS and incubated with LCP samples in Opti-MEM media for 4 hrs. The cells were then washed with PBS and incubated overnight in complete DMEM media. Next, 20 µL of the MTS reagent was added into each well and incubated for another 1 hr at 37ºC before recording the absorbance at 490 nm using a Synergy Biotek plate reader. The absorbance of the wells without any cells was subtracted from the untreated controls and treated sample wells for background correction. The cell viability (%) relative to untreated cells was calculated as [A]sample/[A]untreated × 100%, where [A]untreated is the absorbance of the wells without LCP and [A]sample is the absorbance of the wells treated with LCP. All experiments were performed in triplicate and the cytotoxicity values reported are the mean from three different measurements.
Results And Discussion
Formulation And Characterization Of Lipid-Coated Polyplex
A sonication- and extrusion-free technique for formulating dual loaded LCP was developed. First, we studied the N/P ratio needed for complete complexation of CD2.5kD-PEI and the 5.2 kB plasmid. For all the N/P ratios studied, the flow rate was kept constant at 5 mL/min for each syringe. The positively charged polymer was mixed with the anionic plasmid to produce electrostatically condensed polyplex. The hydrodynamic diameters of the polyplex were 133 ± 9 nm, 97 ± 3, 87 ± 8 and 78 ± 10 nm at N/P of 5, 10, 15 and 20, respectively (Figure 2A). At N/P ratios of 5 and 10, the polydispersities were 0.26 ± 0.01 and 0.27 ± 0.03, respectively. These PDI increased to 0.32 ± 0.03 and 0.33 ± 0.06 for N/P of 15 and 20, respectively (Figure 2B). At N/P of 5, the measured ζ of the polyplex was −18.2 ± 1.1 mV, suggesting that the plasmid was incompletely complexed by the polymer. At an N/P of 10, the polyplex displayed a positive ζ = 14.8 ± 2.9 mV, suggesting complete complexation of the plasmid cargo. For higher N/P ratios, we did not observe significant increases in surface charge, such that ζ = 12.6 ± 3.0 mV and 16.3 ± 0.8 mV at N/P 15 and 20, respectively (Figure 2C). These findings are consistent with our gel-shift assay results (Figure 2D). At N/P 5, there was a significant amount of uncomplexed plasmid as revealed by the migrated electrophoretic bands visible on the agarose gel. At N/P ≥ 10, however, no bands were visible due to the absence of free plasmids. Thus, all subsequent lipid-coated polyplex formulations employed N/P = 10.
When T junctions are used in flow reactor systems, mixing occurs primarily due to diffusion at the contact surface between the two fluid streams, thereby necessitating the use of long mixing channels.32 Flow properties within such microfluidic channels can be predicted by calculating the Reynolds number (Re).33 Re is a dimensionless number and is defined by the following equation:

where 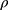 is the density of the fluid (kg/m3),
is the density of the fluid (kg/m3), 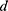 is the diameter of the channel (m),
is the diameter of the channel (m),  is the flow velocity of the fluid (m/s) and µ is the dynamic viscosity of the fluid (kg/m.s).34 Considering the dynamic viscosity and density of water at 25ºC to be 8.9 × 10−4 kg/m•s and 997 kg/m3 respectively, at a total flow rate of 10 mL/min, the Re of the system can be calculated to be 149.7, which is in the laminar flow regime where mixing is diffusion controlled.33 However, higher flow rates and sharp turns along the fluid flow path have been shown to enhance mixing by promoting the formation of eddies and vortices within the fluidic channels.35,36 We evaluated the effect of flow rate on polyplex formation at N/P = 10 by increasing the total flow rate (TFR) from 0.02 mL/min to 10 mL/min while maintaining a flow rate ratio of 1:1. Increasing the TFR produced a polyplex size decrease from 234 ± 57 nm at the lowest flow rate to 97 ± 3 nm at the highest flow rate of 10 mL/min (Figure 3A). Improvement in polyplex polydispersity was also observed from 0.66 at the lowest TFR to 0.28 at a TFR of 10 mL/min (Figure 3B). Additionally, the ζ of the polyplex increased from 8.6 ± 1.5 mV to 14.8 ± 2.9 mV (Figure 3C). These results confirm that higher flow rates ensure efficient mixing between the cationic polymer and the pDNA streams, resulting in smaller and more highly condensed polyplex of greater homogeneity. All subsequent formulation experiments used a TFR of 10 mL/min.
is the flow velocity of the fluid (m/s) and µ is the dynamic viscosity of the fluid (kg/m.s).34 Considering the dynamic viscosity and density of water at 25ºC to be 8.9 × 10−4 kg/m•s and 997 kg/m3 respectively, at a total flow rate of 10 mL/min, the Re of the system can be calculated to be 149.7, which is in the laminar flow regime where mixing is diffusion controlled.33 However, higher flow rates and sharp turns along the fluid flow path have been shown to enhance mixing by promoting the formation of eddies and vortices within the fluidic channels.35,36 We evaluated the effect of flow rate on polyplex formation at N/P = 10 by increasing the total flow rate (TFR) from 0.02 mL/min to 10 mL/min while maintaining a flow rate ratio of 1:1. Increasing the TFR produced a polyplex size decrease from 234 ± 57 nm at the lowest flow rate to 97 ± 3 nm at the highest flow rate of 10 mL/min (Figure 3A). Improvement in polyplex polydispersity was also observed from 0.66 at the lowest TFR to 0.28 at a TFR of 10 mL/min (Figure 3B). Additionally, the ζ of the polyplex increased from 8.6 ± 1.5 mV to 14.8 ± 2.9 mV (Figure 3C). These results confirm that higher flow rates ensure efficient mixing between the cationic polymer and the pDNA streams, resulting in smaller and more highly condensed polyplex of greater homogeneity. All subsequent formulation experiments used a TFR of 10 mL/min.
Next, we studied the concentration of lipid needed for efficient coating of the polyplex (N/P = 10). The effects of 0.5 mM, 1 mM, 2 mM, 4 mM and 8 mM lipid on the size, surface charge and polydispersity of the LCP were evaluated. At a concentration of 0.5 mM lipid, the LCP diameters were 259 ± 68 nm, a larger diameter than expected that we attribute to LCP aggregation. At 1, 2, 4 or 8 mM lipid concentrations, the LCP sizes decreased to an essentially uniform size of 108 ± 10 nm, 118 ± 40 nm, 110 ± 10 nm and 127 ± 20 nm, respectively (Figure 3D). Polydispersities were found to follow a similar trend wherein the LCP had high polydispersity (0.67 ± 0.15) at 0.5 mM, but becoming similar above 1 mM lipid concentration (0.16 ± 0.01 at 1 mM, 0.18 ± 0.01 at 2 mM, 0.15 ± 0.01 at 4 mM and 0.19 ± 0.01 at 8 mM) (Figure 3E). In contrast, ζ were found to display a significant decrease from −14.8 ± 2.2 mV at 0.5 mM to −26.8 ± 1.0 at 4 mM (Figure 3F). Increasing lipid concentration to 8 mM did not result in any significant change in surface charge in the zeta potential. Thus, we conclude that 4 mM lipids were sufficient to produce a complete coating of the polyplex, such that further increases in lipid concentration would only result in depositing multiple lipid coats onto the polyplex core and unloaded empty liposomes.
Finally, we also studied the effect of flow mixing on the size distribution of complexes in comparison to bulk mixing. For this we generated a batch of polyplex at N/P of 10 by bulk phase mixing followed by lipid coating using lipid film hydration method. The polyplex made in bulk phase had a larger hydrodynamic diameter of 154 ± 10 nm, with a polydispersity of 0.32 ± 0.16. However, a much more dramatic difference in size distribution of lipid-coated polyplexes were found between the two formulation techniques. LCP prepared with the lipid film hydration method resulted in much larger sized particles with a hydrodynamic diameter of 381 ± 9 nm and a polydispersity of 0.5 ± 0.05. The frequency distribution plots from DLS analysis (Supplementary Figure 2) demonstrate that flow mixing results in much more uniform lipid coat around the particles, but in lipid film hydration method, the lipid coating causes particles to form large aggregates and produce a wide size distribution of particles.
TEM Images
The morphology of the polyplex and LCP was also evaluated using negative stain transmission electron microscopy. TEM images showed that polyplex formed at N/P = 10 were smaller than 100 nm in diameter, in good agreement with our DLS data (Supplementary Figure 3A). Lipid coating of the polyplex increased the particle diameter. Size analysis of the TEM images using ImageJ revealed that the LCP are made up of about 90 nm diameter polyplex that are coated with a 12 nm lipid layer around the polyplex core, resulting in LCP that are about 114 nm in diameter (Supplementary Figure 3B). Since a single bilayer of lipid is approximately 5 nm in thickness, we can expect that there are about 2 bilayers around each polyplex at 4 mM concentration.37
Quantifying Plasmid, Pyrene And Curcumin Content
Next, we decided to probe the assembly of LCP as envisioned in Figure 1. We used four different treatment conditions to sequentially dissemble the LCP and release the encapsulated pDNA prior to incubation with the pDNA intercalating dye, PicoGreenTM. These were where the dye was added 1) without any treatment, 2) with heparin treatment alone, 3) with Triton X-100 treatment alone, and 4) with both heparin and Triton X-100 treatments. We used heparin at a concentration of 0.15 mg/mL to disassemble polyplex because addition of a higher heparin concentration did not produce increased PicoGreenTM fluorescence. This indicates that 0.15 mg/mL heparin is sufficient to completely decomplex the polyplex and release the entrapped pDNA (Supplementary Figure 4). Triton X-100 (1%) has been shown to disrupt membranes of multilamellar vesicles.38 As seen in Figure 4, for LCP, the intercalating dye is able to interact with released pDNA and fluoresce only after treatment with both heparin and Triton X-100. However, for the polyplex (Figure 4, inset) only heparin treatment is sufficient to release the pDNA and enabling dye interaction. This supports the concept that there is an additional layer of lipids in LCP that needs to be disrupted before the polyplex can be dissembled using heparin. Additionally, the percentage of the pDNA payload that was loaded inside the polymeric core of the LCP was calculated to be 92.0% ± 1.1% using the following formula:
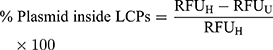
The remaining ~8% of the payload remained exposed to the surface and will likely be degraded in presence of nucleases.
Next, we determined the encapsulation and loading efficiency of the formulation technique. Plasmid and pyrene content analysis showed that there was no significant difference between the encapsulation and loading of the payload amongst the targeted, scrambled and non-targeted controls. The plasmid encapsulation efficiencies for non-targeted, scrambled and targeted formulations were 83.6% ± 0.2%, 86.1% ± 0.6% and 83.7% ± 5.1%, respectively (Figure 5A). Thus, ~76% of the plasmid used in the formulation ends up in the core of the LCP where it is completely protected from possible nuclease degradation. To determine the % mass of plasmid within the LCP, loading efficiencies were calculated and were found to be 0.33% ± 0.01% for all the different formulations (Figure 5B). Similarly, the encapsulation efficiencies of pyrene were found to be 24.3% ± 1.8%, 20.6% ± 0.8% and 21.3% ± 0.5% for the non-targeted, scrambled and targeted LCP, respectively (Figure 5C), indicating that the different formulations were each composed of 0.15% ± 0.01% of pyrene by mass (Figure 5D). For the curcumin loaded targeted LCP formulation, the encapsulation was 62.5% ± 4.7% and loading was at 0.8% ± 0.04% (Supplementary Figure 5). These findings indicate that a 1:2 pyrene:plasmid and 2.5:1 curcumin:plasmid mass ratio can be achieved within the LCP. Thus, for a 5.2 kB EGFP-NLS plasmid, it can be calculated that the LCP particles contain either 8300 pyrene or 23,000 curcumin molecules on average per copy of plasmid loaded within the LCP.
DNase Protection Assay
Since intravesical administration of the LCP would require it to be in contact with urine, we evaluated the ability of the delivery system to protect the plasmid against the nuclease degradation of the most prevalent DNA-hydrolyzing enzyme in urine, i.e., DNAse-I.39,40 LCP containing pDNA were treated with and without 2U DNAase-I at 37ºC for 4 hrs. After exposure to the enzyme, the DNAase was inactivated using EDTA before isolating the nucleic acid contents of the samples. Plasmids extracted from DNase treated and untreated LCP showed identical plasmid band patterns on agarose gels, indicating that the plasmid inside the LCP was protected from enzymatic degradation under these conditions. In contrast, the naked plasmid treated with same amount of DNAase did not show any electrophoretic bands on the gel, indicating that the DNAase enzyme was active and completely digested the naked plasmid (Figure 5E).
Quantifying Cellular Delivery Of Plasmid And Pyrene
The developed delivery system utilizes a novel bacterial adhesin peptide that enables the delivery system to engage exposed fibronectin matrix at the tumor site.41 We anticipate that this interaction will prevent loss of vehicle from the bladder tumor sites due to the urine influx and bladder voiding. Previously, we have reported that empty Cy5.5 labelled liposomes displaying the RWFV targeting peptide was able to associate with murine bladder cancer cells (MB49) is a sequence specific manner.28 Unfortunately, those dispersions were prepared using sonication and extrusion techniques, making it impossible to achieve the delivery of fragile nucleic acid payloads in a functional manner. The formulation technique used here avoids both sonication and extrusion to enable the encapsulation and delivery of functional plasmid. Interestingly, weak transfection activity was observed for CD2.5kD-PEI polyplex alone (i.e., no lipid coating or targeting peptide) (Figure 6). This can be attributed to the fact that although the polyplex associates with cells electrostatically in a non-specific manner due to its positive zeta potential, the transfection efficiency is low because low molecular weight PEI lacks the strong buffering capacity of its high molecular weight counterparts and hence cannot efficiently escape the endosomes.42,43 Additionally, the membrane destabilizing effect of the cyclodextrin may not be sufficient to promote endosomal escape, leading to cargo degradation within the lysosomal compartments. Thus, to facilitate endosomal escape, we utilized CHEMS and DOPE to coat the polyplex. At acidic endosomal pH, CHEMS becomes protonated and loses its ability to stabilize bilayers such that the inverted hexagonal phase forming lipid, DOPE, promotes fusion with the endosomal membrane.44,45 Since coating of the positively charged polyplex with a lipid mixture containing anionic CHEMS introduces a negative surface charge on the LCP, non-targeted LCP are electrostatically repelled from the cell surface, thus limiting entry of the LCP into the target cell population. Introduction of RWFV peptide onto the LCP particle surface, however, overcomes this barrier. To examine the impact of sequence specificity of the targeting peptide on transfection efficiency, we synthesized two versions of the targeting peptide-lipopolymer construct. One contained the critical RWFV tetrapeptide sequence and other contained WVRF (a scrambled version of the key RWFV binding motif) in the targeting peptide. Scrambled peptides are often used as negative controls to show that a specific sequence is critical for function.46,47 Naked DNA and Lipofectamine LTX were used as negative and positive controls, respectively. Cellular internalization of FAP-FBN-integrin complexes, the pathway by which RWFV constructs enter the cells via the caveolar uptake pathway, are known to occur with very slow kinetics in MB49 cells.41 Our results show that at 4 hrs and 12 hrs time points, transfection by RWFV-targeted LCP are significantly lower than the transfection achieved by Lipofectamine LTX. After 24 hrs, however, the presence of the targeting ligand resulted in four times greater GFP expression as compared to the scrambled peptide and comparable to positive controls. These results demonstrate that the RWVF tetrapeptide sequence is crucial for boosting the plasmid expression profile. Similar results were observed for pyrene delivery (Figure 7). After 4-hr incubation, targeted LCP resulted in 2-fold enhancement in pyrene delivery to cells relative to scrambled LCP and 4-fold relative to non-targeted LCP. After 24 hrs, the pyrene fluorescence decreases, possibly due to metabolism by the cellular polycyclic aromatic hydrocarbon (PAH) metabolizing enzymes.48,49 Secondly, these data show that although the internalization of the RWFV-targeted LCP-FBN-integrin complex occurs via slow uptake kinetics compared to cationic LipofectamineTM LTX, the endocytosed targeted LCP can efficiently decomplex and deliver similar quantity of functional payload to the nucleus for protein expression. Additionally, to make sure that the developed formulation was non-toxic to MB49 cells, we performed MTS assays. MTS data suggest that all the developed formulations resulted in similar cellular viability as compared to untreated cells, thus showing that the LCP were non-toxic to cells at concentrations used (Supplementary Figure 6).
Anti-Proliferative Effect Of RWFV-Targeted LCP On Bladder Cancer Cells
Curcumin is a safe, affordable and efficacious drug that has been shown to induce cell death in multiple cancer cell lines.50 Recently, curcumin treatment has been suggested as a potential chemopreventive therapy for intravesical administration in bladder cancer management.16,51,52 Thus, we sought to evaluate the anti-proliferative effect of our RWFV-targeted LCP loaded with curcumin. MTS assay data (Figure 8) show that after 48 hrs of incubation post-treatment, only 45% of MB49 bladder cancer cells are viable when treated with RWFV-targeted formulations at a dose of (5 µg/mL curcumin dose). Non-targeted formulations or free curcumin, however, did not display any significant toxicity at the concentrations tested. We attribute this observation to the removal of the non-targeted formulation or free drug during the washing step post 4-hr treatment (which mimics loss of drug in a clinical setting due to urine influx into bladder and bladder voiding), thus limiting the amount of drug that is internalized. The presence of the RWFV-targeting ligand, however, promotes internalization via the FAP-FBN-integrin pathway, to induce a cytotoxic effect. Additionally, the empty targeted LCP also did not show any significant signs of toxicity, suggesting that the delivery system components are well tolerated.
Conclusion
In conclusion, we have developed a carrier system for co-delivery of plasmid DNA and small hydrophobic molecules to MB49 bladder cancer cells in a targeted manner. The fabrication utilizes an easy microfluidic mixing approach that can be readily scaled-up.29,53 The inner cationic and outer hydrophobic character of the LCP allows for simultaneous entrapment of nucleic acids and small molecules, suggesting that these formulations may be useful for dual delivery approaches. The use of a lipid shell conveniently allowed the incorporation of the RWFV-lipopeptide, a component that confers tumor-targeting and internalization properties to the LCP formulations. The non-toxic nature of the RWFV-targeted LCP, combined with the efficient transfection and small molecule delivery characteristics, warrants further investigation as a co-delivery platform for intravesical therapy in bladder cancer management. Further investigation of the system in an MB49 orthotopic bladder cancer model is currently being pursued.
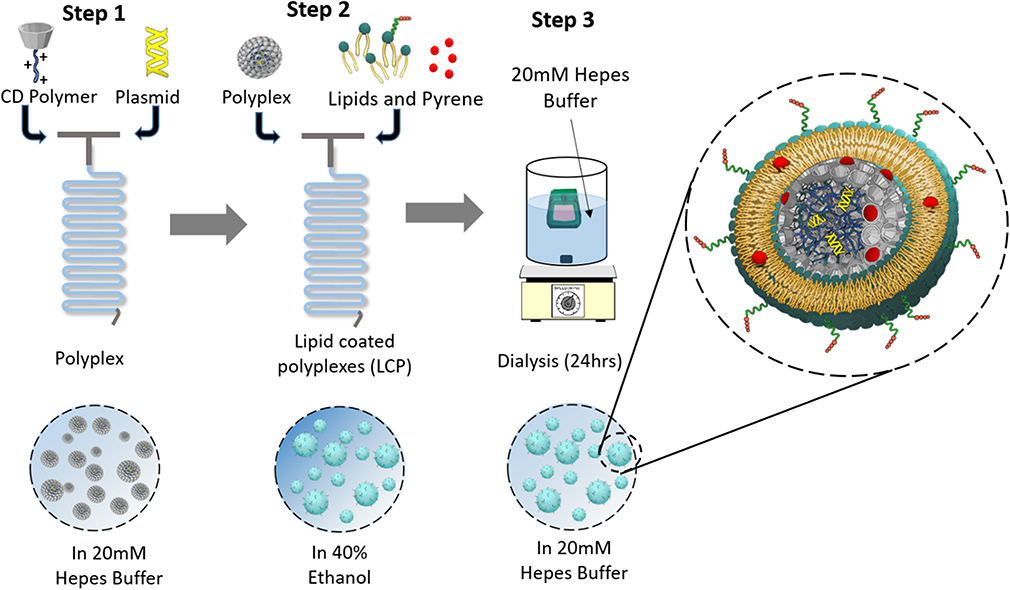 |
Figure 1 A schematic representation of LCP formulation steps. Step 1: cationic CD2.5kD-PEI and plasmid are mixed at the desired N/P ratio within a microfluidic reactor. Step 2: polyplex and a lipid mixture dissolved in 80% ethanol solution are mixed in the same microfluidic reactor. Step 3: the LCP are dialyzed to remove ethanol and free pyrene from the formulation. A total flow rate of 10 mL/min and flow rate ratio of 1:1 was used for mixing. (Enlargement) Conceptual diagram of a single lipid-coated polyplex (LCP) in cross section. |
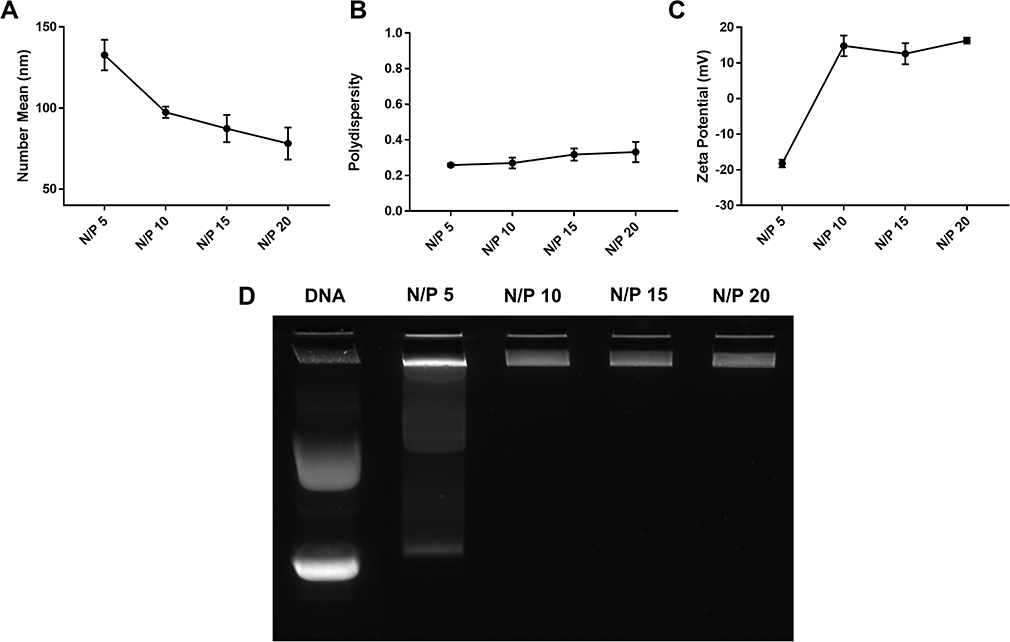 |
Figure 2 Effect of N/P ratio on polyplex properties. (A) Hydrodynamic diameter (number mean); (B) polydispersity; (C) zeta potential; (D) gel-shift assay for polyplex prepared by mixing 60 µg/mL plasmid with CD2.5kD-PEI in 20mM HEPES buffer at different N/P ratios. The total flow rate was 10 mL/min and the flow rate ratio was 1:1. Results are the mean ± SD (n=3). |
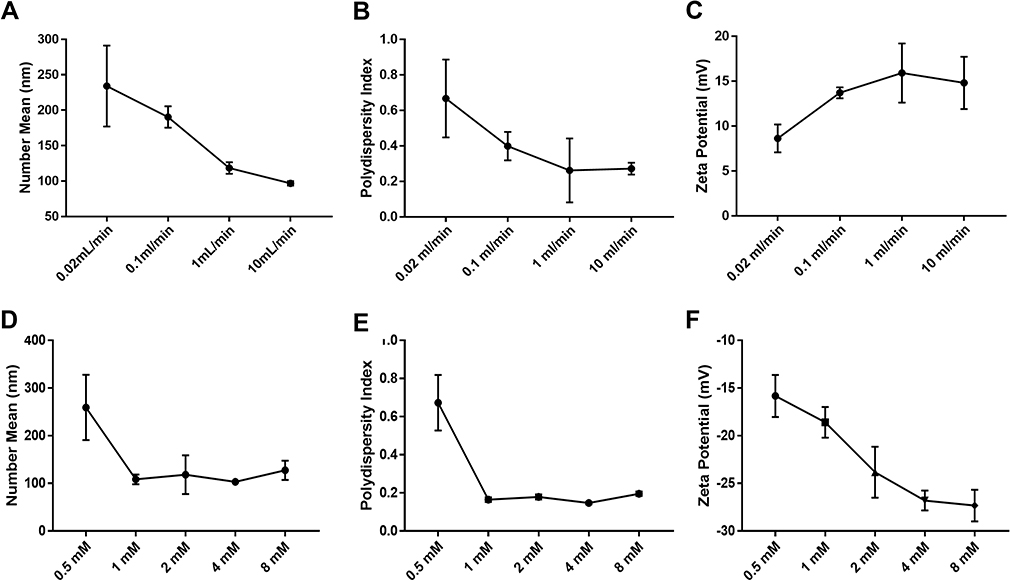 |
Figure 3 Effect of total flow rate and lipid concentrations on properties of polyplex and LCP, respectively. (A, D) Hydrodynamic diameter (number mean); (B, E) polydispersity; and (C, F) zeta potential. Polyplex were prepared by mixing 60 µg/mL plasmid with CD2.5kD-PEI in 20mM HEPES buffer at a N/P ratio of 10. The flow rate ratio was kept constant at 1:1. For LCP, 1 mL of polyplex solution was mixed with 1 mL of ethanolic lipid mixture (DOPE:CHEMS:DSPC:DSPE-mPEG2k = 34:54:10:2) solution of varying concentration. The total flow rate was kept constant at 10 mL/min and flow rate ratio was 1:1. Results are the mean ± SD (n=3). |
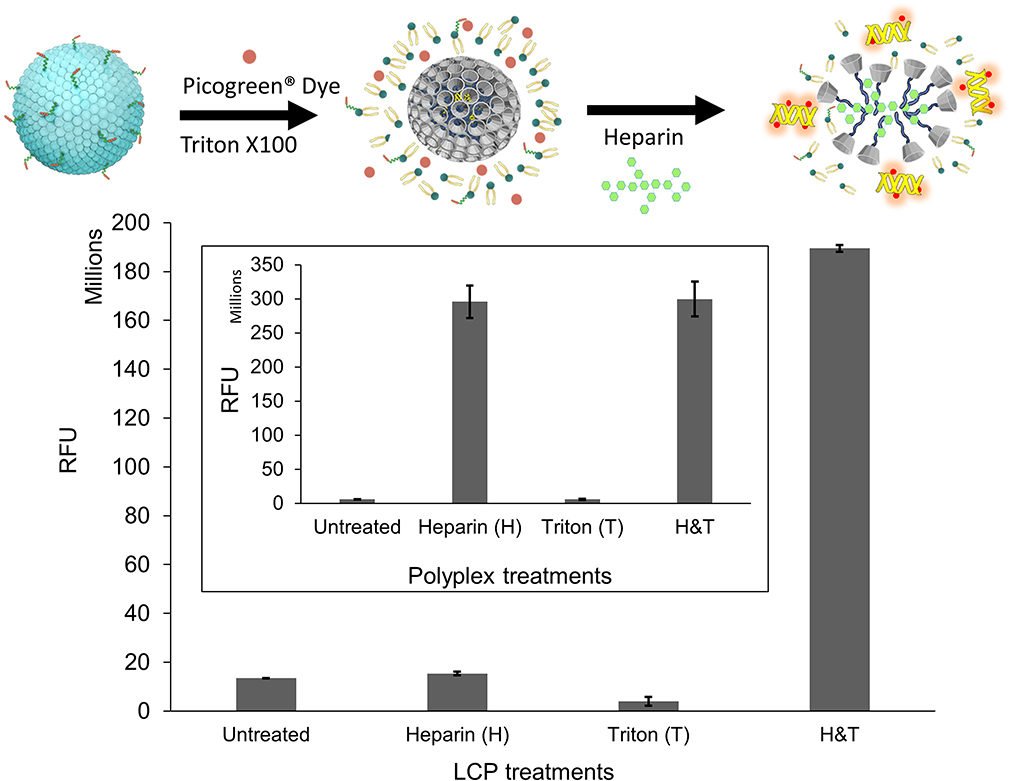 |
Figure 4 Effect of heparin and Triton X-100 treatments on disassembly of LCP and polyplex (Inset). LCP or polyplex were treated at a concentration of 0.15 mg/mL heparin or 1% Triton X-100 or both. Disassembly was studied by PicoGreenTM fluorescence enhancement at 490 nm excitation and 525 nm emission wavelengths. |
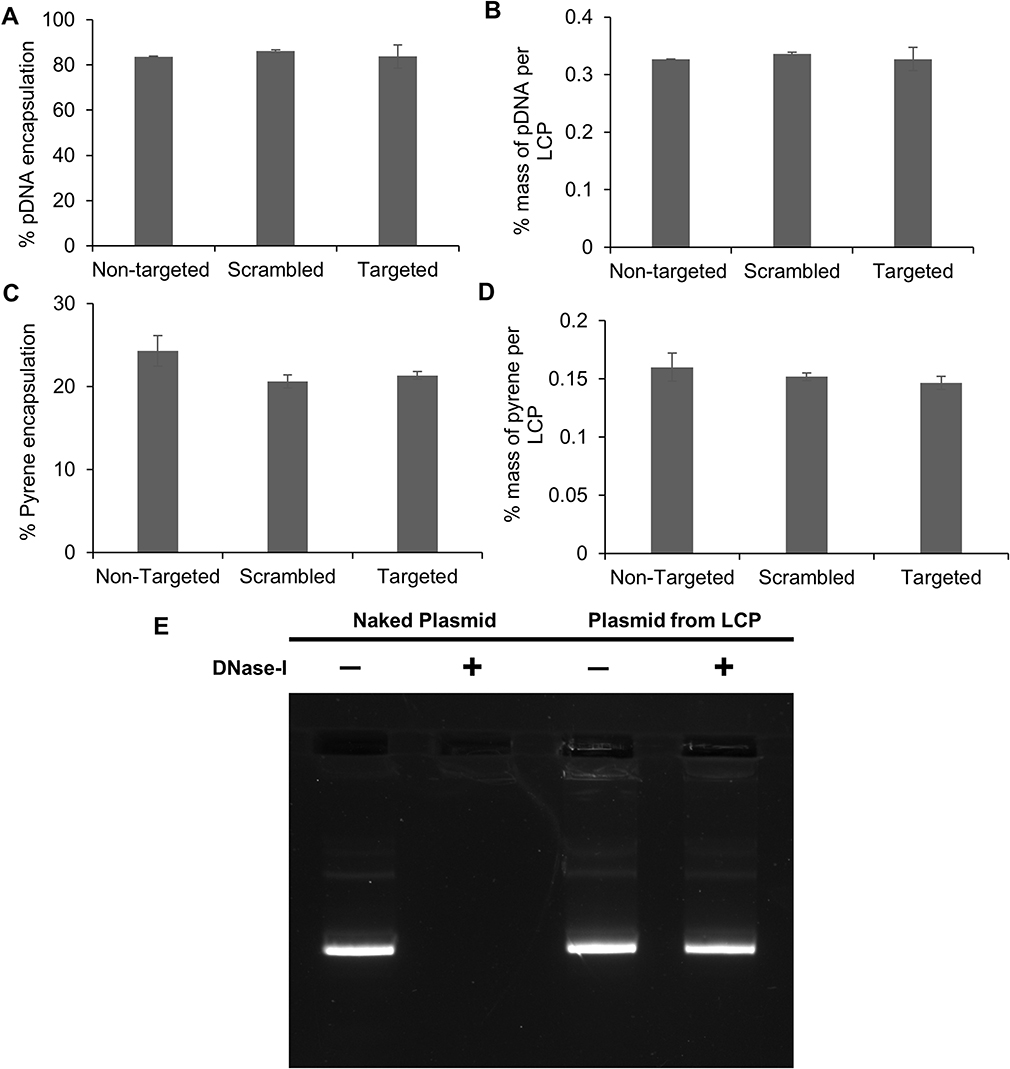 |
Figure 5 Encapsulation and loading efficiency of (A, B) plasmid and (C, D) pyrene. (E) The ability of LCP to protect loaded plasmid from DNAase degradation. Plasmid content was measured via PicoGreenTM assay after decomplexing the LCP using 1% Triton-X100 to disrupt the lipid layer and 0.15 mg/mL heparin to release the plasmid from the polyplex. Pyrene content was measured by dissolving the LCP in 70% ACN solution to release pyrene, followed by HPLC separation and pyrene detection using absorbance at 338 ± 4 nm. Results are reported as mean ± SD (n=3). For analyzing enzymatic degradation of plasmid, LCP were treated for 4 hrs with 2U DNAase-I at 37ºC before quenching the enzymatic reaction with 2.5 mM EDTA and heating at 65ºC for 10 mins. The entrapped pDNA was released using sequential treatment with 1% Triton X-100 and 0.15 mg/mL heparin. The released pDNA was concentrated using vacuum centrifugation before agarose gel (1%) electrophoresis to analyze for plasmid integrity. |
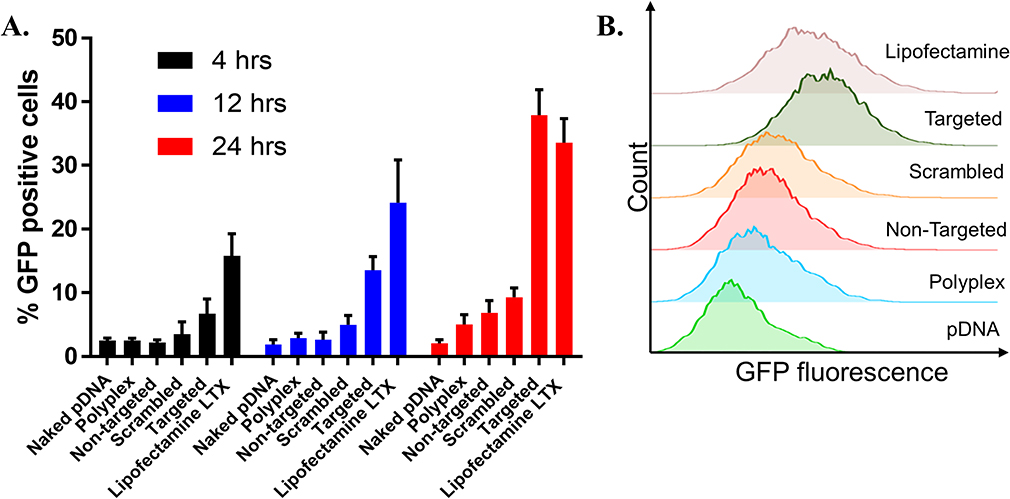 |
Figure 6 (A) EGFP-NLS plasmid transfection efficiency of the non-targeted, scrambled and targeted LCP. (B) Histogram plot for GFP fluorescence distribution in MB49 cell population at 24 hrs. Cells were treated with LCP formulations for 4 hrs at 37°C, followed by PBS washing to remove unbound complexes. The cells were further incubated for 4 hrs, 12 hrs or 24 hrs and then trypsinized, and resuspended in PBS for flow cytometry analysis. Error bars indicate the standard deviation of the mean (n=3). (Two-way ANOVA). p<0.0001. |
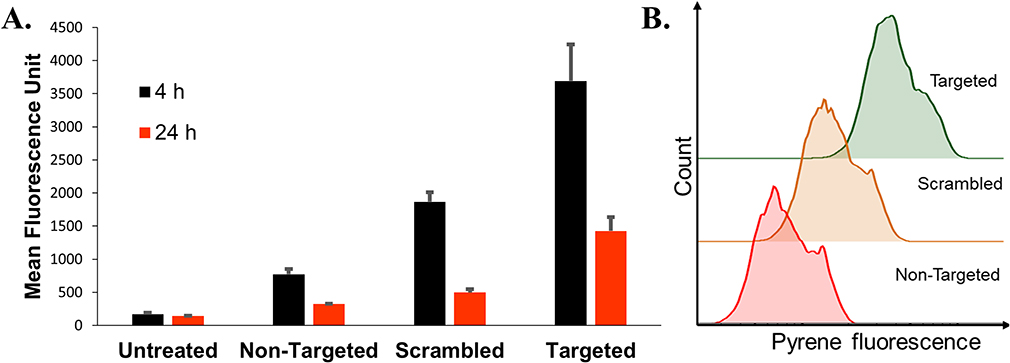 |
Figure 7 (A) Pyrene delivery of the non-targeted, scrambled and targeted LCP. (B) Histogram plot for pyrene fluorescence distribution in MB49 cell population at 4 hrs. MB49 cells were treated with LCP for 4 hrs at 37°C, followed by PBS washing to remove non-specific interactions. After further 4 or 24-hr incubation, cells were trypsinized, and resuspended in PBS for quantification of pyrene fluorescence using flow cytometer. Error bars indicate the standard deviation of the mean (n=3). Two-way ANOVA, p<0.0001. |
 |
Figure 8 Anti-proliferative effect of RWFV-targeted LCP loaded with curcumin. MB49 cells were treated for 4 hrs followed by washing to remove uninternalized particles. The cells were incubated another 48 hrs before adding the MTS reagent. After incubating for 1 hr, the absorbance of soluble formazan formed due to bio-reduction of MTS by live MB49 cells population was measured at 490 nm. All treatments for each formulation were performed in triplicate and error bars represent the standard deviation of the mean (n=3). |
Acknowledgments
The authors would like to thank the Purdue Center for Cancer Research Pilot Grant Program (CCSG P30 CA023168) for the support of this project. NIH Shared Instrumentation Grant S10DO20029.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Anastasiadis A, de Reijke TM. Best practice in the treatment of nonmuscle invasive bladder cancer. Ther Adv Urol. 2012;4(1):13–32.
2. Botteman MF, Pashos CL, Redaelli A, Laskin B, Hauser R. The health economics of bladder cancer. Pharmacoeconomics. 2003;21(18):1315–1330. doi:10.1007/BF03262330
3. Massari F, Santoni M, Ciccarese C, et al. Emerging concepts on drug resistance in bladder cancer: implications for future strategies. Crit Rev Oncol Hematol. 2015;96(1):81–90. doi:10.1016/j.critrevonc.2015.05.005
4. Volpe A, Racioppi M, D’Agostino D, et al. Advanced bladder cancer: new agents and new approaches. A review. Urol Oncol. 2013;31(1):9–16. doi:10.1016/j.urolonc.2010.03.022
5. Roh Y-G, Mun M-H, Jeong M-S, et al. Drug resistance of bladder cancer cells through activation of ABCG2 by FOXM1. BMB Rep. 2018;51(2):98–103. doi:10.5483/BMBRep.2018.51.2.222
6. Sonpavde G, Sternberg CN, Rosenberg JE, Hahn NM, Galsky MD, Vogelzang NJ. Second-line systemic therapy and emerging drugs for metastatic transitional-cell carcinoma of the urothelium. Lancet Oncol. 2010;11(9):861–870. doi:10.1016/S1470-2045(10)70086-3
7. Saika T, Tsushima T, Och J, et al. Over-expression of metallothionein and drug-resistance in bladder cancer. Int J Urol. 1994;1(2):135–139. doi:10.1111/iju.1994.1.issue-2
8. Kunze D, Erdmann K, Froehner M, Wirth MP, Fuessel S. siRNA-mediated inhibition of antiapoptotic genes enhances chemotherapy efficacy in bladder cancer cells. Anticancer Res. 2012;32(10):4313–4318.
9. Li X, Wang H, Wang J, et al. Emodin enhances cisplatin-induced cytotoxicity in human bladder cancer cells through ROS elevation and MRP1 downregulation. BMC Cancer. 2016;16(1):578. doi:10.1186/s12885-016-2640-3
10. Khan M, Ong ZY, Wiradharma N, Attia ABE, Yang -Y-Y. Advanced materials for co-delivery of drugs and genes in cancer therapy. Adv Healthc Mater. 2012;1(4):373–392. doi:10.1002/adhm.201200109
11. Yang Z, Gao D, Cao Z, et al. Drug and gene co-delivery systems for cancer treatment. Biomater Sci. 2015;3(7):1035–1049. doi:10.1039/C4BM00369A
12. Dai X, Tan C. Combination of microRNA therapeutics with small-molecule anticancer drugs: mechanism of action and co-delivery nanocarriers. Adv Drug Deliv Rev. 2015;81:184–197. doi:10.1016/j.addr.2014.09.010
13. Shen Z, Shen T, Wientjes MG, O’Donnell MA, Au JLS. Intravesical treatments of bladder cancer: review. Pharm Res. 2008;25(7):1500–1510. doi:10.1007/s11095-008-9566-7
14. Ariotti N, Hall Thomas E, Rae J, et al. Modular detection of GFP-labeled proteins for rapid screening by electron microscopy in cells and organisms. Dev Cell. 2015;35(4):513–525. doi:10.1016/j.devcel.2015.10.016
15. Takeuchi H, Yamamoto H, Niwa T, Hino T, Kawashima Y. Mucoadhesion of polymer-coated liposomes to rat intestine in vitro. Chem Pharm Bull (Tokyo). 1994;42(9):1954–1956. doi:10.1248/cpb.42.1954
16. Falke J, Parkkinen J, Vaahtera L, Hulsbergen-van de Kaa CA, Oosterwijk E, Witjes JA. Curcumin as treatment for bladder cancer: a preclinical study of cyclodextrin-curcumin complex and BCG as intravesical treatment in an orthotopic bladder cancer rat model. Biomed Res Int. 2018;2018:7. doi:10.1155/2018/9634902
17. Patnaik S, Gupta KC. Novel polyethylenimine-derived nanoparticles for in vivo gene delivery. Expert Opin Drug Deliv. 2013;10(2):215–228. doi:10.1517/17425247.2013.744964
18. Li C, Zhong D, Zhang Y, et al. The effect of the gene carrier material polyethyleneimine on the structure and function of human red blood cells in vitro. J Mater Chem B. 2013;1(14):1885–1893. doi:10.1039/c3tb00024a
19. Petersen H, Fechner PM, Martin AL, et al. Polyethylenimine-graft-poly(ethylene glycol) copolymers: influence of copolymer block structure on DNA complexation and biological activities as gene delivery system. Bioconjug Chem. 2002;13(4):845–854. doi:10.1021/bc025529v
20. Godbey WT, Wu KK, Mikos AG. Size matters: molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J Biomed Mater Res. 1999;45(3):268–275. doi:10.1002/(ISSN)1097-4636
21. Forrest ML, Gabrielson N, Pack DW. Cyclodextrin–polyethylenimine conjugates for targeted in vitro gene delivery. Biotechnol Bioeng. 2005;89(4):416–423. doi:10.1002/(ISSN)1097-0290
22. Mishra S, Webster P, Davis ME. PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles. Eur J Cell Biol. 2004;83(3):97–111. doi:10.1078/0171-9335-00363
23. Bus T, Traeger A, Schubert US. The great escape: how cationic polyplexes overcome the endosomal barrier. J Mater Chem B. 2018;6(43):6904–6918. doi:10.1039/C8TB00967H
24. Zhao W, Schorey JS, Groger R, Allen PM, Brown EJ, Ratliff TL. Characterization of the fibronectin binding motif for a unique mycobacterial fibronectin attachment protein, FAP. J Biol Chem. 1999;274(8):4521–4526. doi:10.1074/jbc.274.8.4521
25. Kulkarni A, VerHeul R, DeFrees K, et al. Microfluidic assembly of cationic-β-cyclodextrin: hyaluronicacid-adamantane host: guest DNA nanoparticles. Biomater Sci. 2013;1(10):1029–1033. doi:10.1039/c3bm00189j
26. Wright KJ, Badwaik VD, Samaddar S, et al. Organocatalytic synthesis and evaluation of polycarbonate pendant polymer:β-Cyclodextrin-based nucleic acid delivery vectors. Macromolecules. 2018;51(3):670–678. doi:10.1021/acs.macromol.7b02293
27. Kulkarni A, Badwaik V, DeFrees K, et al. Effect of pendant group on pDNA delivery by cationic-β-cyclodextrin: alkyl-PVA-PEGPendant polymer complexes. Biomacromolecules. 2014;15(1):12–19. doi:10.1021/bm401096v
28. Lee Y, Kischuk E, Crist S, Ratliff TL, Thompson DH. Targeting and internalization of liposomes by bladder tumor cells using a fibronectin attachment protein-derived peptide–lipopolymer conjugate. Bioconjug Chem. 2017;28(5):1481–1490. doi:10.1021/acs.bioconjchem.7b00153
29. Jeffs LB, Palmer LR, Ambegia EG, Giesbrecht C, Ewanick S, MacLachlan I. A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm Res. 2005;22(3):362–372. doi:10.1007/s11095-004-1873-z
30. Heyes J, Palmer L, Chan K, Giesbrecht C, Jeffs L, MacLachlan I. Lipid encapsulation enables the effective systemic delivery of polyplex plasmid DNA. Mol Ther. 2007;15(4):713–720. doi:10.1038/sj.mt.6300101
31. Duan X, Xiao J, Yin Q, Zhang Z, Mao S, Li Y. Amphiphilic graft copolymer based on poly(styrene-co-maleic anhydride) with low molecular weight polyethylenimine for efficient gene delivery. Int J Nanomedicine. 2012;7:4961–4972. doi:10.2147/IJN.S32069
32. Capretto L, Cheng W, Hill M, Zhang X. Micromixing within microfluidic devices. In: Lin B, editor. Microfluidics: Technologies and Applications. Berlin, Heidelberg: Springer Berlin Heidelberg; 2011:27–68.
33. Ward K, Fan ZH. Mixing in microfluidic devices and enhancement methods. J Micromech Microeng. 2015;25:9. doi:10.1088/0960-1317/25/9/094001
34. Reynolds O. An experimental investigation of the circumstances which determine whether the motion of water shall be direct or sinuous, and of the law of resistance in parallel channels. Philos Trans R Soc London. 1883;174:935–982.
35. Wong SH, Ward MC, Wharton CW. Micro T-mixer as a rapid mixing micromixer. Sens Actuators B Chem. 2004;100(3):359–379. doi:10.1016/j.snb.2004.02.008
36. Kang T, Anderson P. The effect of inertia on the flow and mixing characteristics of a chaotic serpentine mixer. Micromachines. 2014;5(4):1270–1286. doi:10.3390/mi5041270
37. Ding W, Palaiokostas M, Wang W, Orsi M. Effects of lipid composition on bilayer membranes quantified by all-atom molecular dynamics. J Phys Chem B. 2015;119(49):15263–15274. doi:10.1021/acs.jpcb.5b06604
38. Sila M, Au S, Weiner N. Effects of triton X-100 concentration and incubation temperature on carboxyfluorescein release from multilamellar liposomes. Biochim Biophys Acta Biomembr. 1986;859(2):165–170. doi:10.1016/0005-2736(86)90211-7
39. Bryzgunova OE, Laktionov PP. Extracellular nucleic acids in urine: sources, structure, diagnostic potential. Acta Naturae. 2015;7(3):48–54. doi:10.32607/20758251-2015-7-3-48-54
40. Nadano D, Yasuda T, Kishi K. Measurement of deoxyribonuclease I activity in human tissues and body fluids by a single radial enzyme-diffusion method. Clin Chem. 1993;39(3):448–452.
41. Coon BG, Crist S, González-Bonet AM, et al. Fibronectin attachment protein from bacillus Calmette-Guerin as targeting agent for bladder tumor cells. Int J Cancer. 2012;131(3):591–600. doi:10.1002/ijc.v131.3
42. Guo Z, Kong Z, Wei Y, et al. Effects of gene carrier polyethyleneimines on the structure and binding capability of bovine serum albumin. Spectrochim Acta A Mol Biomol Spectrosc. 2017;173:783–791. doi:10.1016/j.saa.2016.10.026
43. Fischer D, Bieber T, Li Y, Elsässer H-P, Kissel T. A novel non-viral vector for DNA delivery based on low molecular weight, branched polyethylenimine: effect of molecular weight on transfection efficiency and cytotoxicity. Pharm Res. 1999;16(8):1273–1279. doi:10.1023/A:1014861900478
44. Lai MZ, Vail WJ, Szoka FC. Acid- and calcium-induced structural changes in phosphatidylethanolamine membranes stabilized by cholesteryl hemisuccinate. Biochemistry. 1985;24(7):1654–1661. doi:10.1021/bi00328a013
45. Hafez IM, Cullis PR. Cholesteryl hemisuccinate exhibits pH sensitive polymorphic phase behavior. Biochim Biophys Acta Biomembr. 2000;1463(1):107–114. doi:10.1016/S0005-2736(99)00186-8
46. Mooney C, Haslam NJ, Pollastri G, Shields DC. Towards the improved discovery and design of functional peptides: common features of diverse classes permit generalized prediction of bioactivity. PLoS One. 2012;7(10):e45012–e45012. doi:10.1371/journal.pone.0045012
47. Mazzucchelli L, Burritt JB, Jesaitis AJ, et al. Cell-specific peptide binding by human neutrophils. Blood. 1999;93(5):1738–1748. doi:10.1182/blood.V93.5.1738
48. Moorthy B, Chu C, Carlin DJ. Polycyclic aromatic hydrocarbons: from metabolism to lung cancer. Toxicol Sci. 2015;145(1):5–15. doi:10.1093/toxsci/kfv040
49. Huberman E, Selkirk JK, Heidelberger C. Metabolism of polycyclic aromatic hydrocarbons in cell cultures. Cancer Res. 1971;31(12):2161–2167.
50. Ravindran J, Prasad S, Aggarwal BB. Curcumin and cancer cells: how many ways can curry kill tumor cells selectively? AAPS J. 2009;11(3):495–510. doi:10.1208/s12248-009-9128-x
51. Shi J, Wang Y, Jia Z, Gao Y, Zhao C, Yao Y. Curcumin inhibits bladder cancer progression via regulation of β-catenin expression. Tumor Biol. 2017;39(7):1010428317702548. doi:10.1177/1010428317702548
52. Watanabe FT, Chade DC, Reis ST, et al. Curcumin, but not Prima-1, decreased tumor cell proliferation in the syngeneic murine orthotopic bladder tumor model. Clinics. 2011;66:2121–2124. doi:10.1590/S1807-59322011001200019
53. Gomez L, Sebastian V, Irusta S, Ibarra A, Arruebo M, Santamaria J. Scaled-up production of plasmonic nanoparticles using microfluidics: from metal precursors to functionalized and sterilized nanoparticles. Lab Chip. 2014;14(2):325–332. doi:10.1039/C3LC50999K
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.