")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Design and prediction of new anticoagulants as a selective Factor IXa inhibitor via three-dimensional quantitative structure-property relationships of amidinobenzothiophene derivatives
Authors Gao J, Tong X, Chang Y, He Y, Mei Y, Tan P, Guo J, Liao G, Xiao G, Chen W, Zhou S, Sun P
Received 2 October 2014
Accepted for publication 17 December 2014
Published 23 March 2015 Volume 2015:9 Pages 1743—1759
DOI https://doi.org/10.2147/DDDT.S75282
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Jia-Suo Gao,1* Xu-Peng Tong,2* Yi-Qun Chang,1 Yu-Xuan He,1 Yu-Dan Mei,1 Pei-Hong Tan,1 Jia-Liang Guo,1 Guo-Chao Liao,3 Gao-Keng Xiao,1 Wei-Min Chen,1 Shu-Feng Zhou,4 Ping-Hua Sun1
1Guangdong Province Key Laboratory of Pharmacodynamic Constituents of Traditional Chinese Medicine and New Drugs Research, College of Pharmacy, Jinan University, Guangzhou, People’s Republic of China; 2College of Pharmacy, China Pharmaceutical University, Nanjing, People’s Republic of China; 3Department of Chemistry, Wayne State University, Detroit, Michigan, USA; 4College of Pharmacy, University of South Florida, Tampa, FL, USA
*These authors contributed equally to this work
Abstract: Factor IXa (FIXa), a blood coagulation factor, is specifically inhibited at the initiation stage of the coagulation cascade, promising an excellent approach for developing selective and safe anticoagulants. Eighty-four amidinobenzothiophene antithrombotic derivatives targeting FIXa were selected to establish three-dimensional quantitative structure–activity relationship (3D-QSAR) and three-dimensional quantitative structure–selectivity relationship (3D-QSSR) models using comparative molecular field analysis and comparative similarity indices analysis methods. Internal and external cross-validation techniques were investigated as well as region focusing and bootstrapping. The satisfactory q2 values of 0.753 and 0.770, and r2 values of 0.940 and 0.965 for 3D-QSAR and 3D-QSSR, respectively, indicated that the models are available to predict both the inhibitory activity and selectivity on FIXa against Factor Xa, the activated status of Factor X. This work revealed that the steric, hydrophobic, and H-bond factors should appropriately be taken into account in future rational design, especially the modifications at the 2'-position of the benzene and the 6-position of the benzothiophene in the R group, providing helpful clues to design more active and selective FIXa inhibitors for the treatment of thrombosis. On the basis of the three-dimensional quantitative structure–property relationships, 16 new potent molecules have been designed and are predicted to be more active and selective than Compound 33, which has the best activity as reported in the literature.
Keywords: CoMFA, CoMSIA, 3D-QSAR, 3D-QSSR, benzothiophene antithrombosis
Introduction
Arterial and venous thromboses are still the major cause of mortality in clinic, although tremendous efforts are taken to prevent and treat thrombotic events. Current anticoagulants such as ticlopidine, clopidogrel, heparin, and warfarin have significant adverse effects like thrombocytopenia, bleeding, and need for careful patient monitoring, making their therapeutic utility restricted.1–3 For the past decade, many Factor Xa (FXa) inhibitors targeting the specific enzymes of the extrinsic common pathway in the coagulation cascade have been advanced to clinical development, but they remain in the potential risk category of bleeding and therefore have a narrow therapeutic window.4–6 Factor IXa (FIXa), a vitamin K-dependent blood coagulation factor, is essential for the amplification or consolidation phase of blood coagulation via activating Factor X (FX) to FXa in the formation of a thrombus with 50 times more efficiency than Factor VIIa/tissue factor.7–11 The selective inhibition of FIXa is believed to be effective in intravascular anticoagulation with maintenance of extravascular hemostasis for FIX activation, both by the stimulation of the intrinsic system and by maintaining a low level of tissue factor.5,12–14 As an alternative therapeutic target, FIXa is specifically inhibited at the initiation stage of the coagulation cascade, promising an excellent approach for developing selective and safe anticoagulants. Recently, 2-amidinobenzothiophene derivatives have been reported as potent FIXa-selective ligands.15,16
Three-dimensional quantitative structure–property relationships (3D-QSPRs) including three-dimensional quantitative structure–activity relationship (3D-QSAR) and three-dimensional quantitative structure–selectivity relationship (3D-QSSR) techniques such as comparative molecular field analysis (CoMFA)17 and comparative similarity indices analysis (CoMSIA)18 are useful methods of ligand-based drug design used to correlate physicochemical descriptors from a set of related compounds to their known molecular activity or molecular property values.19 These computational techniques combining 3D information for the ligands have proved particularly helpful in the design of novel, more potent inhibitors.20,21 Hao et al22 have done some 3D-QSAR work on the binding mode of benzothiophene analogs such as FIXa inhibitors, only by CoMFA method without using CoMSIA, and the predictive CoMFA model they developed with leave-one-out cross-validation q2=0.52, conventional r2=0.97, and r2pred=0.81 still has room for improvement. To our knowledge, there has been no report concerning the application of a 3D-QSSR methodology to selectivity of FIXa, although the 3D-QSSR is also crucial for the development of these FIXa inhibitors. The systematic study on FIXa inhibitors of 3D-QSAR and 3D-QSSR methodologies obtained highly predictive models, by which 16 new potent molecules have been designed. The present paper also explored why and how the target that 2-amidinobenzothiophene derivatives selectively bind with is not FXa, but is actually FIXa. The satisfactory 3D-QSAR and 3D-QSSR models of 84 compounds as serine protease enzyme FIXa inhibitors provide a solid basis for future rational design of more active and selective anticoagulants.
Materials and methods
Database and biological activity
Eighty-four molecules selected for the present study were taken from the works of Wang et al.15,16 In the 3D-QSAR analysis in the current study, 12 compounds were randomly chosen as the test set and the remaining 72 compounds as the training set. In the 3D-QSSR analysis, 73 compounds with the half maximal inhibitory concentration (IC50) values against FIXa and FXa were randomly divided into a test set of nine compounds and a training set of the remaining 64 compounds. For convenience, the IC50 (FIXa) values were converted to their negative logarithm (pIC50) values. The logIC50 (FXa/FIXa) that is the negative logarithm of logIC50 (FIXa/FXA), can be used as an index for the selectivity (FIXa/FXA). The pIC50 values have a span of four log units, from 4.43 to 8.70, while the logIC50 (FXa/FIXa) values have a span of five log units, from −2.80 to 2.73, indicating the reliable 3D-QSAR and 3D-QSSR models. The pIC50 (FIXa) and logIC50 (FXa/FIXa) values and chemical structures of the compounds are shown in Tables 1 and 2.
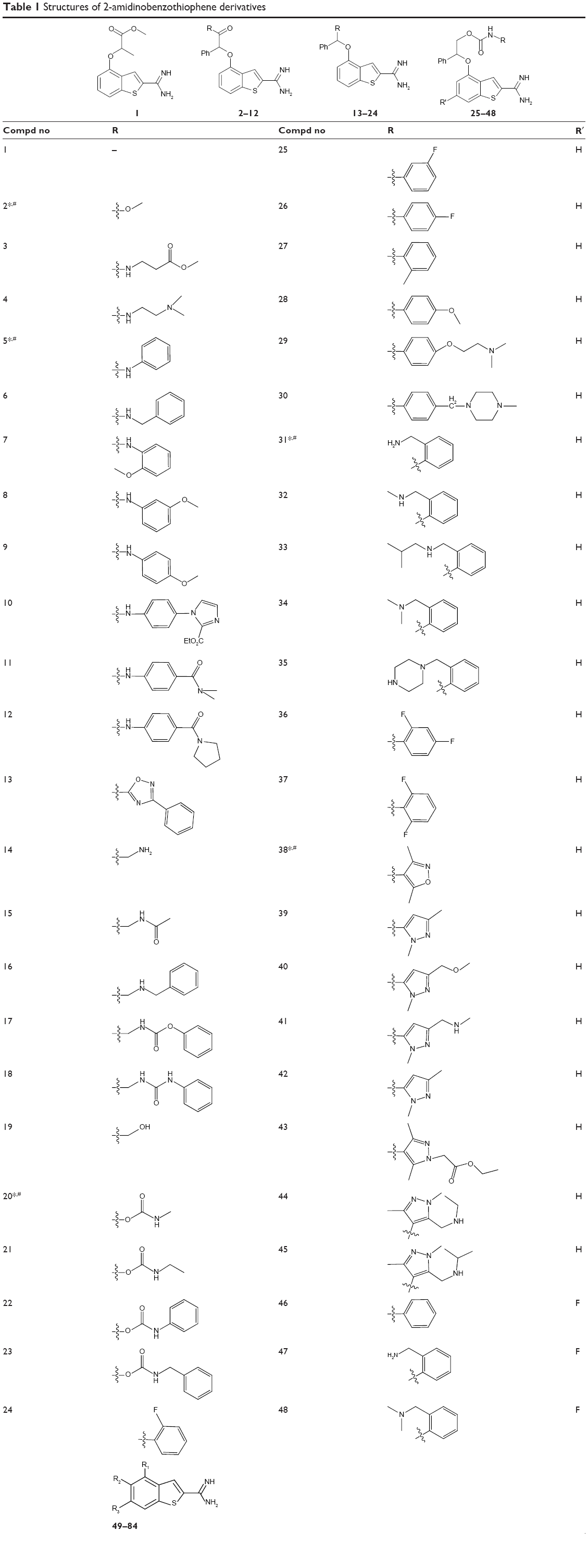  | Table 1 Structures of 2-amidinobenzothiophene derivatives |
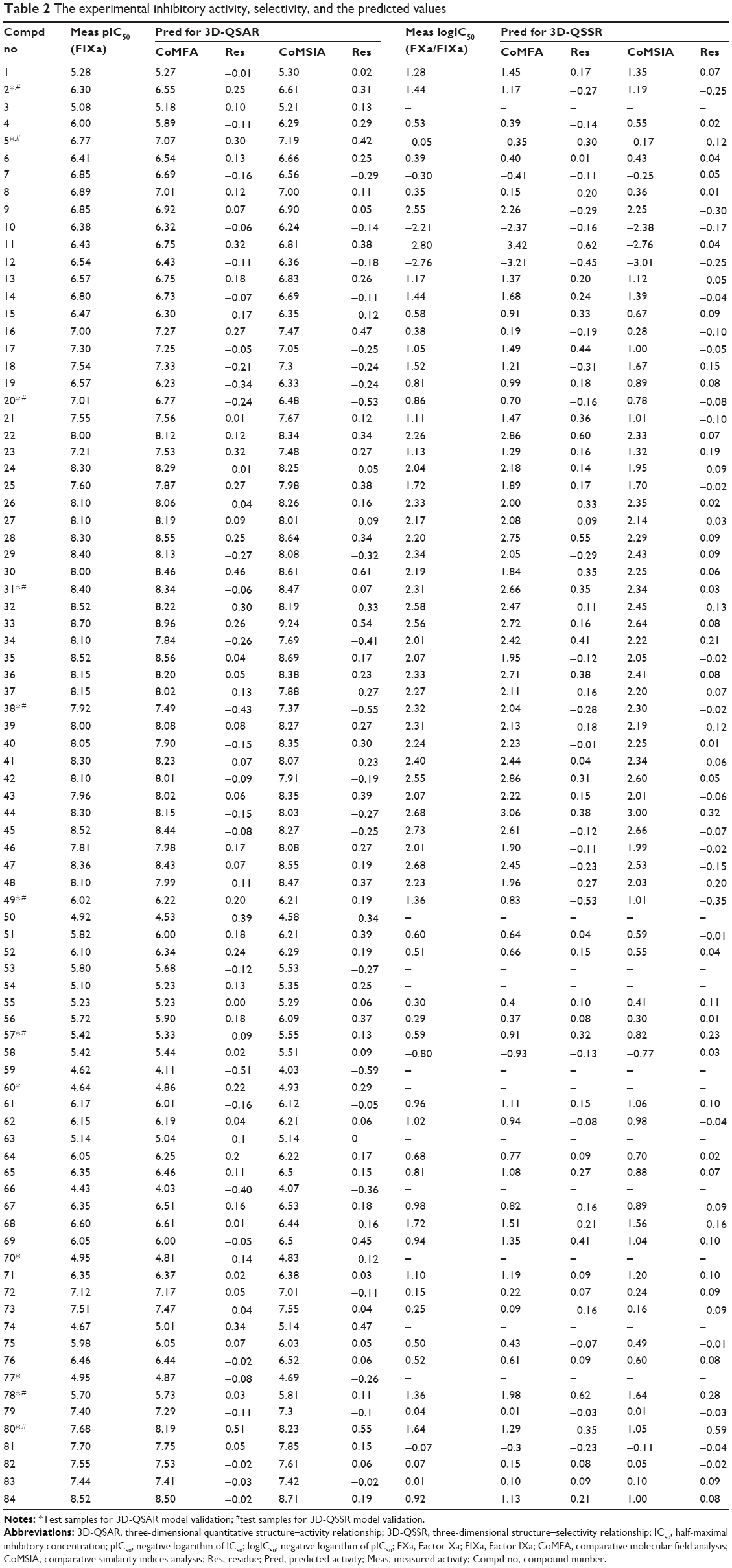 | Table 2 The experimental inhibitory activity, selectivity, and the predicted values |
Molecular structure building and alignment
Molecular alignment is considered to be one of the most essential elements that extensively influence the quality and the predictive ability of the models in CoMFA and CoMSIA analyses, although other factors such as lattice shifting step size, probe atom type, and orientation of the aligned compounds might have influence on the results as well.23,24 A 2-amidinobenzothiophene ring with structural rigidity, comprising the common core of all molecules in the data set, was selected as the common substructure to align all of the molecules (Figure 1). Active conformation selection is a key step for 3D-QSAR analysis, and in the present study, the molecular conformation of Compound 33 (the most active compound) obtained from the molecular docking was used as the template to build molecular structures of all the compounds. It can be seen from Figure 1 that all the compounds studied have similar bioactive conformations.
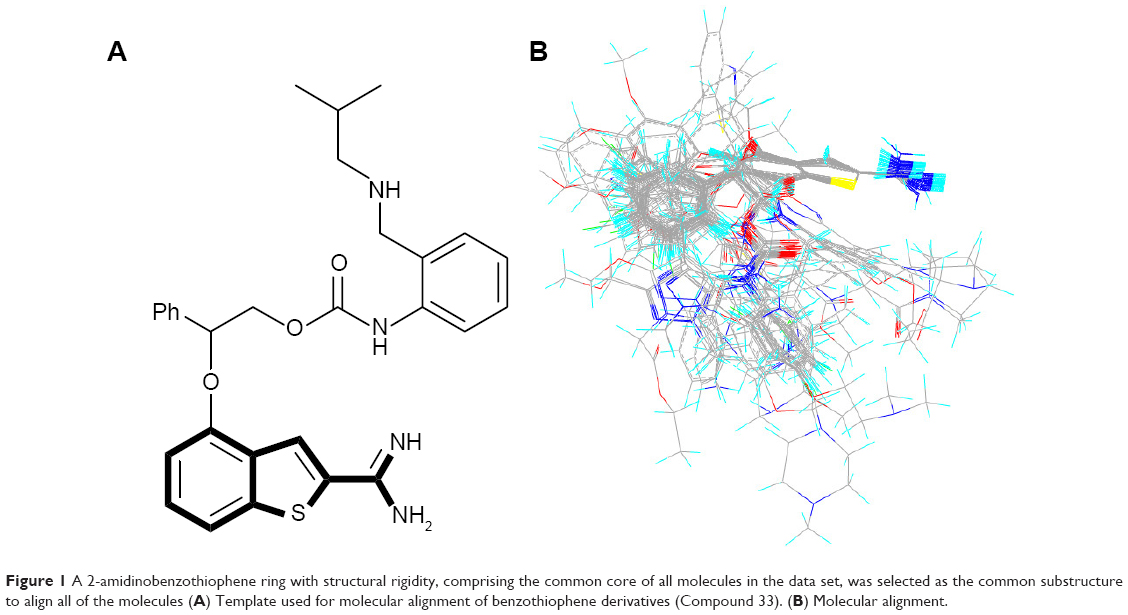 | Figure 1 A 2-amidinobenzothiophene ring with structural rigidity, comprising the common core of all molecules in the data set, was selected as the common substructure to align all of the molecules (A) Template used for molecular alignment of benzothiophene derivatives (Compound 33). (B) Molecular alignment. |
CoMFA methodology
Despite being unable to describe all of the binding forces, CoMFA is still a widely useful tool for QSPR analysis at 3D level.25 In the present study, the grid spacing was set at 2 Å in the x, y, and z directions, and the grid region was automatically generated to be a 3D cubic lattice that extended at least 4 Å beyond van der Waals volume of all aligned molecules on all axes. Lennard-Jones potential and Coulomb potential were employed to calculate steric and electrostatic energies of each molecule, respectively,19,20 and an sp3-hybridized carbon atom with a +1 charge was used as the probe atom to determine the magnitude of the field values. The column filtering value of 2.0 kcal/mol and the cutoff value of 30 kcal/mol improved the signal-to-noise ratio and reduced domination by large steric and electrostatic energies to a minimum.21 The regression analysis was carried out using the partial least squares (PLS) method,20 and the final model was obtained.
CoMSIA methodology
CoMSIA and CoMFA are both based on the same assumption that changes in binding affinities of ligands are related to changes in molecular properties, represented by fields. However, CoMFA only describes the two binding forces of the steric field and electrostatic field, but three other different fields including hydrophobic, hydrogen bond donor, and hydrogen bond acceptor are calculated in CoMSIA.26 For the distance dependence between the probe atom and the molecule atoms, a Gaussian function is used. Because of the different shape of the Gaussian function, the similarity indices can be calculated at all grid points, both inside and outside the molecular surface, while the calculations are not done inside the molecular surface in CoMFA. Equation 1 is used to calculate the similarity indices as follows:
|
|
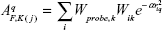
where A is the similarity index at grid point q, summed over all atoms i of the molecule j under investigation. Wprobe,k is the probe atom with radius 1 Å, charge +1, hydrophobicity +1, hydrogen bond donating +1, and hydrogen bond accepting +1. Wik is the actual value of the physicochemical property k of atom i. r2iq is the mutual distance between the probe atom at grid point q and atom i of the test molecule. α is the attenuation factor whose optimal value is normally between 0.2 and 0.4, with a default value of 0.3.27,28
PLS analysis
To linearly correlate the CoMFA and CoMSIA fields to all types of molecular properties, PLS analysis29 was carried out with the leave-one-out and leave-group-out (ten compound groups) cross-validation methods to determine cross-validated r2 (q2) values and the optimal number of components, which usually corresponds to the highest cross-validated coefficient (q2) and the smallest root mean predictive sum of squared errors for the final non-cross-validated models. To avoid over-fitting the models, a higher number of components was accepted only when the q2 differences between two components were larger than 10%. Bootstrapping analysis (ten runs) was also utilized to calculate confidence intervals for the r2 and standard errors of estimate to further assess the robustness of the derived models.20,29 Equations 2 and 3 of q2 and standard errors of estimate, respectively, are given below.
|
|
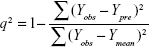
where Yobs, Ypre, and Ymean are experimental, predicted, and mean values of the target property for pIC50 (FIXa) or logIC50 (FXa/FIXa), respectively, and
|
|
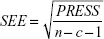
where n, c, and PRESS (predicted sum of squares) represent mean number of compounds, number of components, and 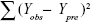 , respectively.
, respectively.
Predictive correlation coefficient
The q2 has been a good indicator of the accuracy of actual predictions: q2>0.6 means a fairly good model, q2=0.4–0.6 means a questionable model, and q2<0.4 means a poor model.30 q2 is a useful but not sufficient criterion for model validation, so external test sets23 were needed to estimate the model’s predictive ability. The predictive correlation coefficient (r2pred), based on the molecules of the test set, was calculated by Equations 4 and 5, shown below:
|
|
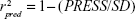
|
|

where Yobs(test) means the observed activity values of the test set, and 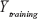 indicates the average activity values of the training set.
indicates the average activity values of the training set.
Results and discussion
CoMFA analysis for 3D-QSAR and 3D-QSSR models
The most active Compound 33 was selected as the template for alignment (Figure 1). The CoMFA model provided a cross-validation q2 value of 0.713 with six components, and the r2 value of 0.945 for the inhibitory activity as characterized by pIC50 (FIXa), whereas a model with the q2 value of 0.689 for five components and the r2 value of 0.942 was obtained for the selectivity characterized by logIC50 (FXa/FIXa) (Table 2).
After region focusing, the new models were generated with the predictive power (q2) value increased, the resolution enhanced, the grid spacing tighter, and the stability improved at a higher number of components. The CoMFA contours for Compound 33 after region focusing are shown in Figure 2. The internal validity were increased from 0.713 to 0.753 for 3D-QSAR, and from 0.689 to 0.770 for 3D-QSSR, and the group cross-validation was increased from 0.740 to 0.768 and 0.581 to 0.765, respectively, with the non-validated r2 increased from 0.942 to 0.965 for the 3D-QSSR model and a negligible effect for the 3D-QSAR model increasing its value from 0.945 to 0.940 (Table 3). The inhibitory activity values predicted for the tested compounds were in good agreement with the experimental values (Figure 3). The r2pred values of 0.757 (3D-QSAR) and 0.678 (3D-QSSR) further confirmed that the two models were both reliable and accurate, with higher predictive capacity. Therefore, the pIC50 (FIXa) and logIC50 (FXa/FIXa) model will be used to predict the activity and guide future synthetic efforts on novel potent and selective FIXa inhibitors.
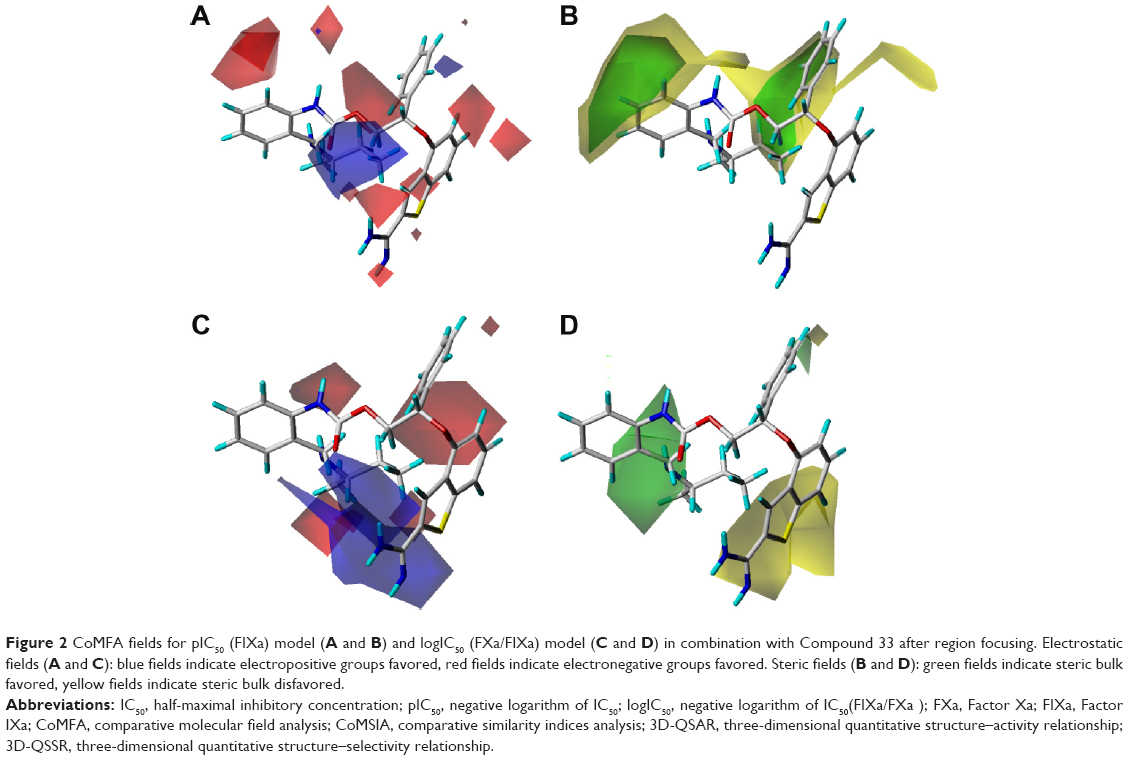 | Figure 2 CoMFA fields for pIC50 (FIXa) model (A and B) and logIC50 (FXa/FIXa) model (C and D) in combination with Compound 33 after region focusing. Electrostatic fields (A and C): blue fields indicate electropositive groups favored, red fields indicate electronegative groups favored. Steric fields (B and D): green fields indicate steric bulk favored, yellow fields indicate steric bulk disfavored. |
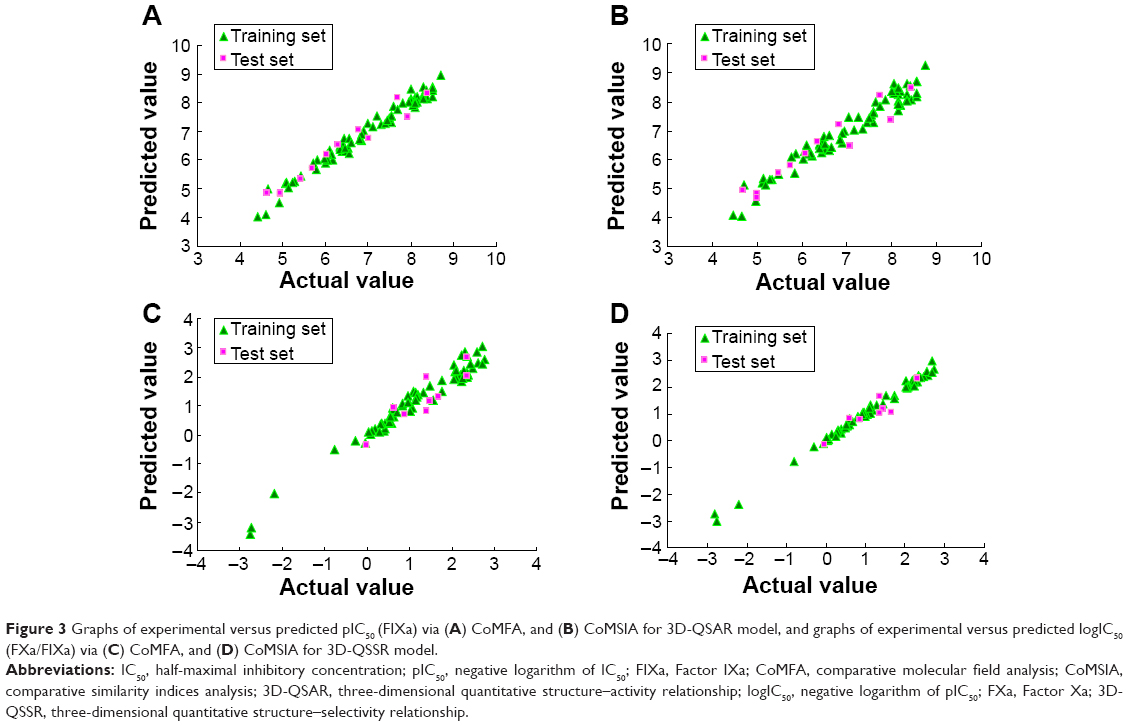 | Figure 3 Graphs of experimental versus predicted pIC50 (FIXa) via (A) CoMFA, and (B) CoMSIA for 3D-QSAR model, and graphs of experimental versus predicted logIC50 (FXa/FIXa) via (C) CoMFA, and (D) CoMSIA for 3D-QSSR model. |
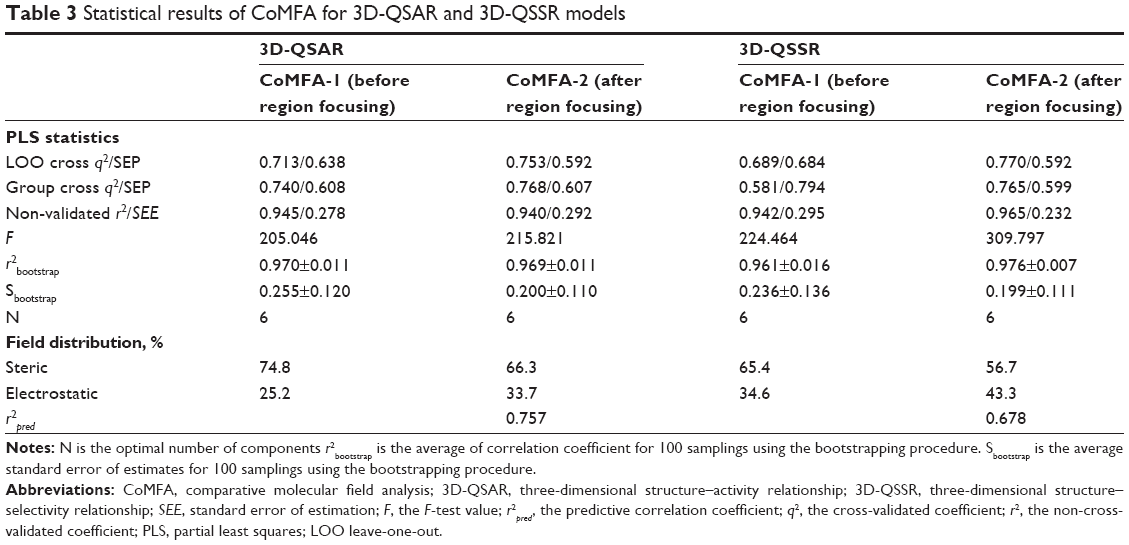 | Table 3 Statistical results of CoMFA for 3D-QSAR and 3D-QSSR models |
CoMSIA analysis for 3D-QSAR and 3D-QSSR models
Eighteen and eleven CoMSIA models were generated for 3D-QSAR and 3D-QSSR, respectively, using combinations of two, three, four, and all five descriptor fields, as shown in Table 4. In 3D-QSAR, Model 11, which was based on steric, hydrophobic, and hydrogen-bond acceptor fields, was found to be the most accurate, yielding a q2 value of 0.735 and an r2 value of 0.966, and the group cross-q2 value of 0.723 and bootstrapped value of 0.980±0.005 confirmed model accuracy. In comparison, Model 9 in 3D-QSSR based on steric, hydrophobic, and hydrogen-bond acceptor and donor fields, was the most accurate, with a q2 value of 0.837, an r2 value of 0.973, a group cross-q2 value of 0.854, and a bootstrapped value of 0.977±0.007 (Table 5). The predicted values were also consistent with the experimental data (Figure 3). These two models were subsequently selected to generate the final CoMSIA models.
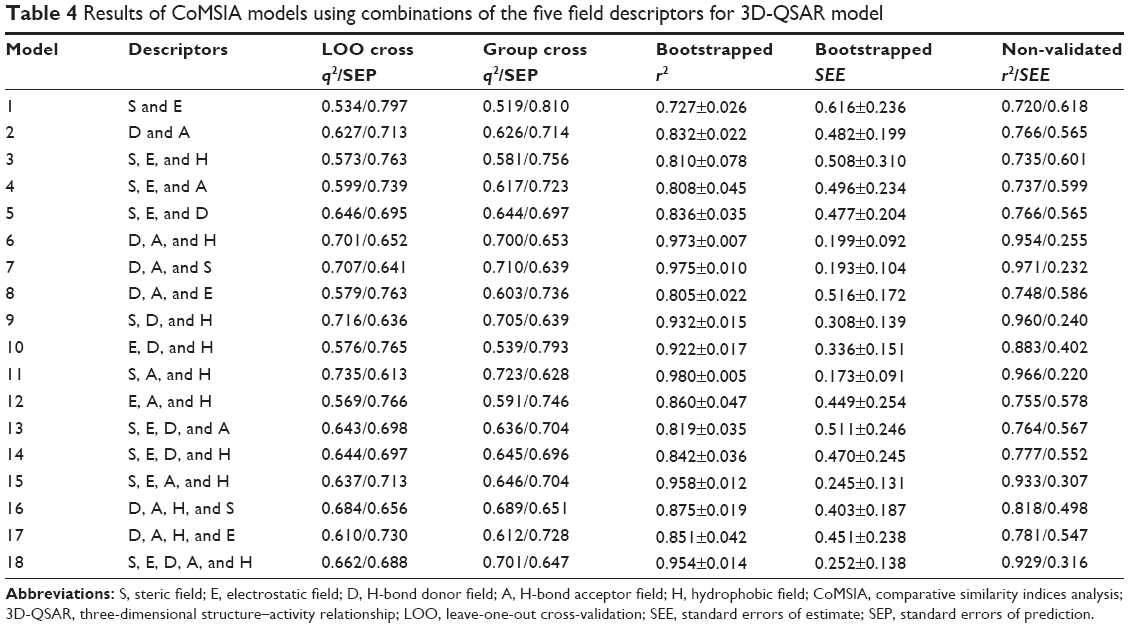 | Table 4 Results of CoMSIA models using combinations of the five field descriptors for 3D-QSAR model |
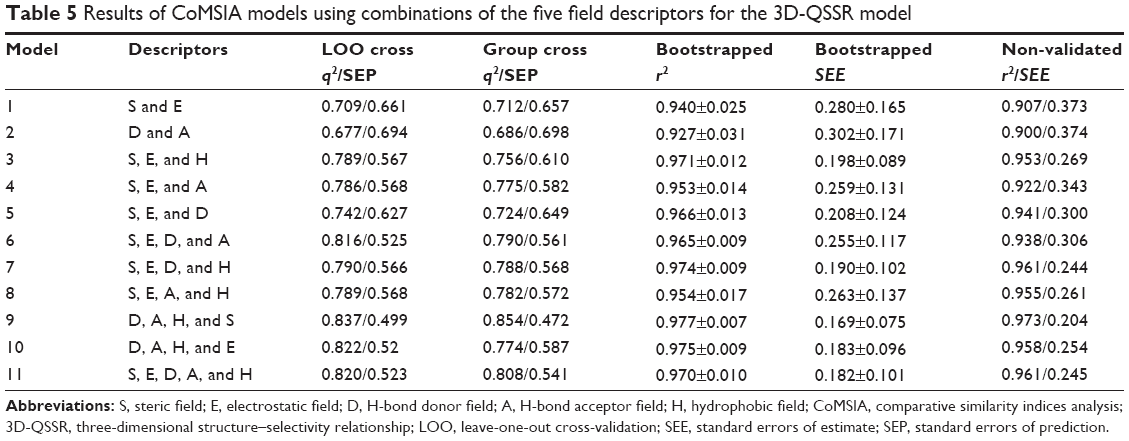 | Table 5 Results of CoMSIA models using combinations of the five field descriptors for the 3D-QSSR model |
3D contour maps of QSAR and QSSR
Scatter plots gave a visual impression of the region as a whole, with the color of each point indicating the field intensity. The results of QSAR models are presented in the contour maps, as shown in Figures 2 and 4, while 3D-QSSR contour maps are shown in Figures 2 and 5.
CoMFA contour maps for pIC50 (FIXa) in the 3D-QSAR model
Figure 2 shows the CoMFA steric and electrostatic fields of Compound 33 for the 3D-QSAR model. The green contours located around the two aromatic rings linked to the side chain of benzothiophene characterized the regions where bulky substituents would increase biological activity, and this could explain Compounds 24-47 consisting of two aromatic rings about 100 times more active than other compounds, such as Compounds 1–4 and Compounds 52–61 without the substituents. That is to say, the two aromatic rings are essential groups of 2-amidinobenzothiophene derivatives for its inhibitory activity on FIXa.
The red and blue contours in Figure 2 depict the favorable sites for electronegative and electropositive groups, respectively. A large blue contour located around the distal side chain at the 2′-position of R group (phenyl ring) in Compound 33 indicated regions where positively charged groups were favorable for the activity, and that is why Compound 22 was less active than the Compound 33 with isobutyl group at the site. Many red contours either encircling the benzothiophene ring or around the side chain indicated that electron-rich substituents for these positions were favorable for the activity. That is the reason why Compound 84, linked with the negatively charged group, showed better activity, only a little lower than the most active Compound 33.
CoMSIA contour maps for pIC50 (FIXa) in the 3D-QSAR model
The best CoMSIA model contour maps of the Compound 33, derived using steric, hydrophobic, and hydrogen bond acceptor fields, are shown in Figure 4. Its steric contour (Figure 4A) was more or less similar to that of CoMFA. Hydrophobic contour maps from CoMSIA are shown in Figure 4B, in which the purple and the gray contours depicted favored and disfavored sites for hydrophobic substituents, respectively. The Compound 33 was surrounded by the gray contours, suggesting that it is essential for the benzothiophene ring to link with some hydrophilic groups such as amino, carboxyl, and oxygen atoms. Therefore, Compounds 81–84 exhibited better activity than Compounds 54–61. Figure 4C shows the CoMSIA hydrogen-bond acceptor fields, denoted by magenta and cyan contours. Magenta contours represent regions where hydrogen-bond acceptor substituents were preferred, and cyan contours indicate unfavorable regions. One large magenta contour around the side chain indicates that the carboxyl group was necessary for potencies, and that is why Compounds 20–48 showed more potency than Compounds 62–73 without carboxyl groups at the side chain. The CoMSIA steric field, hydrogen-bond acceptor, and hydrophobic field explained 27.0%, 26.5%, and 46.5% of the variances, respectively.
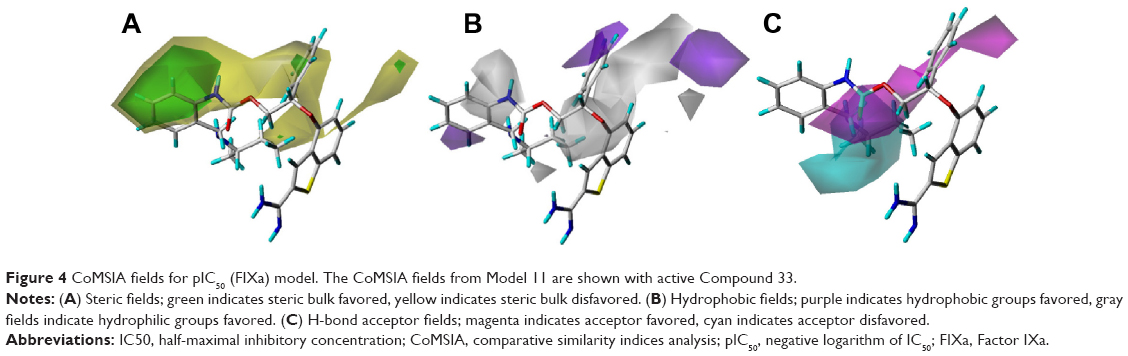 | Figure 4 CoMSIA fields for pIC50 (FIXa) model. The CoMSIA fields from Model 11 are shown with active Compound 33. |
CoMFA contour maps for logIC50 (FXa/FIXa) in the 3D-QSSR model
The CoMFA steric contour map of the Compound 33 displayed a large green polyhedron at the 2′-position of the phenyl in the R group, suggesting that bulky substituents linked to this position would improve the selectivity of compounds on FIXa against FXa (Figure 2). Compound 33 inserted a butylaminomethyl group at the 2′-position into the green area, and Compounds 46 and 47 similarly inserted an ethylaminomethyl and an i-propylaminomethyl group into the same area, respectively, and therefore showed the best selectivity. A large yellow polyhedron around the benzothiophene ring indicated that bulk was disfavored here for the selectivity on FIXa, which could explain why the Compounds 51, 57, and 58 with some bulk groups substituted at the benzothiophene ring showed lower selectivity.
Figure 2 also indicated that the compounds with electropositive groups at the 2′-position increased not only the activity but also the selectivity. Both the Compound 32 with a methylaminomethyl group substituted at 2′-position of the phenyl ring and Compound 33 with a butylaminomethyl substituent showed relatively higher activity and selectivity. These results also could explain why the Compounds 44 and 47 had the best selectivity and much more activity.
CoMSIA contour maps for logIC50 (FXa/FIXa) in the QSSR model
The best CoMSIA model contour maps, derived using steric, hydrophobic, and hydrogen bond donor and hydrogen bond acceptor fields, are shown in Figure 5. The steric contour plots (Figure 5A) correlated well with its CoMFA contour maps. The hydrophobic contour maps with the purple contour indicating favored sites and the gray contour indicating disfavored sites are shown in Figure 5B. The combined information from Figures 4B and 5B indicated that the hydrophobic substituents at the 3′-position and the hydrophilic groups at the 2′-position of the phenyl ring increased not only increased the activity, but also increased the selectivity. That is the reason why Compounds 44 and 47, possessing alkylamino and methyl group at the 2′, 3′-position, respectively, showed the highest selectivity and almost the strongest activity, nearly three times more than Compound 45 with a hydrophilic ethoxyl group at the 3′-position and a hydrophobic methyl group at the 2′-position. Figure 5C and D give the combined information that the amide group at the side chain was an essential group for a high selectivity on FIXa. The contributions of the steric, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor to the final model were 25.5%, 23.0%, 23.6%, and 27.9%, respectively.
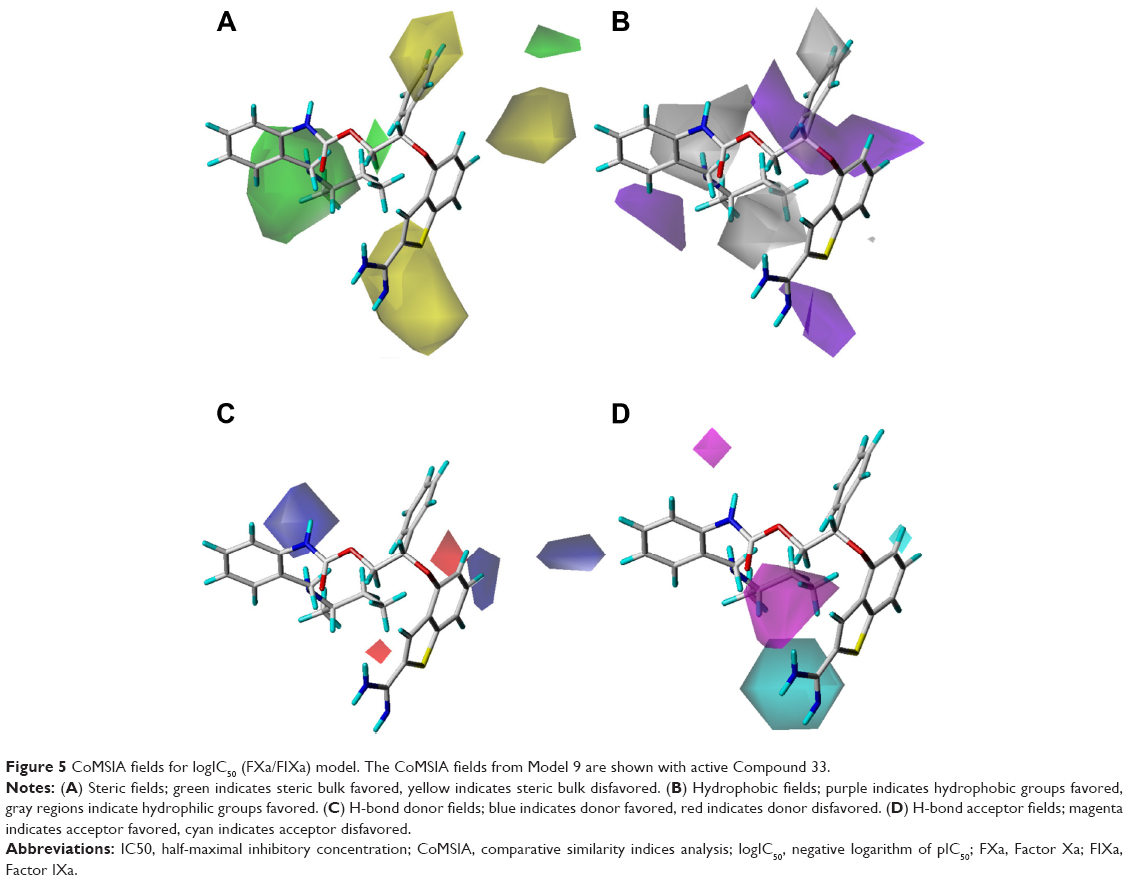 | Figure 5 CoMSIA fields for logIC50 (FXa/FIXa) model. The CoMSIA fields from Model 9 are shown with active Compound 33. |
Designing potent derivatives
Based on the 3D-QSARs and 3D-QSSRs revealed by the present study, 16 novel 2-amidinobenzothiophene derivatives were designed. These molecules were aligned to the database, and their activity and selectivity on FIXa were predicted by the best CoMFA and CoMSIA models previously established in the current study. All of the designed molecules showed better activity and selectivity compared with Compound 33, especially Compounds D2, D8, and D12, which showed 40 times more activity and 60 times more selectivity, respectively. The chemical structures and predicted pIC50 and logIC50 (FXa/FIXa) values of these compounds are shown in Table 6. The predictive results may validate the structure–activity and structure–selectivity relationships in this present work.
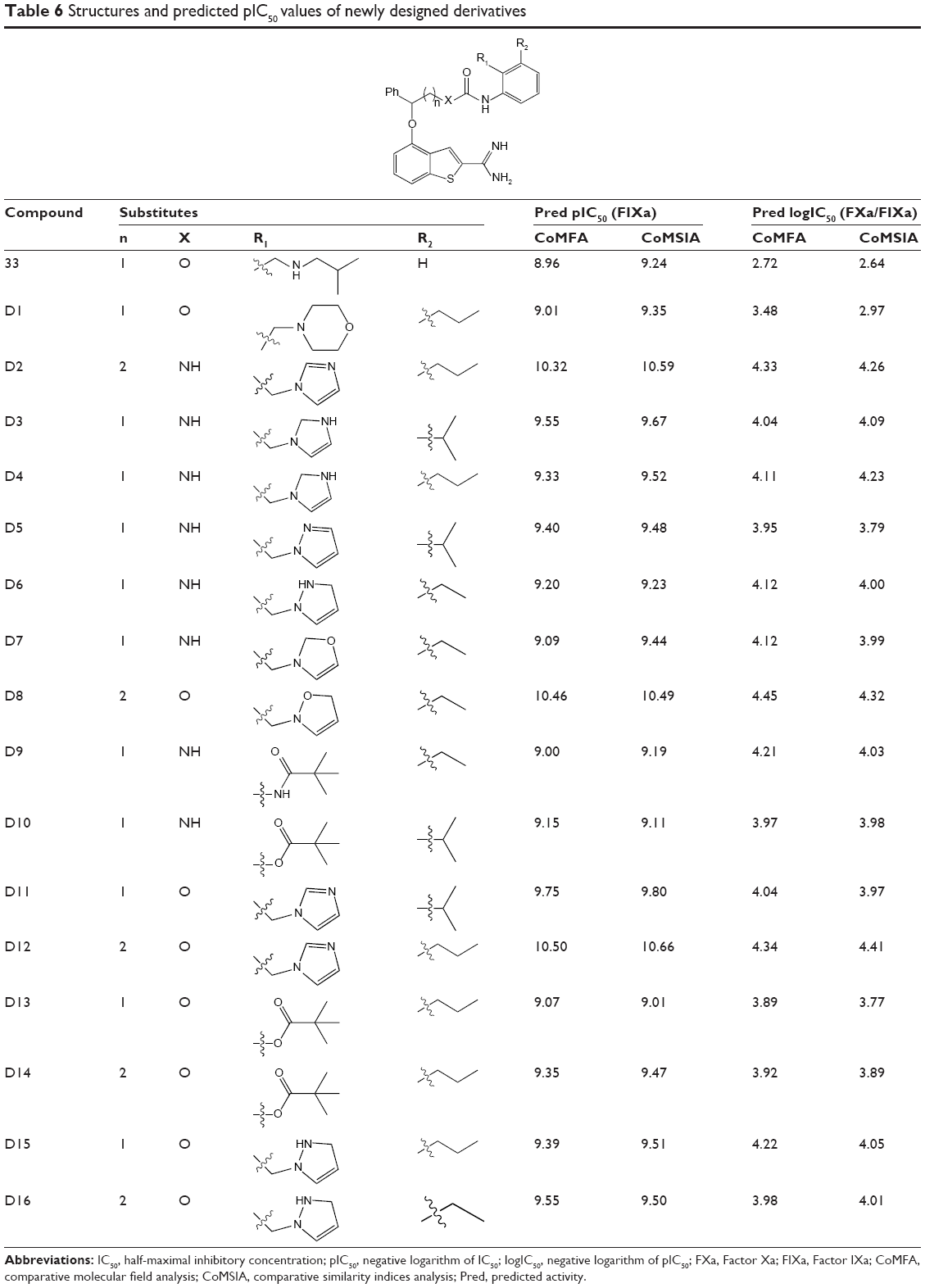 | Table 6 Structures and predicted pIC50 values of newly designed derivatives |
Conclusion
Based on a series of 2-amidinobenzothiophenes with FIXa and FXa inhibitory activity, and the selectivity on FIXa, the predictive 3D-QSAR and 3D-QSSR models with high cross-validation correlation coefficient q2 and non-cross-validation correlation coefficient r2 values have been developed. Along with further testing, the obtained 3D-QSAR and 3D-QSSR models showed their significance in predicting the inhibitory activity and selectivity of amidinobenzothiophene derivatives on FIXa. It was concluded that both the electrostatic and hydrophobic fields should be considered more appropriately; in particular, the electropositive and hydrophilic substituents at the 2′-position and the hydrophobic groups at the 3′-position of the phenyl ring would improve the activity and selectivity of the compounds. On the basis of the 3D-QSPRs, 16 newly designed molecules predicted higher activity and selectivity than the best active Compound 33 described in the literature.
Acknowledgments
This study was supported by a research grant from the National Natural Science Fund of China (No 81373285) and a research project grant from the Industry-Academy-research projects of Zhongshan (No 2012cxy020).
Disclosure
The authors declare no conflicts of interest in this work.
References
Feuerstein GZ, Nichols AJ, Ruffolo RR. Cardiovascular & renal: clopidogrel: a novel antiplatelet drug for prevention and treatment of thrombotic disorders. Expert Opin Inv Drug. 1995;4:425–430. | ||
Feuerstein GZ, Ruffolo RR. The antiplatelet GPIIb/IIIa antibody: TE3 A pioneer drug for the treatment of arterial thrombotic disorders. Expert Opin Inv Drug. 1994;3:1163–1169. | ||
Topol EJ. Toward a new frontier in myocardial reperfusion therapy: emerging platelet preeminence. Circulation. 1998;97:211–218. | ||
Gómez-Outes A, Suárez-Gea ML, Lecumberri R, Rocha E, Pozo-Hernández C, Vargas-castrillón E. New parenteral anticoagulants in development. Ther Adv Cardiovasc Dis. 2011;5:33–59. | ||
Hirsh J, O’Donnell M, Weitz JI. New anticoagulants. Blood. 2005;105:453–463. | ||
Quan ML, Smallheer JM. The race to an orally active Factor Xa inhibitor: recent advances. Curr Opin Drug Discov Devel. 2004;7:460–469. | ||
Frederick R, Pochet L, Charlier C, Masereel B. Modulators of the coagulation cascade: focus and recent advances in inhibitors of tissue factor, factor VIIa and their complex. Curr Med Chem. 2005;12:397–417. | ||
Jesty J, Nemerson Y. The pathways of blood coagulation. In: Beutler E, Lichman MA, Collar BS, Kipps TJ, editors. New York: McGraw Hill; 1995;317–325. | ||
Johnson DJ, Langdown J, Huntington JA. Molecular basis of factor IXa recognition by heparin-activated antithrombin revealed by a 1.7-A structure of the ternary complex. Proc Natl Acad Sci U S A. 2010;107: 645–650. | ||
Lawson JH, Mann KG. Cooperative activation of human factor IX by the human extrinsic pathway of blood coagulation. J Biol Chem. 1991; 266:11317–11327. | ||
Neuenschwander P. Coagulation cascade: intrinsic factors. In: Laurent GL, Shapiro SD, editors. Encyclopedia of Respiratory Medicine. Oxford: Elsevier Ltd; 2006;509–514. | ||
McKean ML, Adelman SJ. Future therapies for the prevention and treatment of venous and arterial thrombosis. Expert Opin Investig Drugs. 1998;7:687–690. | ||
Mizurini DM, Francischetti IM, Andersen JF, Monteiro RQ. Nitrophorin 2, a factor IX(a)-directed anticoagulant, inhibits arterial thrombosis without impairing haemostasis. Thromb Haemost. 2010;104:1116–1123. | ||
Spanier TB, Chen JM, Oz MC, et al. Selective anticoagulation with active site-blocked factor IXA suggests separate roles for intrinsic and extrinsic coagulation pathways in cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1998;116:860–869. | ||
Wang S, Beck R, Blench T, et al. Studies of benzothiophene template as potent factor IXa (FIXa) inhibitors in thrombosis. J Med Chem. 2010; 53:1465–1472. | ||
Wang S, Beck R, Burd A, Blench T, Marlin F, et al. Structure based drug design: development of potent and selective factor IXa (FIXa) inhibitors. J Med Chem. 2010;53:1473–1482. | ||
Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1998;110:5959–5967. | ||
Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem. 1994;37:4130–4146. | ||
Srivastava V, Kumar A, Mishra BN, Siddiqi MI. CoMFA and CoMSIA 3D-QSAR analysis of DMDP derivatives as anti-cancer agents. Bioinformation. 2008;2:384–391. | ||
Hansch C, Smith N, Engle R, Wood H. Quantitative structure-activity relationships of antineoplastic drugs: nitrosoureas and triazenoimidazoles. Cancer Chemother Rep. 1972;56:443–456. | ||
Xiao A, Zhang Z, An L, Xiang Y. 3D-QSAR and docking studies of 3-arylquinazolinethione derivatives as selective estrogen receptor modulators. J Mol Model. 2008;14:149–159. | ||
Hao M, Zhang SW, Yang W. Investigation on the binding mode of benzothiophene analogues as potent factor IXa (FIXa) inhibitors in thrombosis by CoMFA, docking and molecular dynamic studies. J Enzyme Inhib Med Chem. 2011;26:792–804. | ||
Cho SJ, Tropsha A. Cross-validated R2-guided region selection for comparative molecular field analysis: a simple method to achieve consistent results. J Med Chem. 1995;38:1060–1066. | ||
Zheng J, Xiao G, Guo J, et al. Exploring QSARs for 5-lipoxygenase (5-LO) inhibitory activity of 2-substituted 5-hydroxyindole-3-carboxylates by CoMFA and CoMSIA. Chem Biol Drug Des. 2011; 78:314–321. | ||
Huang X, Xu L, Luo X et al. Elucidating the inhibiting mode of AHPBA derivatives against HIV-1 protease and building predictive 3D-QSAR models. J Med Chem. 2002;45:333–343. | ||
Klebe G, Abraham U. Comparative molecular similarity index analysis (CoMSIA) to study hydrogen-bonding properties and to score combinatorial libraries. J Comput Aided Mol Des. 1999;13:1–10. | ||
Böhm M, St rzebecher J, Klebe G. Three-dimensional quantitative structure-activity relationship analyses using comparative molecular field analysis and comparative molecular similarity indices analysis to elucidate selectivity differences of inhibitors binding to trypsin, thrombin, and factor Xa. J Med Chem. 1999;42:458–477. | ||
Woodard B, Saleh MA. Three-dimensional quantitative structure activity relationships of quorum-sensing and biofilm inhibitors in gram-negative bacteria. J Environ Sci Health B. 2008;43:281–287. | ||
Cramer RD 3rd, Patterson DE, Bunce JD. Recent advances in comparative molecular field analysis (CoMFA). Prog Clin Biol Res. 1989; 291: 161–165. | ||
Clark M, Cramer RD 3rd, Jones DM, Patterson DE, Simeroth PE. Comparative molecular field analysis (CoMFA). 2. Toward its use with 3D- structural databases. Tetrahedron Comput Method. 1990;3:47–59. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.