")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Designing biologic selectivity for inflammatory bowel disease – role of vedolizumab
Authors Krupka N, Baumgart D
Received 24 July 2014
Accepted for publication 12 November 2014
Published 17 December 2014 Volume 2015:9 Pages 147—154
DOI https://doi.org/10.2147/DDDT.S50348
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Niklas Krupka, Daniel C Baumgart
Division of Gastroenterology and Hepatology, Department of Medicine, Charité Medical School, Humboldt-University of Berlin, Berlin, Germany
Abstract: Crohn’s disease and ulcerative colitis are two chronic inflammatory bowel conditions. Current approved biologic therapies are limited to blocking tumor necrosis factor alpha. Unfortunately, some patients are primary nonresponders, experiencing a loss of response, intolerance, or side effects. This defines an unmet need for novel therapeutic strategies. The rapid recruitment and inappropriate retention of leukocytes is a hallmark of chronic inflammation and a potentially promising therapeutic target. Here we discuss the clinical trial results of vedolizumab (anti-α4β7, LDP-02, MLN-02, and MLN0002) and its impact on future management of inflammatory bowel disease.
Keywords: ulcerative colitis, Crohn’s disease, vedolizumab, MLN0002, MLN-02, LDP-02, anti-α4β7
Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are idiopathic inflammatory bowel diseases (IBD) that mainly affect the gastrointestinal tract but can manifest in the entire human body. From the current understanding, the pathophysiological events in IBD involve a disturbance of the commensal microbiota, an impairment of mucosal defense mechanisms, an increased permeability of the gut’s epithelial layer, and a dysregulation of the innate and adaptive immune system, all of which eventually promote an aberrant immune response against nonpathogenic gut bacteria and subsequent tissue damage.1–4 Genome-wide association studies have shown a wide range of susceptibility loci for CD and UC, with a significant overlap between both diseases; however, the precise interplay of genetic and environmental factors that underlies disease is still unknown.5–7
One of the histopathological hallmarks of CD, UC, and most chronic inflammatory processes in general is a markedly increased number of leukocytes, specifically memory T cells, in affected tissues, which results from increased cell extravasation and/or retention.8,9 Importantly, the transendothelial migration of leukocytes is a highly regulated process that involves numerous leukocyte and endothelial surface molecules.10,11 Specifically, binding of the leukocyte α4β7 integrin to its principal ligand, the mucosal addressin cellular adhesion molecule 1 (MAdCAM-1), which is expressed in high endothelial venules of the gut lamina propria, gut-associated lymphoid tissue, and mesenteric lymph nodes, has been shown to be pivotal in leukocyte homing to the gastrointestinal tract.12–17 In CD and UC, the expression of MAdCAM-1 is highly upregulated in high endothelial venules of inflammatory sites and promotes an increased capacity to bind leukocytes.18,19 This strongly supports relevance of the MAdCAM-1–α4β7 integrin interaction in disease and makes it a promising therapeutic target.
Current targeted therapies for inflammatory bowel disease
Therapies targeting tumor necrosis factor
Prior to the development of targeted therapies, treatment of CD and UC was based on nonselective modulation or suppression of the immune response, which frequently suffered from limited efficacy or severe side effects associated with immunosuppression. More than a decade ago, infliximab (Remicade®; Janssen Biotech Inc.) – a monoclonal antibody targeting tumor necrosis factor (TNF) – was the first biologic therapy to be approved by the US Food and Drug Administration (FDA) for the treatment of CD and later UC. Large clinical trials and a vast amount of clinical data have proven the efficacy of anti-TNF therapy in CD and UC, and its availability has significantly improved treatment of patients with IBD.20–22 In the last few years, other anti-TNF agents such as adalimumab (Humira®; AbbVie), certolizumab (Cimzia®; UCB), and golimumab (Simponi®; MSD) were approved and now allow clinicians to choose among different application routes and intervals (Table 1). Anti-TNF therapy, however, may be associated with a number of serious and potentially life-threatening adverse events, such as malignancies or opportunistic infections.23,24 Moreover, approximately one third of patients are primary nonresponders to anti-TNF therapy, and another 30%–40% of primary responders eventually lose response to treatment or become intolerant.20,25,26 Hence, new therapeutic strategies are urgently needed.
 | Table 1 Biological therapy for IBD |
Leukocyte migration inhibitors
Based on the pivotal role of leukocyte migration in the pathogenesis of IBD, much basic and clinical research in recent years has focused on identifying and modifying underlying pathways.9,27 Interestingly, the tissue-specificity of the participating ligands and receptors theoretically allows an organ-selective therapy, compared to the rather radical approach of general immunosuppression or immunomodulation. In 2008, natalizumab (Antegren®, Tysabri®; Biogen Idec Inc.), a humanized monoclonal immunoglobulin G4 antibody against the α4 chains of the α4β1 and α4β7 integrin heterodimers, received FDA approval for the treatment of moderate to severe CD. Natalizumab, which had been used before in the treatment of multiple sclerosis,28–31 inhibits interactions of α4β1 integrin with the vascular cell adhesion molecule-1 (VCAM-1) and of α4β7 integrin with MAdCAM-1, respectively, and thereby impairs leukocyte migration to the central nervous system and the gut.12–17 After significant effects of natalizumab on disease activity in patients with CD were reported in early double-blind, randomized controlled trials,32,33 its efficacy as an induction and maintenance therapy for CD was confirmed in the ENACT (International Efficacy of Natalizumab as Active Crohn’s Therapy) and ENCORE (Efficacy of Natalizumab in Crohn’s Disease Response and Remission) trials.34,35 Studies that evaluate natalizumab in patients with UC are rare; however, results of a small pilot trial with ten patients with active UC suggest a positive efficacy.36 Although an acceptable safety profile was first assumed from data of the described clinical trials, case reports of JC (John Cunningham) virus infection with fatal progressive multifocal leukoencephalopathy (PML) after therapy with natalizumab largely restricted its use and damped the early enthusiasm about the potential of leukocyte migration inhibitors.37–39 Nonetheless, proof of concept was shown and, as a consequence, newer gut-specific biologics such as vedolizumab were designed.
Development of vedolizumab for inflammatory bowel disease
Pharmacodynamics
Vedolizumab (MLN0002, MLN02, LDP-02, anti-α4β7) is a humanized monoclonal immunoglobulin G1 antibody targeting α4β7 integrin, which is a potential ligand of VCAM-1 or MAdCAM-1. Pharmacological studies showed that the highest level of vedolizumab binding is to a subset of memory CD4+ cells (including Th17 cells) and eosinophils. Low-to-intermediate-level binding was observed for naïve CD4+ cells, CD8+ cells, B cells, natural killer cells, and basophils. Vedolizumab did not bind to neutrophils, the majority of memory CD4+ lymphocytes, and most monocytes. Importantly, highly specific binding of vedolizumab to α4β7 but not to α4β1 or αEβ7 integrins was confirmed by a series of flow cytometry analyses. Interestingly, although α4β7 integrin is a potential ligand for both MAdCAM-1 and VCAM-1, vedolizumab selectively inhibited adhesion of α4β7 integrin-expressing cells to MAdCAM-1 but not to VCAM-1, even at high concentrations.40 Taken together, these pharmacological properties of vedolizumab suggest that it has the potential to modulate the adaptive immune system of the gastrointestinal tract without causing the systemic side effects that were reported for natalizumab.
Pharmacokinetics
The pharmacokinetic properties of vedolizumab were first assessed in a Phase II dose-ranging study with 46 patients with UC. Infusions of 2 mg/kg, 6 mg/kg, or 10 mg/kg vedolizumab or placebo were administered on day 1, day 15, day 29, and day 85, and evaluations were performed repeatedly through day 253. Maximum serum concentrations and the area under the curve increased proportionally to the administered dose, and serum concentrations declined with a mean half-life of 15–22 days. Maximum saturation of α4β7 integrin on serum lymphocytes was observed at all measurable serum concentrations of vedolizumab.41 Pharmacokinetic data from the Phase III clinical trials that are described in detail below suggest a similar profile in patients with UC or CD, with similar serum trough concentrations at week 6 after administration of 300 mg of vedolizumab at week 0 and week 2 and at steady state using application intervals of 4 weeks or 8 weeks. Importantly, a target saturation of >95% was reported for both dosing intervals and independent of CD of UC. Anti-vedolizumab antibodies were detected in 4.1% (CD) and 3.7% (UC) of patients in at least one sample, and rates were reduced in patients with concomitant immunosuppressive therapy.42,43
Efficacy and safety
After promising results of a study in cotton-top tamarins with chronic colitis, in which administration of an anti-α4β7 monoclonal antibody reduced colonic inflammation and ameliorated diarrhea,44 Feagan et al45 tested the efficacy of vedolizumab in a double-blind, controlled trial of vedolizumab in 181 patients with active UC (for an overview of studies with clinically valuable endpoints, Table 2). Patients were randomly assigned to receive an infusion of vedolizumab (0.5 mg/kg or 2.0 mg/kg) or placebo on day 1 and day 29. The primary outcome measure was clinical remission at week 6, defined as an UC clinical score ≤1 and a modified Baron endoscopic score ≤1, with no evidence of rectal bleeding. Moreover, changes in clinical scores and endoscopic response were evaluated. Clinical remission rates at week 6 significantly differed between patients who received 2.0 mg/kg (32%, n=60) or 0.5 mg/kg (33%, n=58) of vedolizumab, compared with placebo (14%, n=63, P=0.02). Consistently, endoscopic remission was observed in 28% of patients receiving 0.5 mg/kg and 12% of those receiving 2.0 mg/kg of vedolizumab, compared with 8% of those receiving placebo (P=0.007). The prevalence of adverse events was similar in all groups.
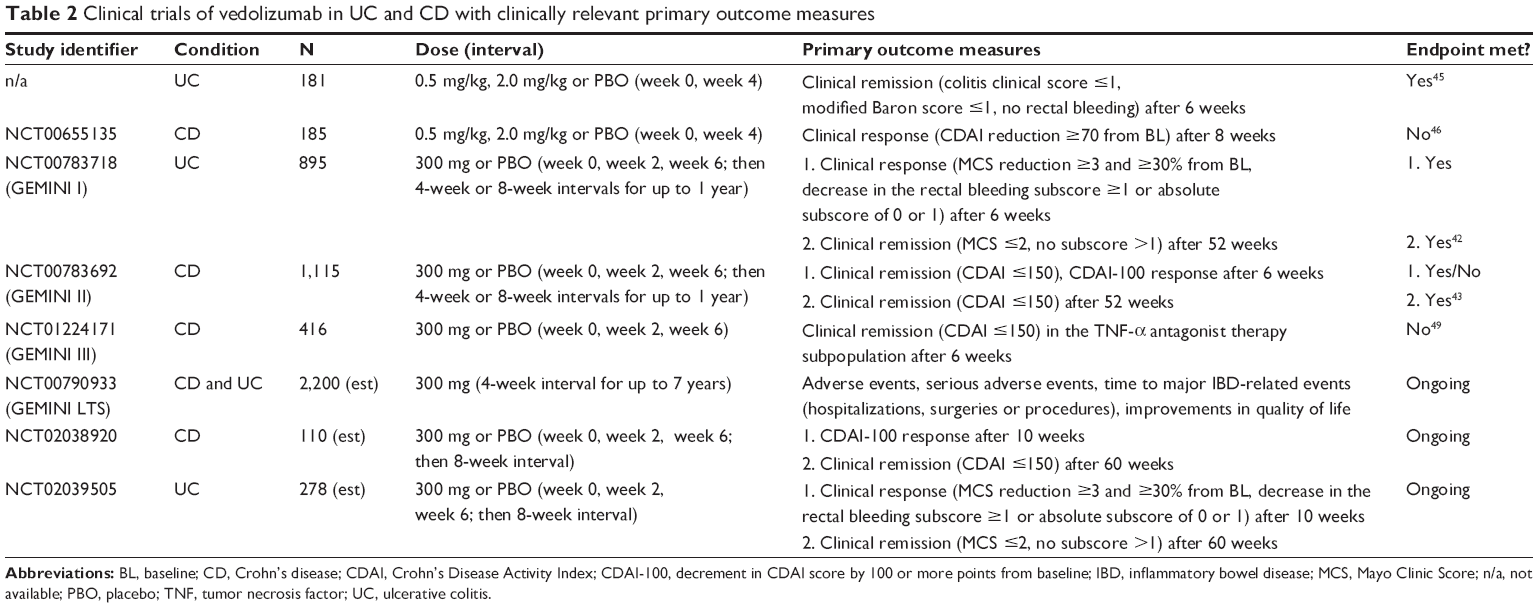 | Table 2 Clinical trials of vedolizumab in UC and CD with clinically relevant primary outcome measures |
In a second double-blind, randomized controlled trial, 185 patients with active CD who were naïve to biologic therapies and had inflammation of the ileum and/or colon were randomized to receive an infusion of vedolizumab (0.5 mg/kg or 2.0 mg/kg) or placebo on day 1 and day 29.46 The primary end point was clinical response at week 8, defined as a decrement in the Crohn’s Disease Activity Index (CDAI)47 by ≥70 points. Additional end points were clinical remission (CDAI ≤150) and CDAI-100 response (decrement in CDAI by ≥100 points from baseline). The primary outcome was not met, as clinical response rates were similar in patients who received 2.0 mg/kg (53%; n=65) or 0.5 mg/kg of vedolizumab (49%; n=62) or placebo (41%; n=58). However, rates of clinical remission and CDAI-100 response differed significantly between the 2.0 mg/kg and placebo group. Rates of adverse events were again similar in all groups.
Most recently, encouraging results were published from a series of large multicenter, double-blind, randomized controlled trials of vedolizumab in UC and CD patients (Table 2).42,43 In the GEMINI I trial, 374 patients with UC were randomized to receive an infusion of 300 mg of vedolizumab or placebo at week 0 and week 2; clinical response (defined as a decrease in the Mayo Clinic Score [MCS]48 ≥3 points and ≥30% from baseline as well as a decrease in the rectal bleeding subscore of ≥1 or an absolute rectal bleeding subscore of 0 or 1) was evaluated at week 6. An additional cohort (521 patients) received open-label vedolizumab. In a maintenance study, patients who had a response to vedolizumab were randomly assigned to continued therapy with vedolizumab every 8 weeks or 4 weeks or to placebo for up to 52 weeks; rates of clinical remission (MCS ≤2 and no subscore >1) were determined.42 Rates of clinical response at week 6 were 47.1% in the vedolizumab group versus 25.5% in the placebo group (P<0.001). Moreover, at week 52, clinical remission was seen in 41.8% and 44.8% of patients who received vedolizumab every 8 weeks or 4 weeks, respectively, compared with 15.9% of patients who were switched to placebo (P<0.001). Rates of adverse events were again similar in all groups.
In a similar study of vedolizumab in CD (GEMINI II trial), the results were less clear.43 While rates of clinical remission (CDAI ≤150) at week 6 differed significantly between patients treated with 300 mg of vedolizumab at week 0 and week 2 (14.5%; n=220) compared with placebo (6.8%; n=148; P=0.02), effects were modest and the other primary endpoint, defined as a CDAI-100 response at week 6, was not met (31.4% versus 25.7%). In the maintenance trial with vedolizumab-responders, clinical remission rates at week 52 were higher in patients assigned to continuous vedolizumab therapy every 8 weeks or 4 weeks (39.0% and 36.4%) than in those who were switched to placebo (21.6%; P<0.001 and P=0.004, respectively). Higher rates of serious adverse events (24.4% versus 15.3%), infections (44.1% versus 40.2%), and serious infections (5.5% versus 3.0%) were observed in the vedolizumab groups compared with placebo.
Comparable results were obtained in the recently published GEMINI III trial, which addressed the efficacy of vedolizumab in patients with moderately to severely active CD and failure or intolerance to prior anti-TNF therapy, a subgroup that reflects a frequent setting in everyday clinical practice.49 Patients were randomized to receive 300 mg of intravenous vedolizumab or placebo at week 0, week 2, and week 6. The primary endpoint, which was defined as clinical remission (CDAI ≤150) at week 6 in the subgroup of patients with prior anti-TNF failure, was not met (15.2% versus 12.1%, P=0.433). However, exploratory analyses of secondary endpoints showed higher rates of clinical remission at week 10 and higher rates of CDAI-100 response at week 6 and week 10 in the anti-TNF failure subgroup when treated with vedolizumab compared with placebo. Rates of adverse events were similar in all groups.
In summary, these studies provide strong evidence that vedolizumab is effective as an induction and maintenance therapy for UC. Vedolizumab also showed promise in non-bionaïve CD patients. The effects of continuous vedolizumab therapy for UC and CD patients who showed an initial nonresponse was addressed in an unpublished analysis that was presented at the Digestive Disease Week 2014.50,51 Interestingly, data suggest that continued application of vedolizumab in initial nonresponders can lead to additional effects on clinical response and remission rates at later time points. Hence, the actual clinical benefit of vedolizumab might be underestimated in the GEMINI trials due to a delayed response.
The long-term safety of vedolizumab is currently assessed in the open-label GEMINI LTS trial (NCT00790933) with pooled data from the Phase II or Phase III trials and new patients who were naïve to vedolizumab. In an early pooled safety analysis of the GEMINI I and II trials, rates of adverse events and serious adverse events were higher in the vedolizumab group when compared with placebo. However, the overall rate of adverse events adjusted for patient-years was higher in the placebo group. The most frequently reported adverse events were nasopharyngitis (13%), headache (12%), arthralgia (12%), nausea (9%), and pyrexia (9%), which suggests an acceptable safety profile.52 Moreover, clinically important infusion reactions were infrequent, and no case of anaphylaxis was observed in the GEMINI I and II trials, which allows the application of vedolizumab in clinical practice without premedications, ie, glucocorticoids, which are frequently used together with anti-TNF therapy.42,43
The European Medicines Agency and FDA approved vedolizumab (trade name Entyvio®; Takeda Pharmaceuticals) for the treatment of adult patients with moderate to severe UC or moderate to severe CD who showed inadequate response to one or more standard therapies.
Clinical status of vedolizumab and future directions
More than 20 years after the identification of the proteins that are involved in leukocyte homing to the gastrointestinal tract, vedolizumab is now available as the first approved member of gut-specific leukocyte migration inhibitors for the treatment of IBD. In the past decade, TNF inhibitors have become an established therapy in severe UC and CD and are more and more used as a first-line medication, especially in fistulizing CD (Table 1).2,3 However, anti-TNF therapy has to be discontinued in many patients due to secondary lack of response or adverse events such as infections or allergic reactions. The results of the large clinical trials presented above raise hope that vedolizumab could become a new drug option for these patients and prevent or delay surgical intervention in some cases.
Apart from its use as a “salvage therapy,” vedolizumab may assume another role in patients who have contraindications to current immunomodulators or anti-TNF biologics. The current label covers its use when one or more standard therapies (which includes corticoids) have failed, so theoretically, it could be used as a first-line biologic therapy. Interestingly, results of a recent network meta-analysis suggest similar efficacy and safety of vedolizumab and anti-TNF therapy (ie, adalimumab, infliximab, or golimumab) for induction and maintenance therapy in bionaïve patients with moderately to severely active UC, which supports its use as an alternative first-line biologic.53 However, to clearly define the role of vedolizumab in the spectrum of currently available biologics, comparison trials are urgently needed.
An important aspect that needs consideration is the difference in observed efficacy of vedolizumab induction therapy in UC and CD: whereas data in UC are convincing, only a modest effect was observed in patients with CD. Sandborn et al43 argue that the severity of disease in the study population could be a potential confounder. Indeed, the high rates of prior surgery (42%) and failure of at least one TNF antagonist (approximately 50%) suggest a CD population with refractory disease. Interestingly, the rate of prior failure of anti-TNF-therapy in the GEMINI I population was lower (approximately 40%), which may underlie the difference in observed efficacy. In accordance with this hypothesis, subgroup analyses in both the GEMINI II and GEMINI III trials show a trend towards higher rates of CDAI-100 response and clinical remission in bionaïve patients.43,49 In addition to confounding, a delayed effect of vedolizumab in CD, possibly due to the transmural nature of disease, may explain the modest results at week 6.43 The marked increase of remission rates between week 6 and week 10 in the GEMINI III trial and the additional effects of continued therapy in initial nonresponders strongly support this assumption and have important implications for the evaluation of response in clinical practice.49,50 However, more trials in different subgroups and with longer induction protocols are necessary to determine the actual effect of vedolizumab in CD.
Another investigation-worthy subject remains the effect of vedolizumab on extraintestinal manifestations of IBD, especially as it could be limited due to the gut-specificity of the drug. More research is needed to address the efficacy of vedolizumab in patients with extraintestinal manifestations of IBD.
The pooled safety analysis of the GEMINI I and GEMINI II trials suggests that vedolizumab has a safety profile that is comparable with or even better than that of anti-TNF therapy.52 However, cautious observation of long-term effects is necessary, especially with respect to opportunistic infections such as PML. Theoretically, the gut-specific design of vedolizumab should not impair binding of α4β1 integrin to VCAM-1, which is critical for the migration of lymphocytes to the central nervous system.54,55 In the more than 3,000 patients who have received vedolizumab as of today, no cases of PML have been reported. However, PML infection is a rare event, with an estimated risk of 0.5–1.4 per 1,000 patients who are treated with natalizumab for 1−24 months after prior immunosuppressant therapy; thus, postmarketing risk-management strategies are justified.56 Moreover, four cases of malignant neoplasms were reported in the GEMINI II study, suggesting that careful monitoring of the occurrence of cancer as a potential adverse event of long-term therapy is necessary.
Apart from vedolizumab, other molecules that target leukocyte migration to the gut are currently investigated in Phase II and Phase III clinical trials.9 Etrolizumab (rhuMAb-β7, anti-β7; Hoffmann–La Roche), a humanized monoclonal antibody that targets the β7 subunit of both the α4β7 and αEβ7 integrin heterodimers, was recently shown to be more efficient than placebo as an induction therapy for UC, and its anti-αEβ7 activity could be favorable as the αEβ7 integrin–E-cadherin interaction is believed to promote lymphocyte retention.57 Other biologics that target the α4β7 integrin–MAdCAM-1 interaction are PF-00547659 (anti-MAdCAM; Pfizer) and AMG 181 (anti-α4β7; Amgen).58–61 Clinical trials need to address whether their efficacy and safety are comparable to that of vedolizumab, and eventually head-to-head studies will be necessary to integrate these new agents into treatment algorithms, especially with regard to the choice of the initial biologic drug.
Conclusion
The gut-specific inhibition of lymphocyte migration with vedolizumab seems to be an effective and safe new treatment option for patients with IBD, although many issues still have to be addressed in additional clinical trials, it will be interesting to see if vedolizumab will change clinical practice and how it performs against established drugs and other new treatment strategies.
Disclosure
Daniel C Baumgart has served as a speaker or paid scientific consultant for Takeda, Genentech, Roche and Pfizer, that are developing anti-adhesion therapies. All of his activities and contracts are in conformity with the “FSA-Kodex Fachkreise” (voluntary self-monitoring code for expert consultants to the pharmaceutical industry), have been checked by the legal Department of Charité - Universitätsmedizin Berlin, and have been approved by the directorate of the Faculty of Medicine Charité - Universitätsmedizin Berlin. Niklas Krupka reports no conflicts of interest in this work.
References
Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369(9573):1627–1640. | ||
Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380(9853):1590–1605. | ||
Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380(9853):1606–1619. | ||
Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474(7351):298–306. | ||
Barrett JC, Lee JC, Lees CW, et al; UK IBD Genetics Consortium; Wellcome Trust Case Control Consortium 2. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41(12):1330–1334. | ||
Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42(12):1118–1125. | ||
Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43(3):246–252. | ||
Selby WS, Janossy G, Bofill M, Jewell DP. Intestinal lymphocyte subpopulations in inflammatory bowel disease: an analysis by immunohistological and cell isolation techniques. Gut. 1984;25(1):32–40. | ||
Thomas S, Baumgart DC. Targeting leukocyte migration and adhesion in Crohn’s disease and ulcerative colitis. Inflammopharmacology. 2012;20(1):1–18. | ||
Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. | ||
Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011;6:323–344. | ||
Streeter PR, Berg EL, Rouse BT, Bargatze RF, Butcher EC. A tissue-specific endothelial cell molecule involved in lymphocyte homing. Nature. 1988;331(6151):41–46. | ||
Hu MC, Crowe DT, Weissman IL, Holzmann B. Cloning and expression of mouse integrin beta p(beta 7): a functional role in Peyer’s patch-specific lymphocyte homing. Proc Natl Acad Sci U S A. 1992;89(17):8254–8258. | ||
Berlin C, Berg EL, Briskin MJ, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74(1):185–195. | ||
Briskin MJ, McEvoy LM, Butcher EC. MAdCAM-1 has homology to immunoglobulin and mucin-like adhesion receptors and to IgA1. Nature. 1993;363(6428):461–464. | ||
Erle DJ, Briskin MJ, Butcher EC, Garcia-Pardo A, Lazarovits AI, Tidswell M. Expression and function of the MAdCAM-1 receptor, integrin alpha 4 beta 7, on human leukocytes. J Immunol. 1994;153(2):517–528. | ||
Wagner N, Löhler J, Kunkel EJ, et al. Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature. 1996;382(6589):366–370. | ||
Briskin M, Winsor-Hines D, Shyjan A, et al. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am J Pathol. 1997;151(1):97–110. | ||
Binion DG, West GA, Ina K, Ziats NP, Emancipator SN, Fiocchi C. Enhanced leukocyte binding by intestinal microvascular endothelial cells in inflammatory bowel disease. Gastroenterology. 1997;112(6):1895–1907. | ||
Hanauer SB, Feagan BG, Lichtenstein GR, et al; ACCENT I Study Group. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359(9317):1541–1549. | ||
Sands BE, Blank MA, Patel K, van Deventer SJ; ACCENT II Study. Long-term treatment of rectovaginal fistulas in Crohn’s disease: response to infliximab in the ACCENT II Study. Clin Gastroenterol Hepatol. 2004;2(10):912–920. | ||
Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462–2476. | ||
Dave M, Purohit T, Razonable R, Loftus EV Jr. Opportunistic infections due to inflammatory bowel disease therapy. Inflamm Bowel Dis. 2014;20(1):196–212. | ||
Targownik LE, Bernstein CN. Infectious and malignant complications of TNF inhibitor therapy in IBD. Am J Gastroenterol. 2013;108(12):1835–1842, quiz 1843. | ||
Sandborn WJ, Hanauer SB, Rutgeerts P, et al. Adalimumab for maintenance treatment of Crohn’s disease: results of the CLASSIC II trial. Gut. 2007;56(9):1232–1239. | ||
Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn’s disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol. 2008;6(6):644–653. | ||
Marsal J, Agace WW. Targeting T-cell migration in inflammatory bowel disease. J Intern Med. 2012;272(5):411–429. | ||
Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha4 beta1 integrin. Nature. 1992;356(6364):63–66. | ||
Miller DH, Khan OA, Sheremata WA, et al; International Natalizumab Multiple Sclerosis Trial Group. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348(1):15–23. | ||
Polman CH, O’Connor PW, Havrdova E, et al; AFFIRM Investigators. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910. | ||
Rudick RA, Stuart WH, Calabresi PA, et al; SENTINEL Investigators. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):911–923. | ||
Gordon FH, Lai CW, Hamilton MI, et al. A randomized placebo-controlled trial of a humanized monoclonal antibody to alpha4 integrin in active Crohn’s disease. Gastroenterology. 2001;121(2):268–274. | ||
Ghosh S, Goldin E, Gordon FH, et al; Natalizumab Pan-European Study Group. Natalizumab for active Crohn’s disease. N Engl J Med. 2003;348(1):24–32. | ||
Sandborn WJ, Colombel JF, Enns R, et al; International Efficacy of Natalizumab as Active Crohn’s Therapy (ENACT-1) Trial Group; Evaluation of Natalizumab as Continuous Therapy (ENACT-2) Trial Group. Natalizumab induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2005;353(18):1912–1925. | ||
Targan SR, Feagan BG, Fedorak RN, et al; International Efficacy of Natalizumab in Crohn’s Disease Response and Remission (ENCORE) Trial Group. Natalizumab for the treatment of active Crohn’s disease: results of the ENCORE Trial. Gastroenterology. 2007;132(5):1672–1683. | ||
Gordon FH, Hamilton MI, Donoghue S, et al. A pilot study of treatment of active ulcerative colitis with natalizumab, a humanized monoclonal antibody to alpha-4 integrin. Aliment Pharmacol Ther. 2002;16(4):699–705. | ||
Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353(4):369–374. | ||
Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353(4):375–381. | ||
Van Assche G, Van Ranst M, Sciot R, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353(4):362–368. | ||
Soler D, Chapman T, Yang LL, Wyant T, Egan R, Fedyk ER. The binding specificity and selective antagonism of vedolizumab, an anti-alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases. J Pharmacol Exp Ther. 2009;330(3):864–875. | ||
Parikh A, Leach T, Wyant T, et al. Vedolizumab for the treatment of active ulcerative colitis: a randomized controlled phase 2 dose-ranging study. Inflamm Bowel Dis. 2012;18(8):1470–1479. | ||
Feagan BG, Rutgeerts P, Sands BE, et al; GEMINI 1 Study Group. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699–710. | ||
Sandborn WJ, Feagan BG, Rutgeerts P, et al; GEMINI 2 Study Group. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2013;369(8):711–721. | ||
Hesterberg PE, Winsor-Hines D, Briskin MJ, et al. Rapid resolution of chronic colitis in the cotton-top tamarin with an antibody to a gut-homing integrin alpha4 beta7. Gastroenterology. 1996;111(5):1373–1380. | ||
Feagan BG, Greenberg GR, Wild G, et al. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N Engl J Med. 2005;352(24):2499–2507. | ||
Feagan BG, Greenberg GR, Wild G, et al. Treatment of active Crohn’s disease with MLN0002, a humanized antibody to the alpha4beta7 integrin. Clin Gastroenterol Hepatol. 2008;6(12):1370–1377. | ||
Best WR, Becktel JM, Singleton JW, Kern F Jr. Development of a Crohn’s disease activity index. National Cooperative Crohn’s Disease Study. Gastroenterology. 1976;70(3):439–444. | ||
Schroeder KW, Tremaine WJ, Ilstrup DM. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N Engl J Med. 1987;317(26):1625–1629. | ||
Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147(3):618–627.e3. | ||
Sandborn WJ, Feagan BG, Reinisch W, et al. Mo1217 Effects of continued vedolizumab therapy for Crohn’s disease in week 6 induction therapy nonresponders. Digestive Disease Week 2014; 2014 May 3–6; Chicago, IL. Gastroenterology. 2014;146(5 Suppl 1):S-588. | ||
Feagan BG, Sandborn W, Smyth MD, Sankoh S, Parikh A, Fox I. Mo1222 Effects of continued vedolizumab therapy for ulcerative colitis in week 6 induction therapy nonresponders. Digestive Disease Week 2014; 2014 May 3–6; Chicago, IL. Gastroenterology. 2014;146(5 Suppl 1):S-590. | ||
Colombel J-F, Sands BE, Feagan BG, et al. 643 Integrated safety analysis of vedolizumab for the treatment of ulcerative colitis or Crohn’s disease. Digestive Disease Week 2013; 2013 May 18–21; Orlando, FL. Gastroenterology. 2013;144(5 Suppl 1):S-113. | ||
Danese S, Fiorino G, Peyrin-Biroulet L, et al. Biological agents for moderately to severely active ulcerative colitis: a systematic review and network meta-analysis. Ann Intern Med. 2014;160(10):704–711. | ||
Fedyk ER, Wyant T, Yang LL, et al. Exclusive antagonism of the α4β7 integrin by vedolizumab confirms the gut-selectivity of this pathway in primates. Inflamm Bowel Dis. 2012;18(11):2107–2119. | ||
Haanstra KG, Hofman SO, Lopes Estêvão DM, et al. Antagonizing the α4β1 integrin, but not α4β7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J Immunol. 2013;190(5):1961–1973. | ||
Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366(20):1870–1880. | ||
Vermeire S, O’Byrne S, Keir M, et al. Etrolizumab as induction therapy for ulcerative colitis: a randomised, controlled, phase 2 trial. Lancet. 2014;384(9940):309–318. | ||
Pullen N, Molloy E, Carter D, et al. Pharmacological characterization of PF-00547659, an anti-human MAdCAM monoclonal antibody. Br J Pharmacol. 2009;157(2):281–293. | ||
Vermeire S, Ghosh S, Panes J, et al. The mucosal addressin cell adhesion molecule antibody PF-00547,659 in ulcerative colitis: a randomised study. Gut. 2011;60(8):1068–1075. | ||
Pan WJ, Hsu H, Rees WA, et al. Pharmacology of AMG 181, a human anti-α4β7 antibody that specifically alters trafficking of gut-homing T cells. Br J Pharmacol. 2013;169(1):51–68. | ||
Pan WJ, Köck K, Rees WA, et al. Clinical pharmacology of AMG 181, a gut-specific human anti-α4β7 monoclonal antibody, for treating inflammatory bowel diseases. Br J Clin Pharmacol. Epub 2014 May 6. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.