")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Design and in vitro/in vivo evaluation of sustained-release floating tablets of itopride hydrochloride
Authors Ahmed SM, Ahmed Ali A, Ali AMA, Hassan OA
Received 27 June 2016
Accepted for publication 16 August 2016
Published 14 December 2016 Volume 2016:10 Pages 4061—4071
DOI https://doi.org/10.2147/DDDT.S115909
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Sayed M Ahmed,1 Adel Ahmed Ali,2 Ahmed MA Ali,2,3 Omiya A Hassan2,4
1Department of Industrial Pharmacy, Faculty of Pharmacy, Assiut University, Assiut, 2Department of Pharmaceutics, Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt; 3Department of Pharmaceutics, Faculty of Pharmacy, Taif University, Taif, Kingdom of Saudi Arabia; 4Department of Pharmaceutics, Faculty of Pharmacy, Deraya University, El-Minia Gadida, Egypt
Purpose: The aim of the present study was to improve the bioavailability of itopride (ITO) and sustain its action by formulating as a floating dosage form.
Materials and methods: Sustained-release floating tablets of ITO hydrochloride (HCl) were prepared by direct compression using different hydrocolloid polymers such as hydroxypropyl methylcellulose and ethylcellulose and/or methacrylic acid polymers Eudragit RSPM and Carbopol 934P. The floating property was achieved using an effervescent mixture of sodium bicarbonate and anhydrous citric acid (1:1 mol/mol). Hardness, friability, content uniformity, and dissolution rate of the prepared floating tablets were evaluated. The formulation F10 composed of 28.5% Eudragit RSPM, 3% NaHCO3, and 7% citric acid provided sustained drug release.
Results: In vitro results showed sustained release of F10 where the drug release percentage was 96.51%±1.75% after 24 hours (P=0.031).The pharmacokinetic results indicated that the area under the curve (AUC0–∞) of the prepared sustained-release floating tablets at infinity achieved 93.69 µg·h/mL compared to 49.89 µg·h/mL for the reference formulation (Ganaton®) and the relative bioavailability of the sustained-release formulation F10 increased to 187.80% (P=0.022).
Conclusion: The prepared floating tablets of ITO HCl (F10) could be a promising drug delivery system with sustained-release action and enhanced drug bioavailability.
Keywords: itopride HCl, oral drug delivery, stability study, bioavailability
Introduction
Itopride (ITO) hydrochloride (HCl) is a prokinetic agent1 and has anticholinesterase activity and dopamine D2-receptor antagonistic action. Gastrointestinal motility disorders can be treated using ITO HCl. ITO can activate the motility of the gastrointestinal tract by synergism of its dopamine D2-receptor antagonistic effect and its acetylcholinesterase inhibitory effect. The drug also acts as an antiemetic due to its dopamine D2-receptor antagonistic effect.2 The mode of action, which includes each of the acetylcholinesterase inhibitory action and dopamine D2 antagonism, is only for this drug, and its action is different from the mode of action of other prokinetic drugs.2
Because the drug can be used for chronic conditions, sustained-release oral dosage forms should increase the compliance of the patient and improve its therapeutic response by decreasing the peak-to-trough variation of ITO HCl plasma concentration. The only available extended-release ITO per-oral dosage forms are effective in gastric motility disorder treatment over 24 hours.3,4
ITO is soluble in water and its solubility is not much affected by the pH.5 Reference ITO is a good choice for formulation as a gastroretentive dosage form because its solubility in the stomach medium is high compared to its solubility in the small intestine medium.6,7
The floating dosage forms have been described by several studies.8,9 These systems are formulated to have a bulk density lower than the density of gastric fluid; hence, the buoyancy time of the floating dosage form was prolonged without any effects on the rate of gastric emptying.1,2 These systems have important advantages that include achieving a greater and prolonged therapeutic effect and thus reducing the frequency of administration periods, providing a more effective treatment of local stomach disorders, and minimizing both lower tract inactivation of the drug and drug effects on the lower intestinal flora. However, many limitations of the floating dosage forms were reported. They were not applicable for irritant drugs for gastric mucosa, andthey were also not suitable for drugs that have either solubility or stability problems in gastric fluids.
In the present work, the formula that provided good floating and extended-release characteristics was chosen for in vivo evaluation in comparison with a commercial immediate-release product of ITO HCl. Therefore, this research was aimed at improving the bioavailability of ITO HCl and reducing the required daily dose of this drug by formulating it as an extended-release tablet formula. An in vivo study was also conducted on rabbits to calculate the pharmacokinetic parameters of the selected floating tablet formula and to estimate the relative bioavailability in comparison to the commercial tablet formula of ITO HCl. A stability study was performed to determine the shelf life of the prepared formula compared to the reference generic product (Ganaton®).
Materials and methods
Materials
ITO HCl was obtained from Hangzhou Uniwise International Co., Ltd. (Shanghai, China). Hydroxypropyl methylcellulose (HPMC) 15000 was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Ethylcellulose (EC) was obtained from EMD Millipore (Billerica, MA, USA). Microcrystalline cellulose (Avicel PH 101) was purchased from FMC BioPolymer International Co. (Philadelphia, PA, USA). Eudragit RSPM was obtained from Rhom Pharma GmbH (Darmstadt, Germany). Sodium bicarbonate and citric acid were supplied from El Gomhoria Co. (Assuit, Egypt). Magnesium stearate was supplied from Alba Chemical Company (Alexandria, Egypt). High-performance liquid chromatography (HPLC)-grade methanol was obtained from Sigma-Aldrich Co. Chloroform was obtained from El-Nasr Pharmaceutical Chemicals Company (Cairo, Egypt). Dibasic ammonium phosphate was obtained from BDH Chemicals Ltd. Co. (Poole, England). Commercial immediate-release ITO HCl tablets were supplied from Abbott Laboratories (Abbott Park, IL, USA). All other chemicals were of analytical grade and used as received.
Preparation of floating tablets
Floating matrix tablets of ITO HCl were prepared using direct compression technique by using the following ingredients: HPMC 15000, EC, Eudragit RSPM, magnesium stearate, and sodium bicarbonate.10 The composition of the prepared formulations is listed in Table 1. Sodium bicarbonate was added as a gas-generating agent to maintain the buoyancy of the tablets by producing carbon dioxide in the gastric environment. Drug and the polymer (HPMC 15000, EC, and Eudragit RSPM) were mixed well, and magnesium stearate was finally added and mixed by geometrical mixing. A single die punch machine (EK/0; Korsch, Berlin, Germany) fitted with flat-faced punches (12 mm diameter) was used.
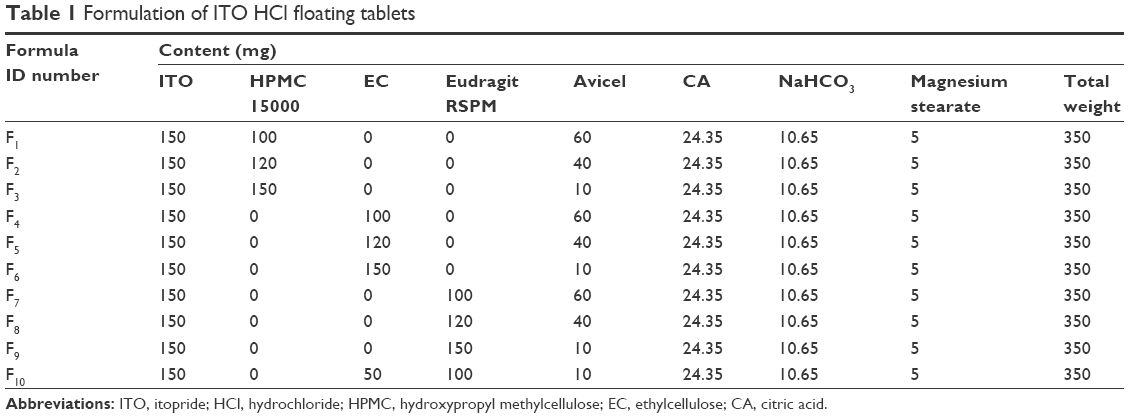 | Table 1 Formulation of ITO HCl floating tablets |
Pharmaceutical evaluation of floating tablet
Floating behavior of the tablets
The buoyancy of the prepared floating tablets was examined by the floating lag time (the time between placing the tablet in the medium and the floating time).
The formulated tablets were placed in a 100 mL beaker containing 0.1 N HCl. The time required for the tablets to rise on the surface and then float was calculated as the floating lag time. However, the floating time is taken as the period of time during which the tablet remains floating on the gastric fluid. The buoyancy determination method was explained by Reddy and Murthy.11
Resultant weight determination
To achieve properly principle of the buoyancy retention, a minimal gastric content was needed. However, the floating force (F) of the dosage form should be able to keep the buoyancy of the drug delivery system on the surface of the gastric content.
The determination of density is not enough to predict the floating force evolution of the drug delivery systems because the dry content of the systems reacts or interacts with the gastric secretion to release their drug contents. To determine the floating force, an accurate method for measuring resultant weight (RW) has been reported in the literature.12,13
RW of the tablet was calculated as indicated in equation 1 and 2:
RW = Fbuoy − Fgrav | (1) |
RW = (Df − Ds)gV | (2) |
here, g is the acceleration of gravity, Df is the fluid density, Ds is the tablet density, Df is the gastric secretion density, and V is the volume of the tablet.
Solubility study
The solubility determinations of pure ITO HCl were performed in a water and acidic medium (pH 1.2) and a phosphate buffer medium (pH 6.8). A sample of 2 g of ITO HCl was added to 25 mL of each aforementioned medium in a glass vial with a cap. The vials were inserted in a shaker incubator and kept at 37°C±0.5°C for 24 hours. The samples were filtered and assayed UV spectrophotometrically at λmax 258 nm.
Determination of hardness
The formulated tablet was placed between the two anvils of hardness tester (Erweka-type TBT; Erweka GmbH, Heusenstamm, Germany), and force (in kg) was gradually increased for getting exact reading. The reading was determined referring to the pressure that was required for breaking the tablets.14
Friability
The formulated tablets (20 tablets) were weighed and then placed in the friabilator (Erweka friabilator apparatus; Erweka GmbH), and the apparatus was rotated at 25 rpm for 4 minutes. After revolutions, the tablets were dedusted and weighed again. The obtained value should not be >1%. The friability percent was determined using the following formula:15
%F = [1− (Wt/Wi)] ×100 | (3) |
where %F = friability percent, Wi = initial weight of the tablet, and Wt = weight of the tablet after revolution.
Drug content
Five tablets of each formula were triturated. The amount of powder equal to 100 mg of the drug was weighed and placed in a 100 mL volumetric flask. HCl (0.1 N) was added to the powder and the flask was shaken for 5 minutes. In all, 0.1 N HCl was added up to 100 mL. The solution was sonicated for 15 minutes and then filtered using a Whatman filter paper. Finally, the solution was diluted with 0.1 N HCl, and the absorbance of the diluted solution was measured using a spectrophotometer (UV-visible) at 258 nm against 0.1 N HCl blank.16
Weight variation
The formulated tablets (20 tablets) were selected from each batch and individually weighed using an electronic balance. The average weight of the 20 tablets was determined.
In vitro dissolution studies
In vitro dissolution studies for the prepared floating tablets of ITO HCl were examined in 0.1 N HCl at 37°C±0.5°C. The dissolution test was carried out for 24 hours, and cumulative drug release was determined for each 1 hour time interval.17
The dissolution examination was carried out using USP Dissolution Apparatus II (paddle type; Takao Manufacturing Co. Ltd, Kyoto, Japan) rotating at 75 rpm. The dissolution medium was heated to 37°C±1°C. Aliquots of dissolution medium (5 mL) were withdrawn at specified time intervals. Then, 5 mL of freshly prepared acid medium was added to the dissolution medium. Determination of the ITO HCl was carried out spectrophotometrically at λmax 258 nm, using the same dissolution medium as a blank. All determinations were conducted three times.
Mechanism of drug release
The mechanism of drug release from matrices can be determined by treating the dissolution value of each formula with different kinetic release models.18 The drug release values were plotted according to zero-order, first-order, and Higuchi diffusion models.
In vivo evaluation of a selected floating formula by comparative bioavailability study
Treatment protocol and sample analysis
The chosen floating formula containing 150 mg of ITO was compared with the commercial immediate-release ITO HCl tablets. The study was achieved using three groups of New Zealand rabbits (2.5 kg), each group consisting of three rabbits. Group I (control group) was starved and only water was allowed. Group II was administered the floating tablet formula F10. Group III was administered the commercial six tablets. The rabbit groups II and III were starved overnight before the administration of the drug and continued fasting until 4 hours postdose. Each group was given a drug dose of 15 mg/kg from the tested preparations (F10) and the commercial tablets. The study was performed as single-dose crossover design, with 7 days washout period. Blood samples (1 mL) were taken from the marginal ear vein and were transferred to tubes containing heparin at time intervals 0, 0.5, 1, 2, 4, 6, 8, 12, 18, and 24 hours after drug administration. Plasma was directly separated using a centrifuge and was kept at −20°C until used for analysis. Before the administration of the drug, samples of the blood were collected and plain plasma was separated by centrifugation and used for the construction of the calibration curve. All experiments were performed following relevant international regulations as per the Guide for the Care and Use of Laboratory Animals and were approved by the Assiut University Ethical Experimentation Committee of Faculty of Medicine.
The plasma samples were treated with 10% perchloric acid to precipitate the protein, followed by centrifugation at 4,000 rpm for 15 minutes. Analyses of ITO HCl were conducted using an HPLC with fluorescence detection. Levofloxacin was used as an internal standard. A reversed phase Hypersil BDS C18 (250×4.6 mm, 5 m) column was used. An isocratic degassed mobile phase was prepared using 0.2 mol/L ammonium acetate–methanol (15:85, v/v), and the flowing rate was 1.1 mL/min. The wavelengths of excitation and emission during the analysis were at 304 nm and 344 nm, respectively. From plasma concentrations of the ITO HCl, the pharmacokinetic parameters were calculated. The determination of the drug was carried out in triplicate.
Calculation and statistical treatment of pharmacokinetic parameters
The pharmacokinetic parameters were determined from the data of plasma level obtained from the individual rabbits and presented as mean ± standard deviation (SD). From the data of plasma concentration, the maximum plasma concentration (Cmax, μg/mL) and the corresponding time (Tmax, hour) were directly obtained for the two treatments in each individual rabbit. A plot of the mean plasma concentration versus time was constructed for each treatment. The area under the curve from time 0 to 24 hours (AUC0–24 μg·h/mL) was obtained by applying the trapezoidal rule. Accordingly, the area under the curve from time 0 hour to infinity (AUC0–∞) was determined by adding the area under the tail to AUC0–24 hour. The area under the tail was calculated by dividing the last determined concentration by the elimination rate constant obtained by linear regression of the elimination phase of the plasma concentration versus time curve. The mean residence time (MRT; hour), which is a noncompartmental pharmacokinetic parameter, was determined using a suitable equation.19 After the estimation of the area under the first-moment curve (AUC0–∞, μg·h/mL), the relative bioavailability (FR) of the examined formula compared with the commercial product was calculated as follows:
|
|
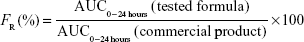
The significance of the difference between the two treatments was evaluated by one-way analysis of variance using a statistical computer package (SPSS Version 13.0; SPSS Inc., Chicago, IL, USA). Differences were considered significant at P<0.05.
Stability study
Effect of aging (shelf storage)
Samples of the selected formulae of ITO HCl tablets, which gave the most sustaining in vitro drug release, were stored in amber glass bottles and kept at room temperature for 6 months. The selected formulae of the prepared tablets were tested for their drug content, release characteristics, and physical properties.
Accelerated stability testing
Samples from the selected tablets were stored in amber-colored glass bottles in closed desiccators containing saturated solution of sodium chloride to attain 75% relative humidity (RH). The desiccators were kept at temperatures of 30°C, 40°C, and 50°C±2.0°C in thermostatically controlled hot air ovens for 6 months. The samples from each of the selected formulae were withdrawn after time intervals of 0.5, 1, 2, 3, 4, 5, and 6 months. The drug content was determined using the HPLC assay method.
The mobile phase consisted of filtered, degassed mixture of HPLC-grade methanol and chloroform 9:1 v/v pumped at a flow rate of 1.5 mL/min. The UV detector was adjusted at 258 nm, and the element peaks were investigated using peak height ratio. All analyses were performed at room temperature.
Determination of drug content in the stored tablets
Drug content in the stored tablets was determined using the HPLC method. At the specified time intervals, three randomly selected tablets were finally powdered. An accurately weighed amount of powder equivalent to 150 mg of ITO HCl was mixed with HPLC-grade methanol in a 100 mL volumetric flask and sonicated for 10 minutes. The solution was filtered, and 1 mL of the filtrate was transferred to a 25 mL volumetric flask. In all, 5 mL of the internal standard stock solution was pipetted into each volumetric flask and the volume was completed in the mobile phase. The obtained clear solutions were filtered through a 0.45 μm disk filter, degassed, and then 20 μL of solutions were injected onto the HPLC column. ITO HCl concentration in each sample was determined utilizing the constructed calibration curve.
Statistical analysis
SPSS Version 18 software (SPSS Inc.) was used to analyze the data. All the trials were carried out in triplicate, and the results were expressed as mean ± SD. The data for various formulations were statistically analyzed using one-way analysis of variance. P<0.05 was considered to be statistically significant. The differences were evaluated for statistical significance using the Student’s t-test.
Results
Solubility study
Solubility of ITO HCl in water, acidic medium (pH 1.2), and alkaline solution (pH 7.4) was 48.4 mg/mL, 50.5 mg/mL, and 47.6 mg/mL, respectively. Although the solubility of the drug in the acidic medium was the highest, it was not much affected by the change in the pH. These findings are in agreement with those reported by Satapathy et al.5
Evaluation of the prepared ITO HCl tablets
Formulations of floating ITO HCl tablets prepared were evaluated for different parameters, eg, thickness, hardness, friability, percent of weight variation, and percent of the drug content (Table 2). All the formulation of tablets showed uniform thickness and diameter. Concerning the test of weight variation, the pharmacopeial limit of the percentage deviation for the prepared ITO HCl floating tablets having a weight of 350 mg was ±5%.
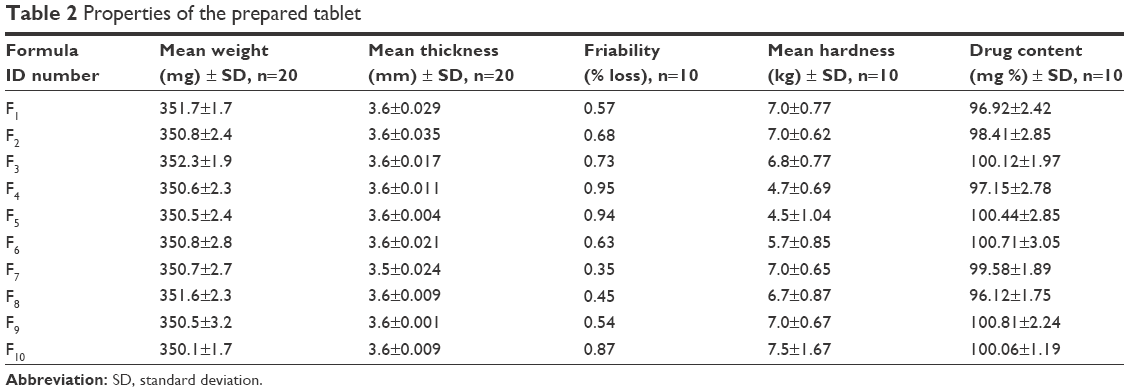 | Table 2 Properties of the prepared tablet |
The mean percentage deviation for each of the prepared ITO HCl floating tablet formulation was found to lie within this limit, and so all ITO HCl floating tablet formulations passed the uniformity test of weight according to official requirements.20 The test of drug content was found to be uniform at different tablet formulations, and the percentage of the drug content was >98%, w/w. The hardness of all batches was found to be between 4.5 kg/cm2 and 7.0 kg/cm2. The friability percent for all the formulations of tablets of <1% indicated that the friability percent is within the official limits. Finally, it could be concluded that each of the tablet formulations had acceptable pharmacopeial properties and complied with the specifications for hardness, friability, weight variation, and drug content.
In vitro buoyancy study
The floating results are shown in Table 3. Sodium bicarbonate generates CO2 in the dissolution medium (0.1 N HCl), and the tablet becomes buoyant, as shown in Figure 1.21 Thus, sodium bicarbonate was essential to achieve optimum in vitro buoyancy (ie, floating lag time of 3–6 minutes and floating duration of 24 hours). A further increase in sodium bicarbonate concentration does not show any significant effect on the floating behavior.22 The floating force was determined as shown in Table 3. Formulae F4, F5, and F6 have negative values. However, the other formulae have positive values.
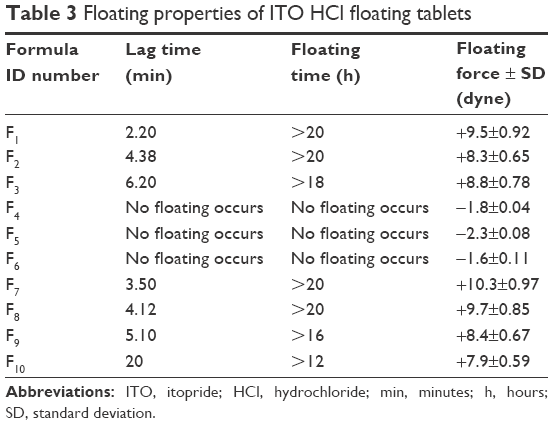 | Table 3 Floating properties of ITO HCl floating tablets |
 | Figure 1 In vitro buoyancy studies. |
In vitro drug release study
In vitro release of ITO HCl from different formulations is shown in Figure 2. Formulations F1, F2, and F3 showed 97.33%±2.11%, 99.57%±2.42%, and 99.05%±3.04% drug release at the end of 6, 8, and 12 hours, respectively. Formulations F4, F5, and F6 showed drug release of 96.81%±2.92%, 95.44%±1.07%, and 96.73%±2.34% at 8, 12, and 16 hours, respectively. Formulations F7, F8, F9, and F10 showed 98.01%±2.45%, 99.07%±2.42%, 95.06%±1.71%, and 96.51%±2.91% drug release at 12, 24, and 24 hours, respectively. F10 was selected as the optimized formulation because it gives sustained release of ITO HCl over a period of 24 hours up to 96.51%±1.75%.
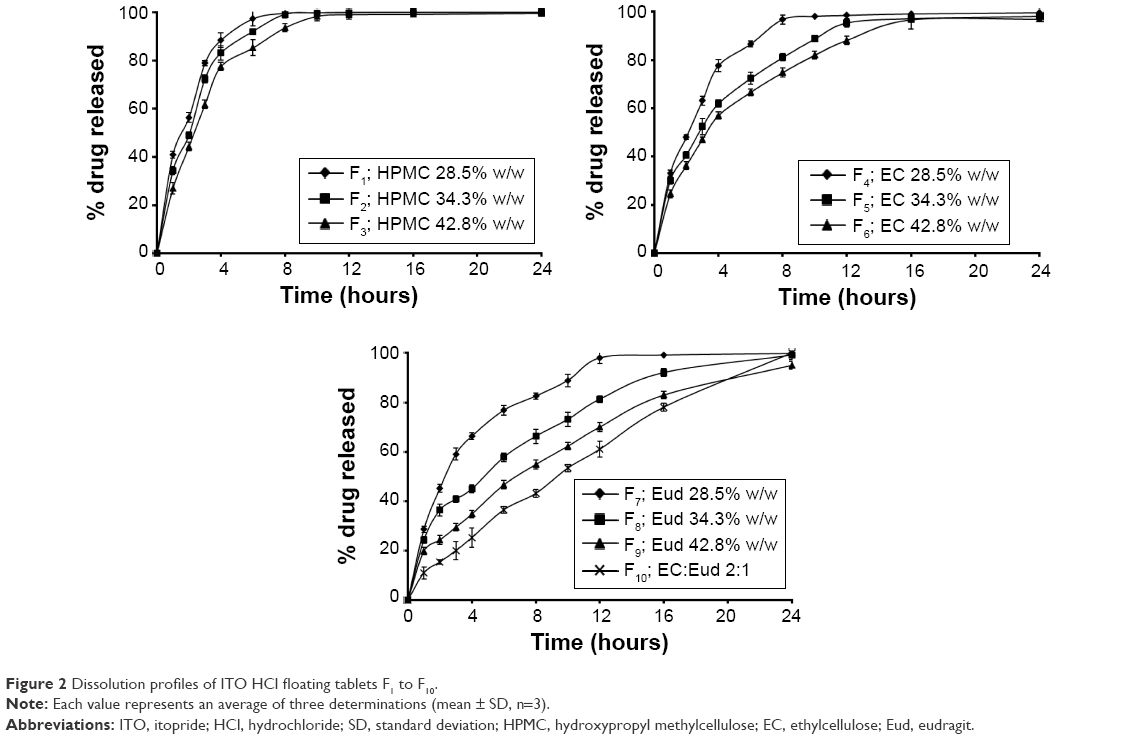 | Figure 2 Dissolution profiles of ITO HCl floating tablets F1 to F10. |
In vitro release data were subjected to various kinetic models to predict the best drug release kinetic mechanism, as shown in Table 4. The kinetic models used were zero-order, first-order, and Higuchi diffusion models. Higuchi diffusion model was fit for the formulations.
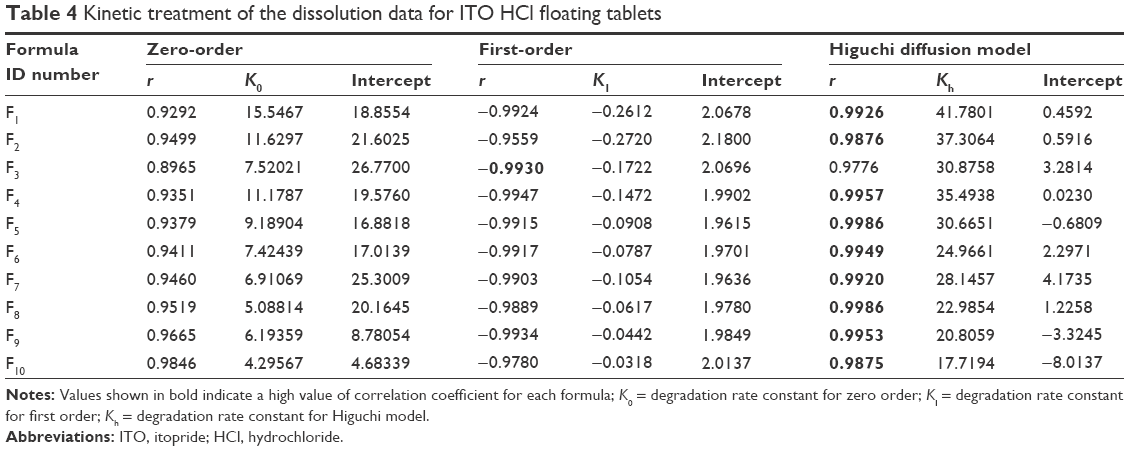 | Table 4 Kinetic treatment of the dissolution data for ITO HCl floating tablets |
Comparative bioavailability of ITO
The plasma concentration change in ITO with time after oral administration of the reference standard and the prepared floating tablet to rabbits was represented, as shown in Figure 3.
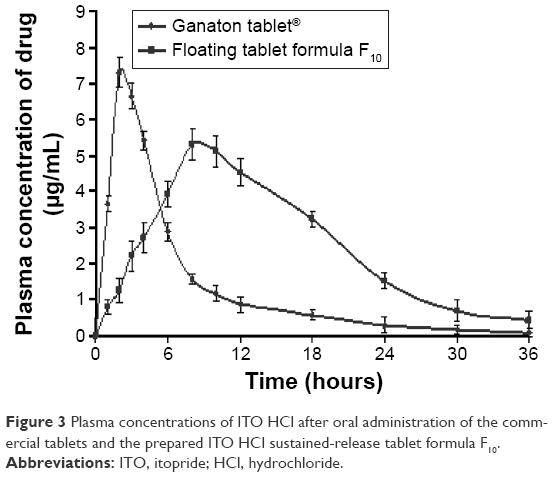 | Figure 3 Plasma concentrations of ITO HCl after oral administration of the commercial tablets and the prepared ITO HCl sustained-release tablet formula F10. |
Table 5 shows the pharmacokinetic parameters with ± SD generated from the individual data analysis.23 The maximum concentration Cmax of the prepared floating tablets (F10) was found to be 5.31 μg/mL, and the corresponding Tmax value was 8 hours. It was noted that the floating tablets exhibited delayed Tmax.
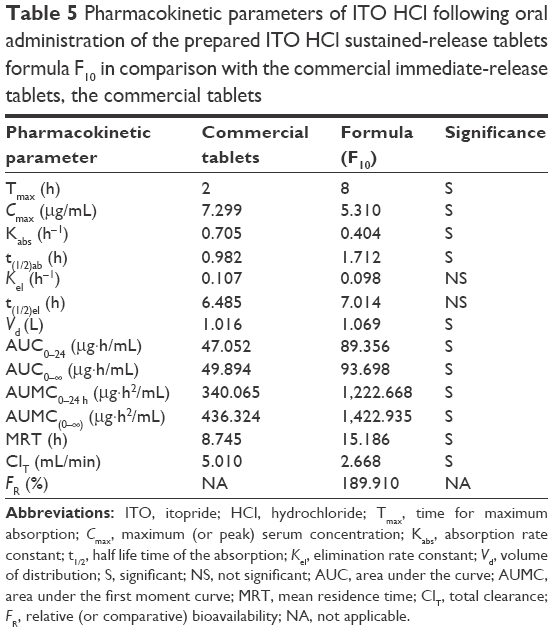 | Table 5 Pharmacokinetic parameters of ITO HCl following oral administration of the prepared ITO HCl sustained-release tablets formula F10 in comparison with the commercial immediate-release tablets, the commercial tablets |
The t1/2 value was found to be 1.712 hours for the floating ITO HCL tablet formula. The value of the MRT, which is the noncompartmental analog of t1/2, was also parallel to that of t1/2. The examined tablet formula showed higher MRT (15.186 hours). AUC0–∞ =93.698 μg·h/mL for the prepared floating formula; the relative bioavailability was 189.910%. ITO is considered as an example of drugs with high pharmacokinetic variability. The maximum relative standard deviation was 2.91%. The floating tablet showed more sustained-release characteristics.
Stability study
Effect of aging (shelf storage)
The changes in the physical properties of the selected formulae of ITO HCl tablets stored at ambient conditions (30°C, 40°C, and 50°C±1°C and 75% RH) for 6 months were measured (Table 6). The results revealed that no change in the color of the tablets has occurred during the storage period. Tablet formulae show good physical stability with respect to hardness, friability, and tablet dimensions (diameter and thickness). The drug content in the aged formulae of tablets was not changed significantly (P<0.05) as that of the corresponding freshly prepared formulae. This indicates chemical stability of the active ingredient in the tested formulae.
 | Table 6 Physical properties of the aged batches of floating tablets after shelf storage for 6 months |
It was found that the release rate of ITO HCl from the aged tablets was slightly decreased in comparison with the corresponding freshly prepared dosage form as shown in Figure 4.
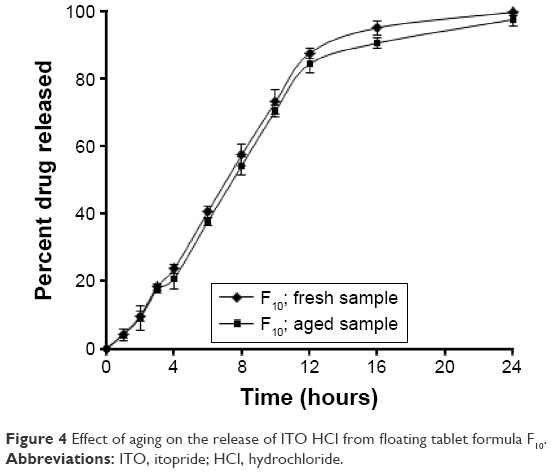 | Figure 4 Effect of aging on the release of ITO HCl from floating tablet formula F10. |
However, there was a decrease in the release rate of the drug from the aged formulae; this was not seen as a practically effective difference in the drug release because it is a common practice to report the release results of sustained-release products in terms of a range than as a single point.
Accelerated stability testing of ITO HCl in the selected formulae.
Table 7 shows the remaining ITO HCl content in the selected floating tablets formula F10 after storage for 6 months at different temperatures (30°C, 40°C, 50°C) and 75% RH. The HPLC analysis showed that the average percentage of ITO HCl content ranged from 96.90% to 100.15%.
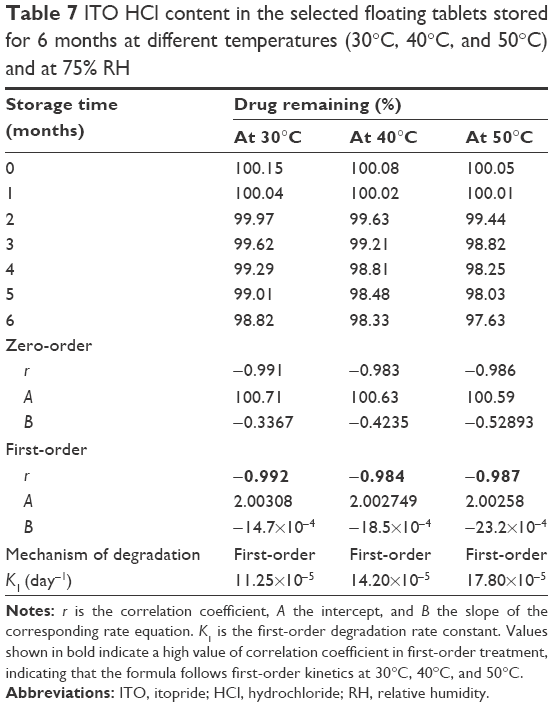 | Table 7 ITO HCl content in the selected floating tablets stored for 6 months at different temperatures (30°C, 40°C, and 50°C) and at 75% RH |
Kinetic analysis of the data was carried out to determine the mechanism of drug degradation and t90% (the time necessary to reach 90% of the labeled potency). Table 8 shows the values of the correlation coefficient of ITO HCl degradation in the selected formulae according to zero- and first-order reaction kinetics. The degradation of ITO HCl in the studied formulae followed first-order kinetics as indicated from the highest values of the correlation coefficient.
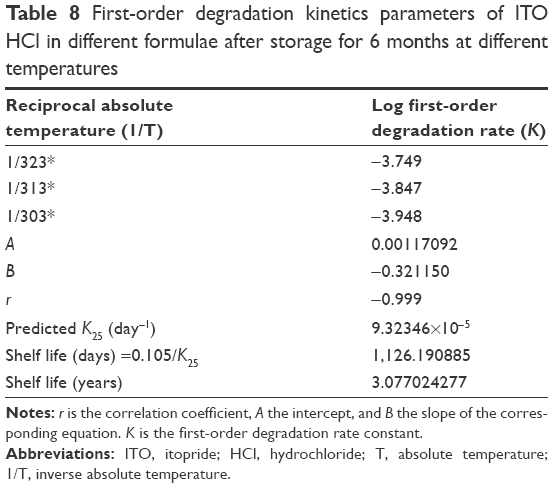 | Table 8 First-order degradation kinetics parameters of ITO HCl in different formulae after storage for 6 months at different temperatures |
Discussion
The common way of controlling delivery is incorporating drug into a polymer. As the drug is freely water soluble, it requires careful selection of synthetic polymers such as Eudragit RSPM, Eudragit RS 100, ethylcellulose, and HPMC 15000.
Flow properties are very important for powder to be compressed into tablets. Materials that do not have good flow properties are difficult to compress into tablets and may lead to improper mixing of drug with other ingredients, which cause compression problems. The flow property of plain ITO HCl was improved by the addition of lubricants and directly compressing agent microcrystalline cellulose.
Floating results showed that as the concentration of polymer increased, the floating lag time, duration of floating, and matrix integrity increased by using sodium bicarbonate as a gas-generating agent for helping the tablets for floatation.24 Sodium bicarbonate generates CO2 in the dissolution medium (0.1 N HCl); the gas liberated is trapped and protected within the gel formed by the hydration of the polymer, leading to decrease in the tablet density <1 g/mL, and the tablet becomes buoyant.21 Moreover, the increased amount of sodium bicarbonate caused a large amount of effervescence, which in turn resulted in pore formation that led to rapid hydration of the polymer matrix and thereby to a rapid release of the drug.
Release profile indicated that increasing the polymer concentration from 34% w/w to 42% w/w has drastically retarded the release of ITO HCl. A direct relationship was observed between concentration and cumulative percent drug release. The formulation containing 32% w/w Eudragit RSPM (F8) showed complete release of drug in 24 hours in a controlled manner as compared to HPMC 15000.
Combination of EC with Eudragit RSPM gave an excellent sustained release up to 24 hours. This is due to the increase in the gel strength of tablets; gel structure is formed around the tablet matrix, which considerably decreases the release rate of the drug since the drug has to diffuse through this gel barrier into the bulk phase.
These results are in accordance with those reported by Sanchez-Lafuente et al,25 who found that incorporation of Eudragit RSPM with hydrophobic ethylcellulose led to desirable modulation of drug release.
A kinetic study revealed that release of ITO from the prepared floating tablets showed diffusion-controlled release mechanism. This finding could be attributed to the formation of the gel structure by placing the tablet into the aqueous media.
The HPLC method used in the assay of ITO HCl is accurate, precise, specific, and a practical tool for the analysis of ITO HCl in plasma samples. The mean plasma concentration–time profiles reflect the sustaining of drug absorption from the tablet formula F10 compared with the immediate release tablets. The increase in AUC0–24 hour and FR% of the prepared sustained-release formulations could be attributed to an increase in the residence time of the drug in the gastrointestinal tract. First, the prepared tablets were designed to release the drug very slowly along for 24 hours; accordingly, these systems should remain in the gastrointestinal tract until all drug cargo is released completely. An increase in the floating time leads to an increase in the residence time of the drug in the gastrointestinal tract, accordingly the greatest chance for drug release and absorption; hence, bioavailability is directly increased. The enhancement of the relative bioavailability of ITO from the oral route was a direct result of the elimination of the hepatic first-pass metabolism on oral delivery of the ITO.
The decrease in the in vitro release behavior of the prepared formula (F10) could be attributed to the storage conditions26 and the variations in the swelling behavior of the matrix polymer.27 This is in agreement with the results of Hosny,28 who reported a minor decrease in the dissolution of tablets after storage at room temperature. The samples evaluated after 1 month showed no change in the in vitro drug release pattern, indicating good similarity of dissolution profiles before and after stability studies.
Conclusion
Floating tablets of ITO HCl were successfully prepared in this study for improving the bioavailability of the drug. The floating technique used should also be suitable for sustaining the release of the drug. The prepared tablets could float on the dissolution medium surface and sustain release of ITO HCl over 24 hours. The tablets prepared with ethylcellulose and Eudragit RSPM at 1:2 ratio showed optimum findings with respect to floating lag time, total floating duration, swelling ability, and sustained drug release profile.
In vivo evaluation of the prepared floating tablet proved that the bioavailability of the drug was increased 1.89-fold and plasma concentration of the drug after 24 hours was equal to that of the commercial tablet after 8 hours. Hence, the amount of the drug and number of administration per day were decreased. In line with recent recommendations to use the lowest amount of chemicals, the clinical study proved that more safety and fewer side effects could be predicted with this technique.
Disclosure
The authors report no conflicts of interest in this work.
References
Kim YS, Kim TH, Choi CS, et al. Effect of itopride, a new prokinetic, in patients with mild GERD: a pilot study. World J Gastroenterol. 2005;11(27):4210–4214. | ||
Iwanaga Y, Miyashita N, Saito T, Morikawa K, Itoh Z. Gastroprokinetic effect of a new benzamide derivative itopride and its action mechanisms in conscious dogs. Jpn J Pharmacol. 1996;71(2):129–137. | ||
Bose A, Wong TW, Singh N. Formulation development and optimization of sustained release matrix tablet of itopride HCl by response surface methodology and its evaluation of release kinetics. Saudi Pharm J. 2013;21(2):201–213. | ||
Holtmann G, Talley NJ, Liebregts T, Adam B, Parow C. A placebo-controlled trial of itopride in functional dyspepsia. N Engl J Med. 2006;354(8):832–840. | ||
Satapathy T, Panda PK, Goyal AK, Rath G. Evaluation of anti-GERD activity of gastro retentive drug delivery system of itopride hydrochloride. Artif Cells Blood Substit Immobil Biotechnol. 2010;38(4):200–207. | ||
Moës AJ. Gastroretentive dosage forms. Crit Rev Ther Drug Carrier Syst. 1992;10(2):143–195. | ||
Sheth PR, Tossounian J. The hydrodynamically balanced system (HBS™): a novel drug delivery system for oral use. Drug Dev Ind Pharm. 1984;10(2):313–339. | ||
Vo AQ, Feng X, Morott JT, et al. A novel floating controlled release drug delivery system prepared by hot-melt extrusion. Eur J Pharm Biopharm. 2016;98:108–121. | ||
Kesarla RS, Vora PA, Sridhar BK, Patel G, Omri A. Formulation and evaluation of floating tablet of H2-receptor antagonist. Drug Dev Ind Pharm. 2015;41(9):1499–1511. | ||
Baumgartner S, Kristl J, Vrecer F, Vodopivec P, Zorko B. Optimisation of floating matrix tablets and evaluation of their gastric residence time. Int J Pharm. 2000;195(1):125–135. | ||
Reddy LH, Murthy RS. Floating dosage systems in drug delivery. Crit Rev Ther Drug Carrier Syst. 2002;19(6):553–585. | ||
Li S, Lin S, Daggy BP, Mirchandani HL, Chien YW. Effect of HPMC and carbopol on the release and floating properties of gastric floating drug delivery system using factorial design. Int J Pharm. 2003;253(1):13–22. | ||
Sauzet C, Claeys-Bruno M, Nicolas M, Kister J, Piccerelle P, Prinderre P. An innovative floating gastro retentive dosage system: formulation and in vitro evaluation. Int J Pharm. 2009;378(1):23–29. | ||
Streubel A, Siepmann J, Bodmeier R. Floating matrix tablets based on low density foam powder: effects of formulation and processing parameters on drug release. Eur J Pharm Sci. 2003;18(1):37–45. | ||
Lachman L, Lieberman HA, Kanig JL. The Theory and Practice of Industrial Pharmacy. Philadelphia, NY: Lea & Febiger; 1986; Vol. 1. | ||
Tadros MI. Controlled-release effervescent floating matrix tablets of ciprofloxacin hydrochloride: development, optimization and in vitro-in vivo evaluation in healthy human volunteers. Eur J Pharm Biopharm. 2010;74(2):332–339. | ||
Srivastava AK, Wadhwa S, Ridhurkar D, Mishra B. Oral sustained delivery of atenolol from floating matrix tablets-formulation and in vitro evaluation. Drug Dev Ind Pharm. 2005;31(4–5):367–374. | ||
Strübing S, Metz H, Mäder K. Characterization of poly (vinyl acetate) based floating matrix tablets. J Control Release. 2008;126(2):149–155. | ||
Wagner JG. Linear pharmacokinetic equations allowing direct calculation of many needed pharmacokinetic parameters from the coefficients and exponents of polyexponential equations which have been fitted to the data. J Pharmacokinet Biopharm. 1976;4(5):443–467. | ||
USP-23, N. United State Pharmacopoeia – XXIII National Formulary- XXIII. Rockville, MD: United State Pharmacopoeia Convention, INC.; 1995:1625–1626. | ||
Machida Y, Inouye K, Tokumura T, Iwata M, Nagai T. Preparation and evaluation of intragastric buoyant preparations. Drug Des Deliv. 1989;4(2):155–161. | ||
Choi BY, Park HJ, Hwang SJ, Park JB. Preparation of alginate beads for floating drug delivery system: effects of CO(2) gas-forming agents. Int J Pharm. 2002;239(1):81–91. | ||
Gerogiannis VS, Rekkas DM, Dallas PP, Choulis NH. Floating and swelling characteristics of various excipients used in controlled release technology. Drug Dev Ind Pharm. 1993;19(9):1061–1081. | ||
Chung M, Chia W, Wan W, Lin Y, Sung H. Controlled release of an anti-inflammatory drug using an ultrasensitive ROS-responsive gas-generating carrier for localized inflammation inhibition. J Am Chem Soc. 2016;137(39):12462–12465. | ||
Sanchez-Lafuente C, Faucci MT, Fernández-Arévalo M, Álvarez-Fuentes J, Rabasco AM, Mura P. Development of sustained release matrix tablets of didanosine containing methacrylic and ethylcellulose polymers. Int J Pharm. 2002;234(1):213–221. | ||
Horhota ST, Burgio J, Lonski L, Rhodes CT. Effect of storage at specified temperature and humidity on properties of three directly compressible tablet formulations. J Pharm Sci. 1976;65(12):1746–1749. | ||
Goskonda VR, Reddy IK, Durrani MJ, Wilber W, Khan MA. Solid-state stability assessment of controlled release tablets containing carbopol 971P. J Control Release. 1998;54(1):87–93. | ||
Hosny EA. Study of accelerated storage conditions affecting physical characteristics, in-vitro dissolution and stability of bioadhesive containing tablets. Boll Chim Farm. 1999;138(6):243–248. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.