")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Design and synthesis of chroman derivatives with dual anti-breast cancer and antiepileptic activities
Received 24 April 2016
Accepted for publication 1 June 2016
Published 2 September 2016 Volume 2016:10 Pages 2779—2788
DOI https://doi.org/10.2147/DDDT.S111266
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Pinki Rawat, Saurabh Manaswita Verma
Department of Pharmaceutical Sciences and Technology, Birla Institute of Technology, Mesra, Ranchi, Jharkhand, India
Abstract: A series of chroman derivatives was designed, prepared, and examined for their anti-breast cancer and antiepileptic activities. All synthesized compounds yielded results that were in good agreement with spectral data. The bioassay showed that some of the resultant compounds exerted remarkable inhibitory effects on growth of human breast cancer cell line MCF-7. In particular, compound 6i (the concentration required for 50% inhibition of cell growth [GI50] =34.7 µM) exerted promising anticancer activity toward MCF-7 cell line. Additionally, compounds 6b, 6c, 6d, 6e, 6g, 6i, and 6l showed advanced antiepileptic activity than reference drugs. None of the compounds showed neurotoxicity, as determined by the rotarod test. The obtained results proved that these distinctive compounds could be relevant as models for future discovery and research, as well as for the production of more number of active derivatives.
Keywords: anti-breast cancer, antiepileptic, chroman, isatin, Schiff base
Introduction
Breast cancer is one of the most commonly diagnosed cancers and is the second leading cause of death among women.1 Currently, a large number of chemotherapeutic agents are available, but toxicity and resistance to standard drug treatments limit the effectiveness of chemotherapy. Therefore, the search for new chemotherapeutic agents is an important task for researchers.
Heterocyclic compounds containing oxygen are presently considered very interesting due to their physicochemical properties being relevant as far as the design of new drugs is concerned. Literature reveals that chromans are an important chemical synthon, associated with a broad range of biological effects, neuroprotective,2 antiestrogens,3 antioxidant,4 anti-HIV,5 and anti-breast cancer agents.6–8
Chromans have attracted a great deal of attention due to their anticancer activities since the discovery of Ormeloxifene (I),9 KBU2046 (Phase II),10 and B43-genistein (II) (Phase II; Figure 1).11 Furthermore, many chromans have contributed to the search for new anticancer agents. It has also been reported that Schiff base (III and IV) and isatin (V) moieties enhanced the anticancer activity of the chroman core.12,13
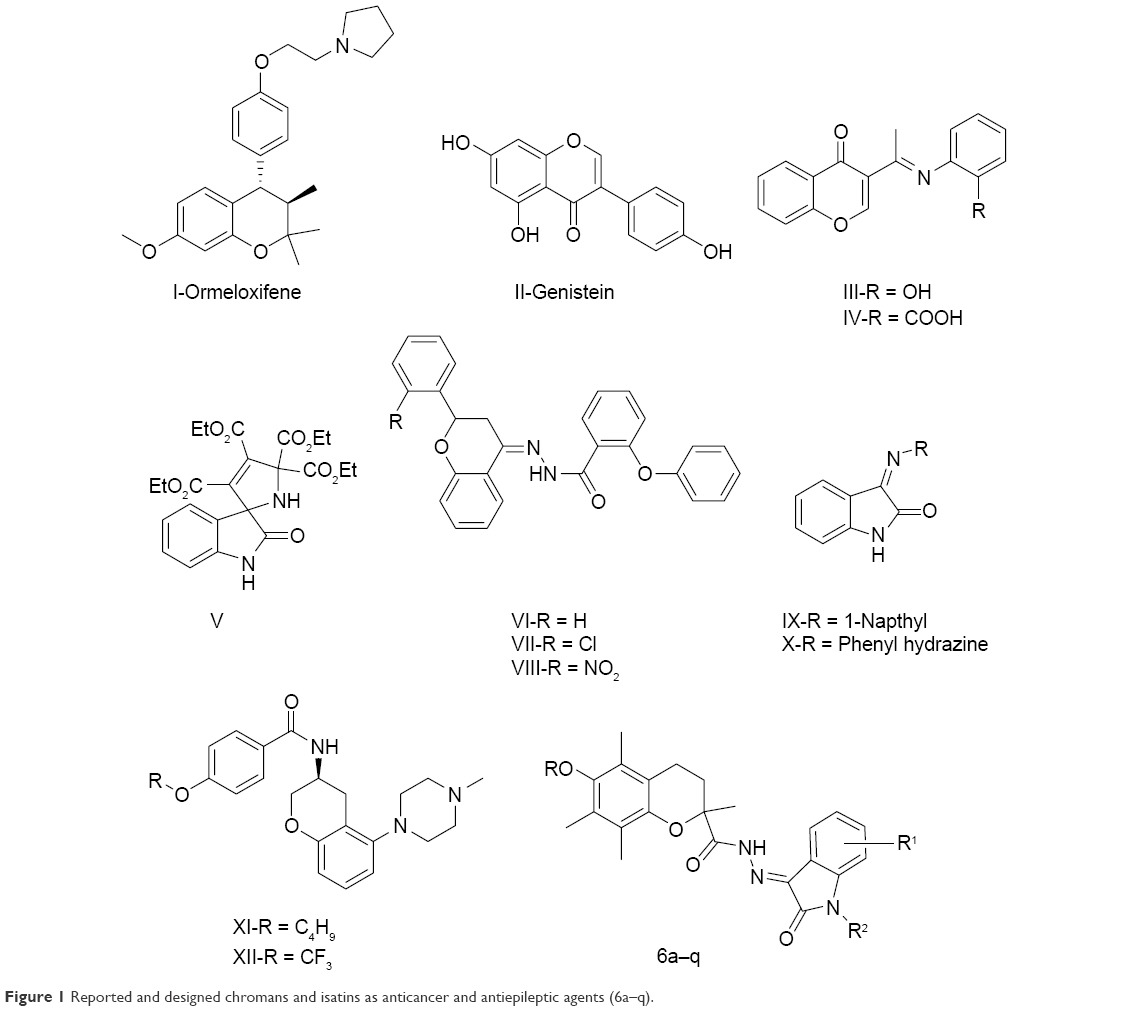 | Figure 1 Reported and designed chromans and isatins as anticancer and antiepileptic agents (6a–q). |
Epilepsy is a common neurological disorder affecting approximately 45–100 million people globally.14 Though many antiepileptic drugs are available in clinical use, neurotoxicity and distinctive adverse effects restrict their clinical use.15 Therefore, it is necessary to discover new chemical pharmacophores as more effective antiepileptics with less neurotoxicity. Literature has revealed that the presence of a Schiff base in a chroman nucleus (VI–VIII) is beneficial for the antiepileptic activity.16 Similarly, isatin (IX, X) being associated with a Schiff base accounts for its antiepileptic activity.17
Reports also suggest that some anticancer compounds have been studied as antiepileptics.18 The anti-breast cancer potential of the voltage-gated sodium channel-blocking antiepileptic drug phenytoin has also been discussed by researchers.19 Chroman derivatives XI and XII have been investigated as voltage-gated sodium channels.20 Therefore, chroman analogs designed as anticancer agents may have antiepileptic activity also.
In this study, advances are made in designing chroman derivatives by adding Schiff base and isatin moieties at the second position. Seven compounds with antimicrobial activity have been reported in a previous work.21 A series of chroman compounds (6a–q) have been designed, synthesized, and their anti-breast cancer and antiepileptic activities evaluated.
Materials and methods
Materials
All commercial chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) and Spectrochem (Mumbai, India). Thin layer chromatography was performed on plates precoated with silica gel GF-254 (Merck Millipore, Billerica, MA, USA), and the identification was done under UV light and spraying with charring solution, followed by heating. Melting points were determined in open glass capillaries and the uncorrected. NMR spectra were recorded on ECX-500 spectrometer (JEOL, Toko, Japan) in dimethyl sulfoxide (DMSO)-d6 and deuterated chloroform (CDCl3) at 400 MHz and 500 MHz for 1H and 125 MHz for 13C. The chemical shifts were recorded in (δ, ppm) relative to tetramethylsilane as an internal standard. The mass spectra were recorded with a Waters-Q-Tof Premier-HAB213 spectrometer and Microscopic II triple-quadrupole mass spectrometer using electron ionization, and the m/z values are indicated in daltons. The infrared (IR) spectra (KBr) were recorded on an FTIR Vector 22 spectrophotometer (Bruker Corporation, Billerica, MA, USA). Supplementary materials show 1H and 13C NMR spectra, FT-IR spectra, and mass spectra of compounds 6a–q associated with this article.
Synthesis of compounds
6-Hydroxy-2,5,7,8-tetramethyl-chroman-2-carboxylic acid methyl ester (3a)
6-Hydroxy-2,5,7,8-tetramethyl-chroman-2-carboxylic acid methyl ester (3a) was prepared by the method provided in the literature.22
6-Methoxy-2,5,7,8-tetramethyl-chroman-2-carboxylic acid methyl ester (3b)
To a solution of 3a (100 mg, 40.0 mmol) in 10 mL anhydrous acetone, 2 g anhydrous K2CO3 was added and the mixture was heated at 50°C. Dimethyl sulfate (0.17 mL, 1.80 mmol) was then added, and the mixture was refluxed for 24 hours. After completion of the reaction, HCl 10% was added until pH 6 and then extracted with diethylether. The organic layer was washed with saturated aqueous NaCl and dried, and the solvent was evaporated in vacuo. Purification by column chromatography (petroleum ether/EtOAc: 90/10) yielded 111 mg (100%) white solid.
Methyl-6-(benzyloxy)-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carboxylate (3c)
To a mixture of 3a (2 g, 7.566 mmol) and K2CO3 (1.568 g, 11.349 mmol) was added with stirring 30 mL of dimethylformamide at 0°C for 20 minutes. Benzyl bromide (1.553 g, 9.080 mmol) was added to the reaction solution and stirred at room temperature overnight. The reaction mixture was diluted with 50 mL cold water, filtered, and washed with cold water to give a white solid (81.67%).
General method for the synthesis of 6-substituted-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (4a–c)
Compounds 3a–c (1 mmol) and 80% hydrazine hydrate (5 mmol) in 10 mL ethanol were refluxed at 80°C for 10 hours. While the reaction proceeded, the ethanol was evaporated. The separated solid was filtered, washed with water, and dried to obtain a dull white solid.
General method for the synthesis of the final compounds (6a–q)
The compounds were prepared through the condensation reaction between 5a–q (1 mmol) and 4a–c (1 mmol) in 10 mL acetic acid under reflux at 120°C for 3–12 hours. Then, 20 mL distilled water was added into the reaction medium. The compounds were filtered and recrystallized in ethanol.
6-Hydroxy-2,5,7,8-tetramethyl-N′-(2-oxoindolin-3-ylidene)-3,4-dihydro-2H-chromene-2-carbohydrazide (6a)
Yield: Yellow solid (74%); melting point (MP): 240°C; IR (KBr, ν, cm−1): 3,426 (NH), 3,227 (OH), 1,694 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.17 (s, 1H, –NH), 7.52 (s, 1H, OH), 7.48 (d, J=7.3 Hz, 1H), 7.32 (t, J=7.6 Hz, 1H), 7.03 (t, J=7.6 Hz, 1H), 6.86 (d, J=7.6 Hz, 1H), 2.59–2.54 (m, 1H), 2.47–2.45 (m, 1H), 2.28–2.25 (m, 1H), 2.18 (s, 3H), 2.04 (s, 3H), 1.96 (s, 3H), 1.86–1.81 (m, 1H), 1.47 (s, 3H); 13C-NMR (125 MHz, DMSO-d6) 12.3 (–CH3), 12.5 (–CH3), 13.3 (–CH3), 20.4 (–CH2), 24.7 (–CH3), 29.6 (–CH2), 78.1 (>C<), 111.6, 117.2, 120.2, 120.8, 121.4, 122.2, 123.1, 123.5, 132.3, 139.1, 143.1, 143.8, 146.7, 162.9 (>C=O), 171.9 (>C=O); high-resolution mass spectrometry (HR-MS): 394.1700 (M+H)+, calculated: 394.1766.
6-hydroxy-2,5,7,8-tetramethyl-N′-(1-methyl-2-oxoindolin-3-ylidene)-3,4-dihydro-2H-chromene-2-carbohydrazide (6b)
Yield: Yellow solid (72%), MP: 237°C; IR (KBr, ν, cm−1): 3,406 (OH), 3,261 (NH), 1,683 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 7.54 (s, 1H, OH), 7.50 (d, J=7.3 Hz, 1H), 7.40 (t, J=7.6 Hz, 1H), 7.10–7.05 (m, 2H), 3.13 (s, 3H), 2.59–2.54 (m, 1H), 2.50–2.44 (m, 1H), 2.29–2.23 (m, 1H), 2.21 (s, 3H), 2.05 (s, 3H), 1.96 (s, 3H), 1.87–1.81 (m, 1H), 1.47 (s, 3H); 13C-NMR (125 MHz, DMSO-d6) 12.3 (–CH3), 12.5 (–CH3), 13.3 (–CH3), 20.4 (–CH2), 24.6 (–CH3), 26.1 (>N–CH3), 29.6 (–CH2), 78.1 (>C<), 110.2, 117.3, 119.5, 120.8, 121.0, 122.2, 123.5, 123.6, 132.2, 138.2, 143.8, 144.3, 146.83, 161.0 (>C=O), 171.9 (>C=O); HR-MS: 408.1926 (M+H)+, calculated, 408.4702.
N′-(5-chloro-2-oxoindolin-3-ylidene)-6-hydroxy-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6c)
Yield: Light Yellow solid (87%), MP: 255°C; IR (KBr, ν, cm−1): 3,598 (NH), 3,151 (OH), 1,680 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.28 (s, 1H, –NH), 7.54 (s, 1H, OH), 7.46 (d, J=2.1 Hz, 1H), 7.35 (dd, J=2.1, 8.2 Hz, 1H), 6.88 (d, J=8.2 Hz, 1H), 2.60–2.54 (m, 1H), 2.47–2.43 (m, 1H), 2.30–2.24 (m, 1H), 2.17 (s, 3H), 2.04 (s, 3H), 1.96 (s, 3H), 1.86–1.80 (m, 1H), 1.47 (s, 3H); 13C-NMR (125 MHz, DMSO-d6) 12.3 (–CH3), 12.4 (–CH3), 13.3 (–CH3), 20.3 (–CH2), 24.6 (–CH3), 29.5 (–CH2), 78.1 (>C<), 113.1, 117.2, 120.8, 120.9, 122.0, 122.2, 123.5, 127.2, 131.7, 138.2, 141.8, 143.8, 146.8, 162.7 (>C=O), 172.0 (>C=O); HR-MS: 450.1377 (M+H)+, calculated, 450.1196.
N′-(5-bromo-2-oxoindolin-3-ylidene)-6-hydroxy-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6d)
Yield: Yellow solid (56%), MP: 260°C; IR (KBr, ν, cm−1): 3,594 (NH), 3,149 (OH), 1,680 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.30 (s, 1H, –NH), 7.56 (s, 1H,), 7.54 (s, 1H, OH), 7.48 (d, J=8.0 Hz, 1H), 6.83 (d, J=8.0 Hz, 1H), 2.60–2.54 (m, 1H), 2.47–2.43 (m, 1H), 2.29–2.24 (m, 1H), 2.17 (s, 3H), 2.03 (s, 3H), 1.96 (s, 3H), 1.86–1.80 (m, 1H), 1.47 (s, 3H); 13C-NMR (125 MHz, DMSO-d6) 12.3 (–CH3), 12.4 (–CH3), 13.3 (–CH3), 20.3 (–CH2), 24.6 (–CH3), 29.5 (–CH2), 78.1 (>C<), 113.5, 114.8, 117.2, 120.8, 122.2, 122.4, 123.5, 123.7, 134.5, 138.1, 142.1, 143.8, 146.8, 162.6 (>C=O), 172.0 (>C=O); HR-MS: 494.0690 (M+Na)+, calculated, 494.0691.
N′-(6-chloro-2-oxoindolin-3-ylidene)-6-hydroxy-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6e)
Yield: Yellow solid (57%), MP: 290°C; IR (KBr, ν, cm−1): 3,443 (NH), 3,141 (OH), 1,678 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.20 (s, 1H, –NH), 7.54 (s, 1H, OH), 7.29 (dd, J=2.5, 8.0 Hz, 1H), 7.19–7.14 (m, 1H), 6.87–6.85 (dd, J=4.3, 8.5 Hz, 1H), 2.60–2.54 (m, 1H), 2.50–2.40 (m, 1H), 2.29–2.24 (m, 1H), 2.17 (s, 3H), 2.03 (s, 3H), 1.96 (s, 3H), 1.85–1.80 (m, 1H), 1.47 (s, 3H); HR-MS: 428.1372 (M+H)+, calculated, 428.1377.
N′-(5-fluoro-2-oxoindolin-3-ylidene)-6-hydroxy-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6f)
Yield: Yellow solid (51%), MP: 290°C; IR (KBr, ν, cm−1): 3,566 (NH), 3,206 (OH), 1,686 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.18 (s, 1H, –NH), 7.52 (s, 1H, OH), 7.29 (dd, J=2.5, 8.0 Hz, 1H), 7.18–7.14 (m, 1H), 6.86 (dd, J=4.3, 8.6 Hz, 1H), 2.59–2.54 (m, 1H), 2.50–2.42 (m, 1H), 2.30–2.25 (m, 1H), 2.18 (s, 3H), 2.04 (s, 3H), 1.96 (s, 3H), 1.86–1.80 (m, 1H), 1.47 (s, 3H); 13C-NMR (125 MHz, DMSO-d6) 12.3 (–CH3), 12.4 (–CH3), 13.3 (–CH3), 20.4 (–CH2), 24.6 (–CH3), 29.6 (–CH2), 78.1 (>C<), 108.4, 108.6, 112.6, 112.7, 117.2, 118.6, 118.8, 120.8, 121.5, 121.6, 122.2, 123.5, 138.7, 139.4, 143.8, 146.8, 157.8, 159.7, 163.1 (>C=O), 172.0 (>C=O); HR-MS: 412.1681 (M+H)+, calculated, 412.1672.
N′-(6-chloro-1-methyl-2-oxoindolin-3-ylidene)-6-hydroxy-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6g)
Yield: Yellow solid (59%), MP: 257°C; IR (KBr, ν, cm−1): 3,479 (NH), 3,223 (OH), 1,697 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 7.54 (s, 1H, OH), 7.47–7.44 (m, 2H), 7.10 (d, J=8.2 Hz, 1H), 3.13 (s, 3H), 2.60–2.54 (m, 1H), 2.50–2.41 (m, 1H), 2.29–2.24 (m, 1H), 2.20 (s, 3H), 2.04 (s, 3H), 1.96 (s, 3H), 1.87–1.81 (m, 1H), 1.47 (s, 3H); 13C-NMR (125 MHz, DMSO-d6) 12.2 (–CH3), 12.5 (–CH3), 13.3 (–CH3), 20.3 (–CH2), 24.6 (–CH3), 26.3 (>N–CH3), 29.5 (–CH2), 78.1 (>C<), 111.8, 117.2, 120.5, 120.8, 121.2, 122.2, 123.5, 127.7, 131.5, 137.3, 143.0, 143.7, 146.8, 160.8 (>C=O), 172.0 (>C=O); HR-MS: 442.1537 (M+H)+, calculated, 442.1530.
6-Methoxy-2,5,7,8-tetramethyl-N′-(2-oxoindolin-3-ylidene)-3,4-dihydro-2H-chromene-2-carbohydrazide (6h)
Yield: Light Yellow solid (58%), MP: 256°C; IR (KBr, ν, cm−1): 3,184 (NH), 1,683 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.19 (s, 1H, –NH), 7.49 (d, J=7.3 Hz, 1H), 7.32 (t, J=7.6 Hz, 1H), 7.03 (t, J=7.6 Hz, 1H), 6.87 (d, J=8.0 Hz, 1H), 3.49 (s, 3H, –OCH3), 2.62–2.58 (m, 1H), 2.51–2.46 (m, 1H), 2.29–2.23 (m, 1H), 2.19 (s, 3H), 2.08 (s, 3H), 2.01 (s, 3H), 1.89–1.84 (m, 1H), 1.48 (s, 3H); 13C-NMR (125 MHz, DMSO-d6) 11.9 (–CH3), 12.4 (–CH3), 12.9 (–CH3), 20.1 (–CH2), 24.5 (–CH3), 29.3 (–CH2), 60.3 (–OCH3), 78.4 (>C<), 111.6, 118.0, 120.2, 121.5, 123.1, 123.1, 126.0, 128.0, 132.4, 139.2, 143.1, 146.4, 150.5, 162.9 (>C=O), 171.6 (>C=O); HR-MS: 408.1924 (M+H)+, calculated, 408.1923.
N′-(5-bromo-2-oxoindolin-3-ylidene)-6-methoxy-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6i)
Yield: Yellow solid (51%), MP: 290°C; IR (KBr, ν, cm−1): 3,194 (NH), 1,681 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.31 (s, 1H, –NH), 7.57 (s, 1H), 7.49 (d, J=8.2 Hz, 1H), 6.84 (d, J=8.2 Hz, 1H), 3.48 (s, 3H, –OCH3), 2.61–2.57 (m, 1H), 2.50–2.45 (m, 1H), 2.30–2.23 (m, 1H), 2.19 (s, 3H), 2.08 (s, 3H), 2.01 (s, 3H), 1.88–1.86 (m, 1H), 1.48 (s, 3H); 13C-NMR (125 MHz, DMSO-d6) 11.9 (–CH3), 12.4 (–CH3), 12.9 (–CH3), 20.1 (–CH2), 24.5 (–CH3), 29.2 (–CH2), 60.3 (–OCH3), 78.4 (>C<), 113.6, 114.8, 118.0, 122.4, 123.0, 123.7, 126.1, 128.0, 134.5, 138.2, 142.1, 146.3, 150.6, 162.6 (>C=O), 171.7 (>C=O); HR-MS: 486.1027 (M+H)+, calculated, 486.1028.
N′-(5-fluoro-2-oxoindolin-3-ylidene)-6-methoxy-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6j)
Yield: Yellow solid (66%), MP: >300°C; IR (KBr, ν, cm−1): 3,487 (NH), 1,684 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.21 (s, 1H, –NH), 7.30 (dd, J=2.8, 8.0 Hz, 1H), 7.19–7.15 (m, 1H), 6.87 (dd, J=4.3, 8.5 Hz, 1H, 1H), 3.49 (s, 3H, –OCH3), 2.62–2.57 (m, 1H), 2.50–2.45 (m, 1H), 2.27–2.22 (m, 1H), 2.19 (s, 3H), 2.08 (s, 3H), 2.01 (s, 3H), 1.88–1.85 (m, 1H), 1.48 (s, 3H); 13C-NMR (125 MHz, DMSO-d6) 11.9(–CH3), 12.4 (–CH3), 12.9 (–CH3), 20.1 (–CH2), 24.4 (–CH3), 29.3 (–CH2), 60.3 (–OCH3), 78.4 (>C<), 108.5, 108.7, 112.7, 112.8, 118.0, 118.6, 118.8, 121.5, 121.6, 123.1, 126.1, 128.0, 138.9, 139.4, 146.3, 150.6, 157.8, 159.7, 163.1 (>C=O), 171.7 (>C=O); HR-MS: 426.1822 (M+H)+, calculated, 426.1829.
N′-(6-chloro-2-oxoindolin-3-ylidene)-6-methoxy-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6k)
Yield: Yellow solid (52%), MP: 270°C; IR (KBr, ν, cm−1): 3,207 (NH), 1,683 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.32 (s, 1H, –NH), 7.48 (d, J=8.0 Hz, 1H), 7.07 (d, J=8.0 Hz, 1H), 6.90 (s, 1H), 3.49 (s, 3H, –OCH3), 2.63–2.57 (m, 1H), 2.51–2.42 (m, 1H), 2.29–2.24 (m, 1H), 2.19 (s, 3H), 2.08 (s, 3H), 2.01 (s, 3H), 1.90–1.84 (m, 1H), 1.48 (s, 3H); HR-MS: 442.1539 (M+H)+, calculated, 442.1539.
6-(benzyloxy)-2,5,7,8-tetramethyl-N′-(2-oxoindolin-3-ylidene)-3,4-dihydro-2H-chromene-2-carbohydrazide (6l)
Yield: Light Yellow solid (74%), MP: 258°C; IR (KBr, ν, cm−1): 3,186 (NH), 1,679 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.20 (s, 1H, –NH), 7.50 (d, J=8.0 Hz, 1H), 7.43 (d, J=7.3 Hz, 2H), 7.36 (t, J=7.3 Hz, 2H), 7.33–7.30 (m, 2H), 7.04 (t, J=7.3 Hz, 1H), 6.87 (d, J=8.0 Hz, 1H), 4.60 (s, 2H), 2.65–2.60 (m, 1H), 2.54–2.47 (m, 1H), 2.30–2.26 (m, 1H), 2.22 (s, 3H), 2.13 (s, 3H), 2.06 (s, 3H), 1.92–1.86 (m, 1H), 1.49 (s, 3H); 13C-NMR (125 MHz, DMSO-d6) 12.3 (–CH3), 12.5 (–CH3), 13.2 (–CH3), 20.3 (–CH2), 24.5 (–CH3), 29.4 (–CH2), 74.6 (–CH2), 78.5 (>CH<), 111.6, 118.1, 120.3, 121.5, 123.2, 126.4, 128.3, 128.9, 132.4, 138.1, 139.2, 143.1, 146.6, 149.3, 163.0 (>C=O), 171.7 (>C=O); HR-MS: 484.2235 (M+H)+, calculated, 484.2236.
6-(benzyloxy)-2,5,7,8-tetramethyl-N′-(1-methyl-2-oxoindolin-3-ylidene)-3,4-dihydro-2H-chromene-2-carbohydrazide (6m)
Yield: Yellow-orange solid (92%), MP: 170°C; IR (KBr, ν, cm−1): 3,236 (NH), 1,696 (CO); 1H-NMR (500 MHz, CDCl3, ppm): 7.77 (d, J=7.3 Hz, 1H), 7.47 (d, J=6.8 Hz, 2H), 7.39–7.36 (m, 2H), 7.35–7.30 (m, 2H), 7.10 (t, J=7.3 Hz, 1H), 6.82 (d, J=8.0 Hz, 1H), 4.68 (s, 2H), 3.23 (s, 3H), 2.70–2.62 (m, 2H), 2.53–2.49 (m, 1H), 2.37 (s, 3H), 2.25 (s, 3H), 2.13 (s, 3H), 2.03–1.99 (m, 1H); 13C-NMR (125 MHz, CHCl3) 12.1 (–CH3), 12.2 (–CH3), 13.0 (–CH3), 20.4 (–CH2), 24.6 (–CH3), 25.7 (>N–CH3), 29.5 (–CH2), 74.7 (–CH2), 78.5 (>C<), 108.8, 117.9, 119.6, 112.0, 123.4, 123.7, 126.3, 127.7, 127.9, 128.5, 128.6, 131.7, 137.9, 138.2, 143.8, 146.6, 149.3, 161.0 (>C=O), 172.9 (>C=O); HR-MS: 498.2399 (M+H)+, calculated, 498.2392.
6-(benzyloxy)-N′-(5-bromo-2-oxoindolin-3-ylidene)-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6n)
Yield: Yellow solid (68%), MP: 270°C; IR (KBr, ν, cm−1): 3,189 (NH), 1,677 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.32 (s, 1H, –NH), 7.57 (s, 1H), 7.50 (d, J=8.2 Hz, 1H), 7.43 (d, J=7.3 Hz, 2H), 7.37 (t, J=7.3 Hz, 2H), 7.33–7.30 (m, 1H), 6.84 (d, J=8.2 Hz, 1H), 4.59 (s, 2H), 2.66–2.58 (m, 1H), 2.54–2.44 (m, 1H), 2.32–2.25 (m, 1H), 2.22 (s, 3H), 2.13 (s, 3H), 2.06 (s, 3H), 1.94–1.86 (m, 1H), 1.50 (s, 3H); 13C-NMR (125 MHz, DMSO-d6): 12.2 (–CH3), 12.4 (–CH3), 13.2 (–CH3), 20.2 (–CH2), 24.4 (–CH3), 29.3 (–CH2), 74.6 (–CH2), 78.5 (>C<), 113.6, 118.1, 121.5, 123.1, 123.7, 126.4, 128.3, 128.9, 135.0, 138.2, 142.2, 146.5, 149.3, 162.7 (>C=O), 171.7 (>C=O); HR-MS: 562.1349 (M+H)+, calculated, 562.1341.
6-(benzyloxy)-N′-(5-fluoro-2-oxoindolin-3-ylidene)-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6o)
Yield: Yellow solid (85%), MP:>300°C; IR (KBr, ν, cm−1): 3,195 (NH), 1,681 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.21 (s, 1H, –NH), 7.43 (d, J=7.4 Hz, 2H), 7.37 (t, J=7.0 Hz, 2H), 7.31–7.30 (m, 2H), 7.18 (t, J=9.0 Hz, 1H), 6.87 (dd, J=4, 8.6 Hz, 1H), 4.59 (s, 2H), 2.65–2.60 (m, 1H), 2.53–2.47 (m, 1H), 2.31–2.25 (m, 1H), 2.22 (s, 3H), 2.12 (s, 3H), 2.06 (s, 3H), 1.92–1.87 (m, 1H), 1.50 (s, 3H); HR-MS: 502.2146 (M+H)+, calculated, 502.2142.
6-(benzyloxy)-N′-(5-chloro-1-methyl-2-oxoindolin-3-ylidene)-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6p)
Yield: Dark Yellow solid (81%), MP: 220°C; IR (KBr, ν, cm−1): 3,260 (NH), 1,712 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 7.49–7.46 (m, 2H), 7.44–7.43 (m, 2H), 7.37 (t, J=7.3 Hz, 2H), 7.33–7.29 (m, 1H), 7.12 (d, J=8.5 Hz, 1H), 4.59 (s, 2H), 3.14 (s, 3H), 2.65–2.60 (m, 1H), 2.54–2.47 (m, 1H), 2.30–2.25 (m, 1H), 2.24 (s, 3H), 2.13 (s, 3H), 2.06 (s, 3H), 1.93–1.89 (m, 1H), 1.50 (s, 3H); HR-MS: 532.1992 (M+H)+, calculated, 532.2003.
6-(benzyloxy)-N′-(5-chloro-2-oxoindolin-3-ylidene)-2,5,7,8-tetramethyl-3,4-dihydro-2H-chromene-2-carbohydrazide (6q)
Yield: Yellow solid (94%), MP: 280°C; IR (KBr, ν, cm−1): 3,188 (NH), 1,678 (CO); 1H-NMR (500 MHz, DMSO-d6, ppm): 11.31 (s, 1H, –NH), 7.46 (d, J=2.5 Hz, 1H), 7.44–7.42 (m, 2H), 7.38–7.35 (m, 3H), 7.32–7.29 (m, 1H), 6.89 (d, J=8.0 Hz, 1H), 4.60 (s, 2H), 2.66–2.60 (m, 1H), 2.53–2.47 (m, 1H), 2.31–2.27 (m, 1H), 2.21 (s, 3H), 2.12 (s, 3H), 2.06 (s, 3H), 1.92–1.87 (m, 1H), 1.50 (s, 3H); HR-MS: 516.1685 (M-H)−, calculated, 516.1690.
In vitro anticancer activity determination by sulforhodamine B assay
The anticancer activity was measured in vitro in the human breast cancer cell line (MCF-7) using sulforhodamine B stain assay applying the method of Skehan et al.23 Human breast cancer cell line MCF-7 was obtained from the Advanced Centre for Treatment Research and Education in Cancer (ACTREC), Navi Mumbai, Maharashtra, India, and were grown in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum, 100 μg/mL streptomycin, and 100 U/mL penicillin at 37°C in a humidified atmosphere with 5% CO2. All compounds (6a–q) were dissolved in DMSO to give a stock solution of 10 μg/mL, from which further dilutions (10−7 M, 10−6 M, 10−5 M, and 10−4 M) in culture medium were prepared. Control cultures were treated with DMSO alone. Cells were plated in 96-well plates (104 cells per well) for 24 hours before treatment with the compounds. Different concentrations of the test compounds (10−7 M, 10−6 M, 10−5 M, and 10−4 M) were added to the cell monolayer. Triplicate wells were prepared for each individual dose. Monolayer cells were then incubated with test compounds for 48 hours at 37°C in an atmosphere of 5% CO2. After 48 hours, cells were fixed, washed, and stained with sulforhodamine B stain. Excess stain was washed with acetic acid and the attached stain was recovered with Tris-EDTA buffer. Color intensity was measured using an enzyme-linked immunosorbent assay reader. The relation between surviving fraction and drug concentration was plotted and the GI50 was calculated for each compound.
Antiepileptic activity
The synthesized compounds (6a–q) obtained from the reaction sequence were dissolved in polyethyleneglycol (PEG-400) administered intraperitoneally into male albino mice (CF-1 strain, 18–25 g) and tested in the MES,24 PTZ seizure,25 and neurotoxicity screens,26 using 30 mg/kg, 100 mg/kg, and 300 mg/kg doses; and observations were recorded at two different time intervals. All experimental protocols were carried out with permission from the Institutional Animal Ethics Committee (IAEC), Birla Institute of Technology, Mesra, Ranchi (CPCSEA approval no: BIT/PH/IAEC/13/2014, dated April 30, 2014), and the experiments were in compliance with the animal care guidelines issued by the IAEC.
The MES test
Albino mice were divided into groups of six animals each. Maximal seizures were induced by the application of alternating current of 60 Hz (50 mA) for 0.2 seconds using corneal electrodes primed with an electrolyte solution containing an anesthetic agent (0.5% tetracaine HCl). During the preliminary screening, the test compounds were administered by intraperitoneal injection at 0.01 mL/kg body weight at doses of 30 mg/kg, 100 mg/kg, and 300 mg/kg prior to testing, and anticonvulsant activity was evaluated after 0.5 hours and 4 hours of administration. Abolition of the hind limb tonic extensor spasm was recorded as anticonvulsant activity in the MES method.27
The PTZ seizure test
The synthesized compounds were administered to mice (n=6) by intraperitoneal injection. After 30 minutes, mice were treated with a subcutaneous injection of PTZ (60 mg/kg). Then, the mice were placed singly in isolated plastic cages and observed for 60 minutes. Protection was referred to as the failure to detect an episode of clonic spasms of at least 5-second duration during this time period.
Neurotoxicity screening
Neurotoxicity was measured by the rotarod test. Mice (n=6) were given intraperitoneal injection of the synthesized compounds at dose levels of 30 mg/kg, 100 mg/kg, and 300 mg/kg. Then, after 30 minutes, the mice were placed on the rotating rod. Neurotoxicity was determined by the inability of the mice to maintain equilibrium on the rod for 1 minute in each of the three trials.16
Results and discussion
Chemistry
The chroman derivatives (6a–q) described in this study were prepared as depicted in Figure 2. The synthesis of 6-substituted-2,5,7,8-tetramethyl-chroman-2-carboxylic acid methyl ester (3a–c) as a key intermediate was accomplished by the reaction of trimethylhydroquinone and methyl methacrylate to produce 3a,22 followed by methylation (yielding 3b)28 and benzylation (to yield 3c).11 The ester moieties of 3a–c endured the nucleophilic attack by hydrazine hydrate, resulting in 4a–c.29 The latter compounds were subjected to condensation reaction with different substituted isatins to produce 6a–q with 51%–94% yield.21 The properties of the synthesized compounds 6a–q are shown in Table 1.
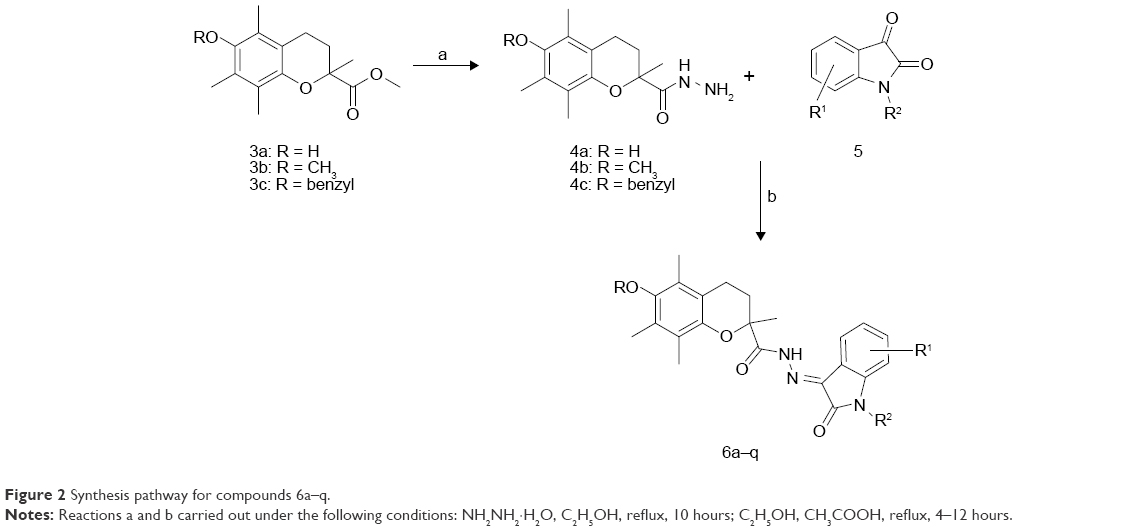 | Figure 2 Synthesis pathway for compounds 6a–q. |
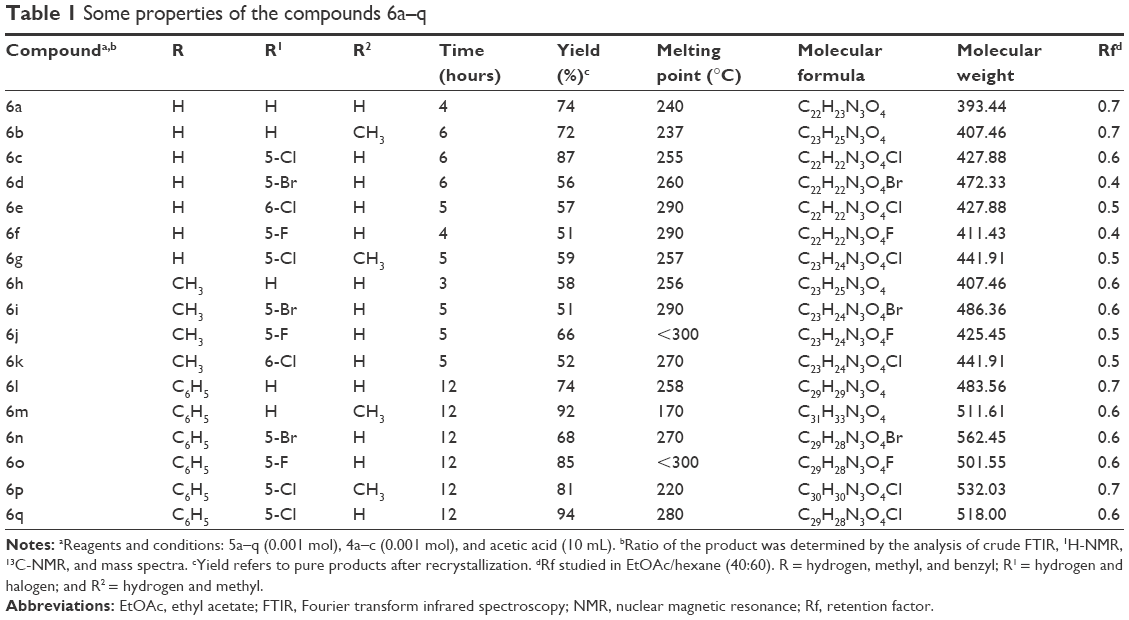 | Table 1 Some properties of the compounds 6a–q |
The compounds were characterized by Fourier transform infrared spectroscopy (FTIR), 1H-nuclear magnetic resonance (NMR), 13C-NMR, mass spectroscopy, and melting point analysis. The analytical data were fully consistent with the proposed structures. The retention factor (Rf) value of all synthesized compounds (6a–q) was calculated in ethyl acetate (EtOAc) and hexane (40:60) and ranged between 0.4 and 0.7.
In the FTIR spectrum, bands at 3,284–3,598 cm−1 and 1,600–1,700 cm−1 confirm the presence of –NH and –CO stretching vibrations, respectively. Compounds 6a–g showed broad absorption bands at 3,141–3,406 cm−1, indicative of a hydroxy group (–OH stretch). The 1H-NMR spectrum revealed a singlet at δ=11.19–11.32 ppm due to NH2 protons (−CONH2). The presence of a hydroxy group (–OH) in the compounds 6a–g was confirmed by the sharp singlet at δ=7.52–7.55 ppm. The methoxy group (–OCH3) showed a singlet at δ=3.48–3.49 ppm in compounds 6h–k. Compounds 6l–q displayed a singlet peak at δ=4.59–4.67 ppm, corresponding to benzylic methyl (-CH2-). In 13C–NMR, the carbonyl group of amide (C=O) was observed at approximately δ=160–172 ppm. The mass spectra showed (M+H)+ and (M+Na+)+ (m/z) peaks, respectively confirming their purity and molecular weight.
In vitro anticancer activity screening
The synthesized compounds (6a–q) were screened for in vitro anticancer activity against human mammary adenocarcinoma (MCF-7) by using sulforhodamine B assay.23 The concentration required for 50% inhibition of cell growth (GI50) for each compound was calculated with reference to a control sample. For each compound, GI50 was calculated from the sigmoidal dose–response curves and the values are presented in Table 2. The data for adriamycin are included as reference.
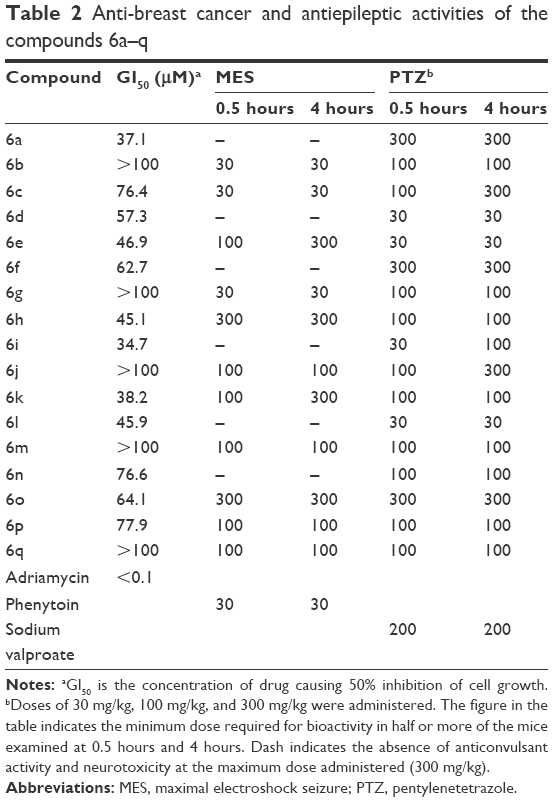 | Table 2 Anti-breast cancer and antiepileptic activities of the compounds 6a–q |
The resultant data showed that the chroman derivatives (6a-q) produced significant cytostatic effects against the human breast cancer MCF-7 cell line. The results revealed that compound 6i was the most active member of the series, with GI50=34.7 μM. Among all the compounds, compounds 6a, 6i, and 6k showed promising activity, with GI50 values of 37.1 μM, 34.7 μM, and 38.2 μM, respectively; compounds 6d, 6e, 6h, and 6l showed GI50 values of 57.3 μM, 46.9 μM, 45.1 μM, and 45.9 μM, respectively; compounds 6c, 6f, 6n, 6o, and 6p showed GI50 values of 76.4 μM, 62.7 μM, 76.6 μM, 64.1 μM, and 77.9 μM, respectively; the remaining five compounds 6b, 6g, 6j, 6m, and 6q showed GI50 values >100 μM in the MCF-7 breast cancer cells. The activity was attributed to the presence of chlorine or bromine group at the fifth position on the phenyl ring of the isatin ring system, whereas in the presence of fluorine, the phenyl group of the isatin ring system induced reduced activity. However, the chlorine group at the sixth position accounts for more activity than the chlorine group at the fifth position. Benzyl substitution in the chroman ring system appeared to be less active than methoxy and hydroxyl substitution.
Antiepileptic activity
The antiepileptic activity and the acute neurotoxicity of all synthesized compounds (6a–q) were evaluated by the use of standard techniques.24–26 Results are presented in Table 2. Phenytoin and carbamazepine were used as the standard drugs for the comparisons.
The most potent compounds 6b, 6c, and 6g resembled the standard drug phenytoin at the dose level of 30 mg/kg after 0.5 hours and 4 hours by the maximal electroshock seizure (MES) method. On the other hand, compounds 6j, 6m, 6p, and 6q displayed activity at the dose level of 100 mg/kg against seizure spread at both the time intervals, whereas compounds 6e and 6k displayed protection at the dose level of 300 mg/kg after 4 hours, suggesting rapid onset but shorter duration of action. Compounds 6h and 6o exhibited protection at both the time intervals 0.5 hours and 4 hours after the dose level of 300 mg/kg.
In the pentylenetetrazole (PTZ) screen, the most active of these compounds were 6d, 6e, and 6l, which provided protection at the dose level of 30 mg/kg after both the time intervals 0.5 hours and 4 hours, excluding compound 6i, which showed protection at the 100 mg/kg dose level after 4 hours. Compounds 6b, 6g, 6h, 6k, 6m, 6n, 6p, and 6q exhibited protection at the 100 mg/kg dose level after both time intervals, whereas compounds 6c and 6j showed protection at 100 mg/kg after 4 hours. The remaining compounds had activity at the dose level of 300 mg/kg at both the time intervals.
Compounds that afforded protection against seizures in the MES and PTZ screens were further tested for their neurotoxicity. All compounds (6a–q) were devoid of neurotoxicity, as determined by the rotarod test.
Gamma-aminobutyric acid (GABA) is the principal inhibitory neurotransmitter substance in the brain and it is widely implicated in epilepsy. It has been reported that PTZ produces seizures by inhibiting GABA neurotransmission. In order to inhibit seizures, enhancement of GABAergic neurotransmission is useful. All newly synthesized compounds (6a–q) showed protection against seizure in the PTZ model (Table 2). These findings suggest that the compounds may have inhibited or attenuated PTZ-induced seizure in mice by enhancing GABAergic neurotransmission.30
The antiepileptic activity correlation of the synthesized compounds (6a–q) revealed that compounds containing a hydroxyl group in the chroman ring system, such as 6b, 6c, 6d, 6e, and 6g, were more potent as compared to the compounds containing a methyl group (6h, 6i, and 6k). Compounds having a benzyl group, such as 6l, 6n, and 6o, in the chroman ring system were the least active. The presence of electron-withdrawing substituents (Cl, Br, and F) in the isatin ring system might have increased the activity, as compounds 6c, 6g, 6j, 6m, and 6q were more active than compounds 6a, 6h, and 6l. However, compounds, such as 6f, 6j, and 6o, which have a fluorine substituent, were found to be less active than compounds with the other electron-withdrawing substituents (Cl and Br).
Conclusion
In this study, the design and synthesis of a series of chroman derivatives have been described. All synthesized compounds were evaluated for their anti-breast cancer and antiepileptic activities. The results of the study demonstrated that compound 6i exerted the most potent activity (GI50=34.7 μM) among the compounds in the series. Additionally, all synthesized compounds were evaluated for their antiepileptic efficacy. In the antiepileptic evaluation, compounds 6b–e, 6g, 6i, and 6l were found to be more active than the reference drugs. This work adds new data to the relationship between chromans and their anti-breast cancer and antiepileptic activities. This provides a platform for further development of related compounds into more active compounds.
Acknowledgments
The authors thank the Advanced Centre for Treatment Research and Education in Cancer (ACTREC), Navi Mumbai, Maharashtra, India, for evaluation of the in vitro anticancer activity. Ms Pinki Rawat is thankful to University Grants Commission – Rajiv Gandhi National Fellowship for providing financial support in the form of National Doctoral Fellowship.
Disclosure
The authors report no conflicts of interest in this work.
References
Viswas KC, Solomon R, Lee H, Trivedi P. Design, synthesis and biological evaluation of some isatin-linked chalcones as novel anti-breast cancer agents: a molecular hybridization approach. Biomed Prev Nutr. 2013;3(4):325–330. | ||
Koufaki M, Kiziridi C, Alexi X, Alexis MN. Design and synthesis of novel neuroprotective 1,2 dithiolane/chroman hybrids. Bioorg Med Chem. 2009;17(17):6432–6441. | ||
Kanbe Y, Kim MH, Nishimoto M, et al. Discovery of thiochroman and chroman derivatives as pure antiestrogens and their structure-activity relationship. Bioorg Med Chem. 2006;14(14):4803–4819. | ||
Lee H, Lee K, Jung JK, Cho J, Theodorakis EA. Synthesis and evaluation of 6-hydroxy-7-methoxy-4-chromanone- and chroman-2-carboxamides as antioxidants. Bioorg Med Chem Lett. 2005;15(11):2745–2748. | ||
Kraus GA, Mengwasser J, Maury W, Oh C. Synthesis of chroman aldehydes that inhibit HIV. Bioorg Med Chem Lett. 2011;21(5):1399–1401. | ||
Gupta A, Dwivedy A, Keshri G, et al. Rapid synthesis of 4-benzylidene and 4-[bis-(4-methoxyphenyl)-methylene-2-substituted phenyl-benzopyrans as potential selective estrogen receptor modulators (SERMs) using McMurry coupling reaction. Bioorg Med Chem Lett. 2006;16(23):6006–6012. | ||
Reddy BVS, Divya B, Swaina M, Rao TP, Yadav JS, Vishnu Vardhan MV. A domino Knoevenagel hetero-Diels-Alder reaction for the synthesis of polycyclic chromene derivatives and evaluation of their cytotoxicity. Bioorg Med Chem Lett. 2012;22(5):1995–1999. | ||
Wang D, Chuang HC, Weng SC, et al. α-Tocopheryl succinate as a scaffold to develop potent inhibitors of breast cancer cell adhesion. J Med Chem. 2009;52(18):5642–5648. | ||
Lal J. Clinical pharmacokinetics and interaction of centchroman – a mini review. Contraception. 2010;81(4):275–280. | ||
Li X, Rebecca F, Xiaoke H, et al. Discovery of a novel drug KBU2046 that inhibits conversion of human prostate cancer to a metastatic phenotype. Cancer Prev Res. 2010;3:B58. | ||
Chen CL, Levine A, Rao A, et al. Clinical pharmacokinetics of the CD19 receptor-directed tyrosine kinase inhibitor B43-genistein in patients with B-lineage lymphoid malignancies. J Clin Pharmacol. 1999;39(12):1248–1255. | ||
Kavitha P, Chary MR, Singavarapu BVVA, Reddy KL. Synthesis, characterization, biological activity and DNA cleavage studies of tridentate Schiff bases and their Co(II) complexes. J Saudi Chem Soc. 2016;20(1):69–80. | ||
Collins I, Jones AM. Diversity-oriented synthetic strategies applied to cancer chemical biology and drug discovery. Molecules. 2014;19(11):17221–17255. | ||
Bell GS, Sander JW. The epidemiology of epilepsy: the size of the problem. Seizure. 2002;11(suppl A):306–314. | ||
Bhat MA, Siddiqui N, Khan SA. Synthesis of novel 3-(4-acetyl-5h/methyl-5-substitutedPhenyl-4,5-dihydro-1,3,4-oxadiazol-2-yl)-2h-chromen-2-ones As potential anticonvulsant agents. Acta Pol Pharm. 2008;65(2):235–239. | ||
Kumar N, Chauhan LS. Synthesis and anticonvulsant activity of some flavones incorporated hydrazide derivatives. Int J Pharm Clin Res. 2015;7(4):317–322. | ||
Verma M, Pandeya SN, Singh KN, Stables JP. Anticonvulsant activity of Schiff bases of isatin derivatives. Acta Pharm. 2004;54(1):49–56. | ||
El-Azab AS, ElTahir KE. Design and synthesis of novel 7-aminoquinazoline derivatives: antitumor and anticonvulsant activities. Bioorg Med Chem Lett. 2012;22(5):1879–1885. | ||
Nelson M, Yang M, Dowle AA, Thomas JR, Brackenbury WJ. The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol Cancer. 2015;14:13. | ||
Kers I, Csjernyik G, Macsari I, et al. Structure and activity relationship in the (S)-N-chroman-3-ylcarboxamide series of voltage-gated sodium channel blockers. Bioorg Med Chem Lett. 2012;22(17):5618–5624. | ||
Rawat P, Verma SM. Synthesis and pharmacological evaluation of 6-hydroxy-2,5,7,8-tetramethyl-N′-(2-oxoindolin-3-ylidene)chroman-2-carbohydrazide derivatives as antimicrobial agents. J Chem Pharm Res. 2016;8(3):149–154. | ||
Hyatt JA. Convenient preparation of 2,7,8-trimethyl-6-hydroxychroman-2-carboxylic Acid (Ɣ-Trolox). Synth Commun. 2008;38(1):8–14. | ||
Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13):1107–1112. | ||
Krall RL, Penry JK, White BG, Kupferberg HJ, Swinyard EA. Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia. 1978;19(4):409–428. | ||
Porter RJ, Cereghino JJ, Gladding GD, et al. Antiepileptic drug development program. Cleve Clin Q. 1984;51(2):293–305. | ||
Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc. 1957;46(3):208–209. | ||
Malik S, Ahuja P, Sahu K, Khan SA. Design and synthesis of new of 3-(benzo[d]isoxazol-3-yl)-1-substituted pyrrolidine-2, 5-dione derivatives as anticonvulsants. Eur J Med Chem. 2014;84:42–50. | ||
Koufaki M, Kiziridi C, Papazafiri P, et al. Synthesis and biological evaluation of benzopyran analogues bearing class III antiarrhythmic pharmacophores. Bioorg Med Chem. 2006;14(19):6666–6678. | ||
Lopez GV, Blanco F, Hernandez P, et al. Second generation of α-tocopherol analogs-nitric oxide donors: synthesis, physicochemical, and biological characterization. Bioorg Med Chem. 2007;15(18):6262–6272. | ||
Fang Y, Sun C, Liu D, Wang S, Quan Z. Synthesis and anticonvulsant activity evaluation of 3-alkoxy-4-(4-(hexyloxy/heptyloxy)phenyl)-4H-1,2,4–triazole. Iran J Pharm Res. 2015;14(1):77–87. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.