")
Back to Journals » Drug Design, Development and Therapy » Volume 12
CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia
Authors Vairy S , Lopes Garcia J, Teira P, Bittencourt H
Received 12 April 2018
Accepted for publication 5 September 2018
Published 12 November 2018 Volume 2018:12 Pages 3885—3898
DOI https://doi.org/10.2147/DDDT.S138765
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sukesh Voruganti
Stephanie Vairy,* Julia Lopes Garcia,* Pierre Teira, Henrique Bittencourt
Division of Haematology and Oncology, Department of Pediatrics, CHU Sainte-Justine, Université de Montréal, Montréal, QC, Canada
*These authors contributed equally to this work
Abstract: Over the past decades, survival of patients with acute lymphoblastic leukemia (ALL) has dramatically improved, but the subgroup of patients with relapsed/refractory ALL still continues to have dismal prognosis. As an emerging therapeutic approach, chimeric antigen receptor-modified T-cells (CAR-T) represent one of the few practice-changing therapies for this subgroup of patients. Originally conceived and built in Philadelphia (University of Pennsylvania), CTL019 or tisagenlecleucel, the first CAR-T approved by the US Food and Drug Administration, showed impressive results in refractory/relapsed ALL since the publication on two pediatric patients in 2013. It is in this context that we provide a review of this product in terms of manufacturing, pharmacology, toxicity, and efficacy studies. Evaluation and management of toxicities, particularly cytokine release syndrome and neurotoxicity, is recognized as an essential part of the patient treatment with broader use of IL-6 receptor inhibitor. An under-assessed aspect, the quality of life of patients entering CAR-T cells treatment, will also be reviewed. By their unique nature, CAR-T cells such as tisagenlecleucel operate in a different way than typical drugs, but also provide unique hope for B-cell malignancies.
Keywords: CTL019, tisagenlecleucel, B-cell acute lymphoblastic leukemia
Pediatric and adult acute lymphoblastic leukemia (ALL): the unmet needs
ALL represents the most common cancer among children with 25% of cancer diagnoses in people under age 15.1 Dramatic improvement in survival has been achieved over the past decades for this subgroup, leading to a 5-year survival rate of 90% for all subtypes combined among children and adolescents.2 Therefore, most recent pediatric trials now aim to reduce long-term toxicity and focus on refractory/relapsed (r/r) ALL that has a much worse prognosis. Current overall survival (OS) for this population is approximately 20% at 5 years.3,4 In adults, ALL is much less frequent and represents only 0.2% of all cancers.1 Prognosis is also less encouraging, with an expected 5-year OS between 20% and 40% despite complete remission (CR) rates of 85%–90%.5–7 This is partly explained by the reduced tolerance to chemotherapy and the different genetic profiles: a large proportion of patients with Philadelphia t(9;22) positive and Ph-like profile,8 a greater number of patients with MLL gene rearrangement t(4;11), monosomy 7, or trisomy 8.9 Among adult patients with Philadelphia-negative ALL, outcome after relapse remained extremely poor, with 5-year OS under 15%.5
These specific challenges in both the pediatric and adult population led to the emergence of innovative therapies, such as targeted therapy with monoclonal antibodies or bispecific T-cell engagers, personalized vaccines, and immunocellular therapy.
Immunocellular therapy aims to harness the power of a patient’s own immune system to fight malignancy. One of those therapeutic approaches involves the use of engineered and activated cytotoxic T cells. Chimeric antigen receptor-modified T-cells (CAR-T cells) with B-cell antigen specificity are a promising therapy for B-cell malignancies and demonstrated impressive clinical efficacy to date.
The idea of adoptive immunotherapy using lymphocytes to attack leukemia was developed in the early 1990s. After cloning the zeta-chain of T cell antigen receptor, the first chimeric antigen receptor was conceived by Eshhar et al.10,11 Many molecular and configurational modifications have been attempted with this product in order to optimize its antitumor efficacy.12
Many North American groups have developed CAR-T products and started clinical trials with anti-CD19 therapies for B-cell malignancies such as non-Hodgkin lymphoma (NHL), chronic lymphoid leukemia (CLL), and ALL. These groups include, among others, Memorial Sloan Kettering Cancer Center (MSKCC), University of Pennsylvania (UPenn) and the Children’s Hospital of Philadelphia (CHOP), Fred Hutchinson Cancer Research Center (FHCRC), and the National Cancer Institute (NCI). In 2010, Kochenderfer et al published the first case report of a patient with refractory and relapsed stage IVB follicular lymphoma showing an impressive response to anti-CD19 CAR-T cells.13 Later, in 2011, results in CLL were published in heavily treated patients showing an overall response rate (ORR) of 57%–100% with 29%–66% complete remission (CR) rate.14,15
In 2012, the University of Pennsylvania was the first to create a research alliance with a pharmaceutical company, Novartis, aiming to develop CAR-T cells for commercialization after its initial clinical success. The product from this alliance, CTL019, later known as tisagenlecleucel, was the first CAR-T treatment approved by the US Food and Drug Administration (FDA). The initial results of CTL019 in ALL were published in 2013 and will be reviewed in this paper.16 Since then, many trials are ongoing with various CAR-T products for different indications, and with promising results. In this article, we will focus on the manufacturing and pharmacology aspects of CTL019, as well as side effects management and efficacy studies for r/r ALL.
Pharmacology of CAR-T cells – CTL019
CD19 CAR-T design
CARs for hematological malignancies have been first designed to recognize CD19 antigen on the surface of B-cells, including normal lymphocytes and leukemic cells. The choice of CD19 for target in immunotherapy comes from its appealing characteristics: being uniformly expressed in B-cell leukemia/lymphomas and healthy B-cells but not on other normal tissues.17,18 Furthermore, targeting normal B-cell lymphocytes is an acceptable on target/off tumor toxicity, as B-cell aplasia can be managed in the clinic with intravenous or subcutaneous immunoglobulins, which will be detailed later.
As mentioned previously, the idea of adoptive cell therapy was developed more than 30 years ago. Before the first CAR-T cells, the concept of lymphokine-activated killer was developed,19 followed by tumor-infiltrating lymphocytes (TILs).20 One major advantage of the CAR-T cells over the TIL is their ability to be human leucocytes antigen (HLA)/T cell receptor (TCR) independent. By obviating the need for presentation of antigen in HLA, it makes this technology a more accessible and universal one.
Basic CAR structure includes an intracellular T-cell activation domain, an extracellular hinge region, a transmembrane domain, and an extracellular antigen-recognition moiety that is usually derived from an antibody (single-chain variable fragment [scFv]).21 The hinge, or spacer element, is conceived to optimize the accessibility of the epitope. Second-generation CAR-T cells encode for a co-stimulatory domain, such as CD2822 or members of the tumor-necrosis factor receptor family such as CD137 (4-1BB).23 This addition of co-stimulation domain provides better cytokine production and proliferation, and enhances persistence of CAR-T cells.22,24 Third-generation CAR-T includes a second co-stimulatory domain using the abovementioned co-stimulatory domains and/or others such as CD27, ICOS, or CD134 (OX40).25
Regarding CTL019, the structure is composed of an anti-CD19 scFv (FMC63) for antigen recognition, a CD8-α hinge region, 4-1BB co-stimulatory domains, and CD3ζ as a signaling domain, with an activation through intracellular immunoreceptor tyrosine-based activation motifs (ITAM; Figure 1).23,26 As described in Table 1, CTL019 from CHOP/UPenn and CAR-T cells built by FHCRC in Seattle have a very similar structure, with 4-1BB costimulatory domain, and the use of a lentivirus vector. The hinge is different, with CTL019 using CD8 and FHCRC an IgG4. Of note, the latter team also uses a manipulated composition ratio of CD4:CD8. Other teams reported in Table 1 use mostly CD28 as co-stimulatory domain and gamma-retrovirus as vector. Clinical results among different products will be discussed further.
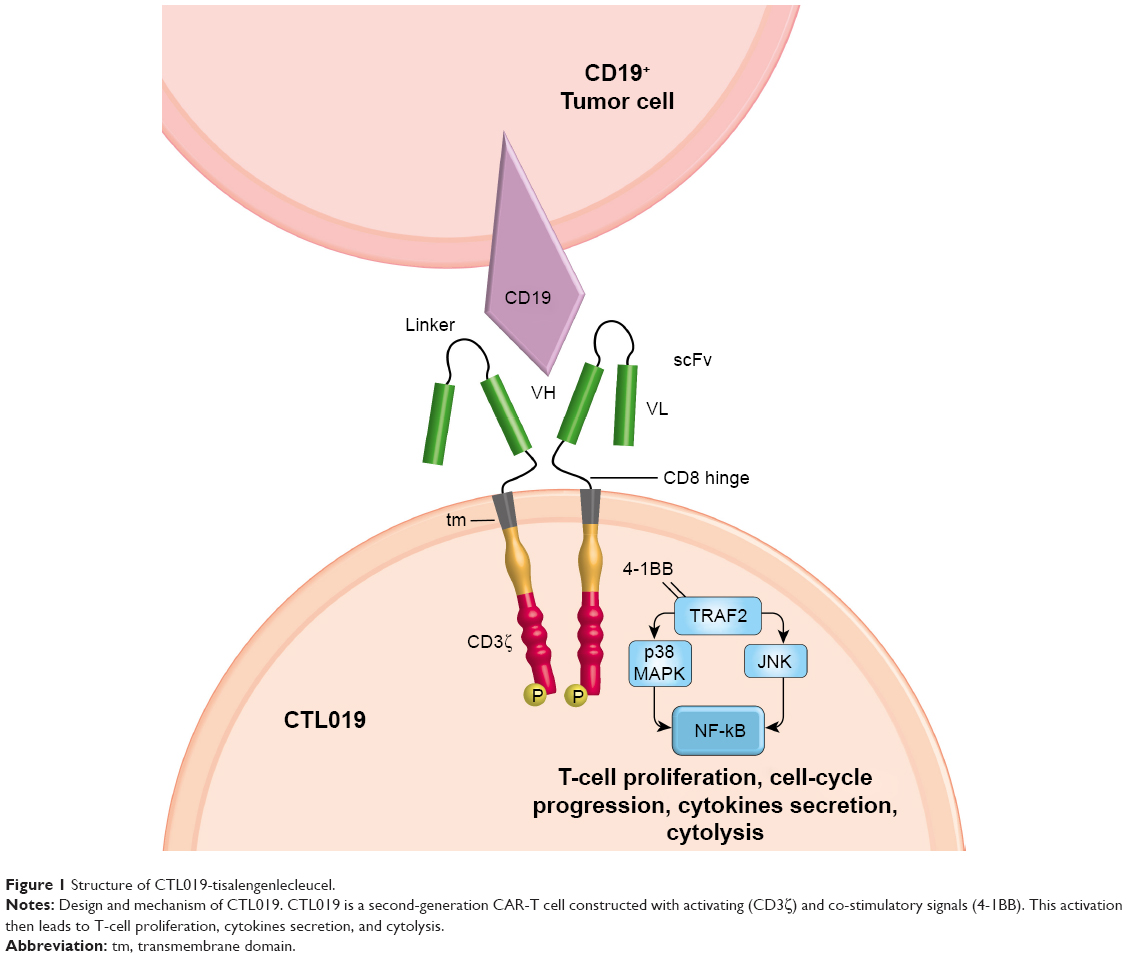 | Figure 1 Structure of CTL019-tisalengenlecleucel. |
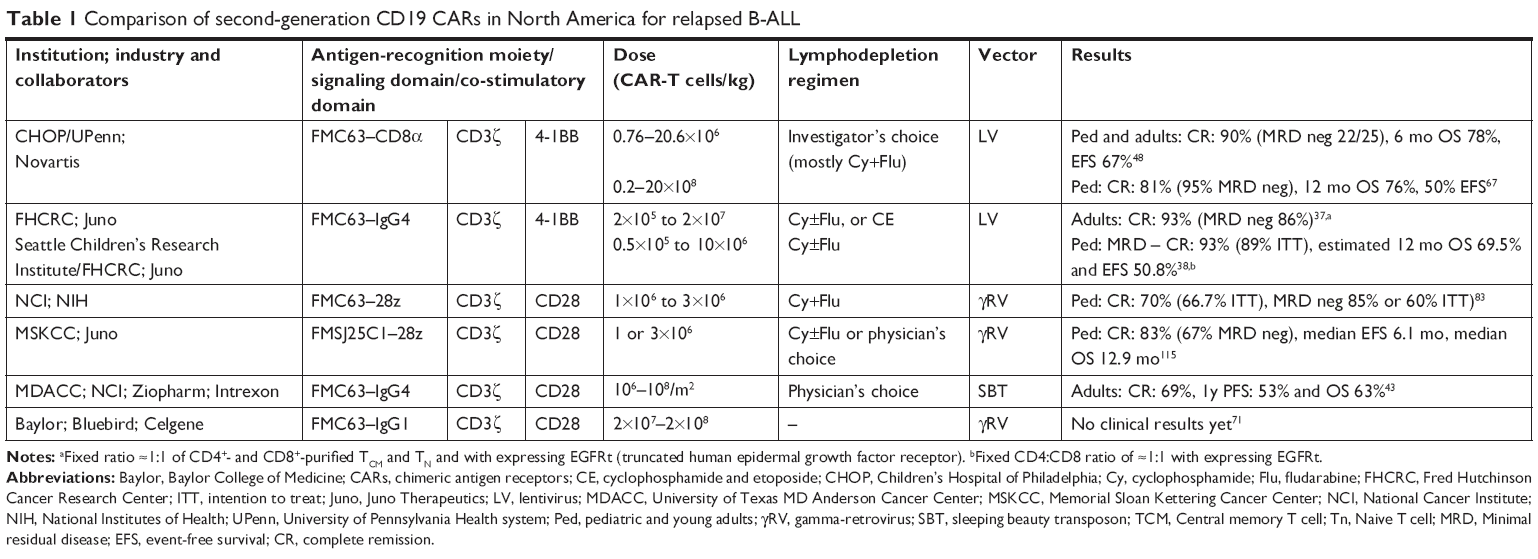 | Table 1 Comparison of second-generation CD19 CARs in North America for relapsed B-ALL |
Manufacturing CTL019
The process begins with the collection of a sufficient number of effector T cells from the patient’s peripheral blood through leukapheresis. The process is similar to peripheral blood stem cell (PBSC) collection, but aiming for mononuclear cells with the goal of obtaining a maximum of CD3+T cells. A minimum of absolute lymphocyte count (ALC) of 500 cells/μL or a CD3+ cell count of 150 cells/μL (if ALC is under 500 cells/μL) is required to start apheresis. The time to harvest the required number of cells is usually between 2 and 3 hours.27,28 Considerations enter into account regarding pediatric vs the adult leukapheresis technique. The smaller blood volume in children needs to have a slower rate of leukapheresis and priming the apheresis volume, in order to reduce the perfusion-related adverse effects such as hypovolemia, hypothermia, and hypocalcemia.29,30 For both age populations, cytotoxic and immunomodulator therapies must have been stopped for at least 2 weeks before collection (except for asparaginase and donor lymphocyte infusions that should be stopped at least 4 weeks, intrathecal methotrexate for 1 week, and steroids for 72 hours).30 Moreover, patients should be at least 3 months after an allogeneic stem cell transplant (allo-SCT) and 2 months from a T-cell lytic agent such as clofarabine.28 These requirements imply that the disease be stable during this time-lapse, and therefore can lead to exclusion of highly aggressive leukemia and/or active Graft-versus-host disease. This represents a fragile balance between good quality and quantity of T effector cells and the urge to treat a refractory patient.
The autologous material from leukapheresis is then shipped to a manufacturing facility. Novartis has currently two facilities to produce CTL019, one in Morris Plains, NJ, USA and the other in Leipzig, Germany. UPenn produces CTL019 for its clinical trials at the Clinical Cell and Vaccine Production Facility (CVPF) in Philadelphia, USA. Counterflow centrifugal elutriation allows the enrichment for the lymphocytes from the leukapheresis product and residual cells to be removed. These are then expanded with beads coated anti-CD3/anti-CD28 monoclonal antibodies (Life Technologies™) for positive selection and activation, creating the effect of autologous antigen-presenting cells (APCs). At the step of cells activation, the transduction is achieved when adding a lentiviral vector coding the CAR. The vector is washed away after 3 days of culture. Culture takes place in a rocking motion sterile bioreactor to reach a volume of 5 L.31 At the end of the culture, the beads are removed through magnetic separation; then cells are concentrated and washed. The latter are cryopreserved in an infusible medium to ensure viability during the transport to the treatment center. This entire phase takes places over 8–12 days.15,27 This information process is retrieved from FDA and CTL019 manufacturing details currently available.27,32
Regarding culture time, CAR-T cells are a lot shorter compared to 5–6 weeks of TIL.27,33 This culture process should be balanced for the memory T-cell differentiation, in order to select the more efficient lymphocytes. T-cells exhibit clear differences in effector and proliferation function and it is the rationale behind the selection of defined subset for an optimal CAR-T cells product performed by the FHCRC group. In fact, increasing levels of stimulation induce transition from naïve T cells (TN) into stem-cell memory T (TSCM), then into central-memory T (TCM), effector-memory cell (TEM), and finally into terminally differentiated effector-T cell (TEFF).34 The latter has been shown to have a decreased production of IL-2 and decreased expression of receptors for homeostatic cytokines and proliferation.35 Moreover, in both CD4+ and CD8+ CAR-T cells derived from TCM or TN, a more potent antitumor activity in vivo compared with TEM is observed.36 CD8+ TCM-derived CAR-T cells seem to be more potent in vivo, but as CD4+ CAR-T cells produced more cytokines, a synergistic effect is observed.36 However, selecting defined T-cell subset, although appealing, can be costly and difficult in severely lymphopenic patients, with complex manufacturing and lacks, until now, data arguing for clinical superiority. Heterogeneity in T lymphocytes populations can contribute to the differences in efficacy and toxicity profiles of the different CAR-T cells. In CTL019 manufacturing, there is no selection of subpopulation on the T-cell content. The FHCRC group, however, has added the step of selection of T-cells subset in order to have a defined composition for its CAR-T product. In their study in adults with r/r B-ALL, they aim to obtain a 1:1 ratio of CD4+ and CD8+T cells and enrich the CD8+ lymphocytes population to target CD8+ TCM. The latter is achieved through a two-step method with depletion of CD4+, CD14+, and CD45RA+ followed by selection of CD62L+.37 In the pediatric counterpart study, they also fixed the ratio at 1:1 for CD4+ and CD8+T cells, but without the enrichment part.38 On their side, City of Hope group has led two trials with CAR-T cells enriched in TCM from either CD8+ alone or CD4/8+ combined, in patients with relapsed NHL. Infusion of CAR-T cells occurred 2–3 days following autologous hematopoietic stem cell infusion.39 Furthermore, Baylor College of Medicine (BCM) group also conceived an allogeneic CD19 CAR-T cells virus specific for patients with B-cell malignancies relapsing after allo-hematopoietic stem cell transplant (allo-HSCT). Mononuclear cells from transplant donors are used to generate EBV-transformed lymphoblastoid B-cell to become APCs for subsequent viral stimulations. After this process, there is a transduction in CD19 CAR-T cells. This approach aimed to provide both antiviral and antitumor effects on the recipient.40
Lentiviruses deliver RNA that is reversed-transcribed into DNA in the target cell. This DNA coding the CAR then integrates into the host genome through a process catalyzed by the vector integrase enzyme and several key sequences.41 Other methods of nonviral gene transfer such as Sleeping Beauty transposon are being investigated.42,43 The integration of vector DNA near an oncogene of the host cells is a concern for all vectors. However, it seems that lentivirus carries a lower risk of mutagenesis compared with retrovirus because of its sites of integration away from the cellular promoter.44,45 Previous experience of the last decade reported from Scholler et al has shown no cases of oncogenicity even with retroviral vector in the context of CAR-T manufacturing.46
The production of CAR-T-cell therapies has largely increased over the past few years, to serve many patients distributed among treatment sites, internationally. It has a highly controlled process to ensure the quality of this product. The vector producer company is also subjected to these quality control tests and has been described by Levine et al in a recent review.27 The viral vector encoding the CAR can be frozen at −80°C and stay stable for 4 years, and up to 9 years in some reports.27,47 During the manufacturing time, bridging chemotherapy is often required to control the leukemia until the CTL019 manufacturing process is complete.
Once the CAR-T cells product is ready, it is shipped back to the treatment center. At the same time, lymphodepleting regimen is administered to the patient to optimize the therapy.48,49 The latter has been shown to facilitate the T-cell expansion in vivo and reduce the disease burden and, therefore, the risk of cytokine release syndrome (CRS). The combination of cyclophosphamide (CY) and fludarabine (Flu) vs CY alone for lymphodepletion regimen seems to provide better results in terms of CAR-T cells persistence and disease-free survival, although no randomized study has been designed for this purpose.37
Mechanism of action
The first-generation CAR had a powerful cytotoxicity, but a poor tumor control and limited persistence, due to premature exhaustion.50 Next, investigators tried to overcome this problem by mimicking physiologic T-cell activation.51 They therefore discovered the 2-signal rule in order to have complete activation, persistence, and anergy prevention. This was achieved by adding a co-stimulator such as 4-1BB, CD28, CD27, OX40, or ICOS to the TCR CD3ζ chain.23,24,52–54 In CTL019, this sustained activation is achieved through the TNF receptor superfamily member 9: 4-1BB, also known as CD137. The latter is a type II transmembrane protein that binds to the TNF-receptor-associated factor 2 (TRAF2), essential to activate NF-kB via p38 mitogen-activated protein kinase (MAPK) and Jun N-terminal kinase (JNK) (Figure 1).55–57 Downstream to this activation of NF-kB, phosphoinositide 3 kinase (PI3K) and protein kinase B (PKB), 4-IBB then upregulates anti-apoptotic factors such as Bcl-2, Bcl-xl, and Bfl-1.57,58 It also acts through interaction with endogenous TCR signaling.59 This leads to potent T-cell proliferation, cell-cycle progression via extracellular signal-regulated kinase (ERK) and PI3K, cytokine secretion such as Interleukin-2 (IL-2), IL-4, IL-5, and IFN-gamma, increased cytolytic potential, prevention of clonal deletion and activation-induced cell death, and decreased sensitivity to transforming-growth-factor beta (TGF-ß) inhibition.57,60–62 All these mechanisms are extrapolated from physiologic roles, and so the exact function into CAR-T cells remains to be elucidated.
Upon CARs multi-homodimerization with CD19 epitope, CD3ζ containing ITAMs serve as substrate for the Src-family kinase lymphocyte-specific protein tyrosine kinase (Lck).63 Intracellular signaling leads to phosphorylation of the endogenous CD3 complex, interaction with TCR, and activation of MAPK cascade and nuclear factor of activated T-cells (NFAT).55,64 In fact, a recent study in xenografted syngeneic murine models demonstrates that TCR signaling impairs the ability of CD8 CAR-T cells to expand, leading to their exhaustion and clearance. This effect was not observed in CD4 CAR-T cells, underlining a significant difference for future CAR models.65
Pharmacokinetics
“By its peculiar immunocellular mechanism”, standard pharmacokinetic components such as biodistribution, excretion, and metabolism are difficult to apply.
Once injected into the patient, there is an expansion of CAR-T cells, notably CTL019, from 1,000 to more than 10,000-fold. The peak is achieved between 10 and 14 days and coincides or follows the onset of clinical symptoms and CRS, between 1 and 21 days after the first infusion.15,66,67 In fact, for CTL019, this expansion often correlates with the severity of the CRS and the preinfusion tumor burden in children. A small decline is observed after peak expansion, probably due to redistribution in tissues, bone marrow, and peripheral blood before the maximal expansion.66
Efficient migration is a key factor for antitumor efficacy, as malignant cells are known to remodel their microenvironment. The CAR-T cells were proved to be trafficked in the bone marrow,15 and decreased to 60%–70% of peripheral blood levels 3–6 months later in pediatric B-ALL.66 CTL019 was also found in the cerebrospinal fluid, maybe explaining the absence of CNS relapses among children with ALL.16,66 Interestingly, no relationship between neurological events and CAR-T cells load into CNS was established.66
Different CAR-T cells persistence has been observed among studies. Of note, longer persistence was correlated with longer event-free survival (EFS) in the studies of CTL019 and other CARs.37,66,68 Recent studies have confirmed CTL019 levels measurable in up to 780 days among patients with ALL.66,67 Kochenderfer et al from NCI described long-term persistence in adult patients with NHL and CLL lasting up to 180 days after anti-CD19 CAR with FMC63-CD28Z.68 On their side, MSKCC demonstrated that their CAR-T-cell (19-28z) had persistence between 3 and 8 weeks in adults with r/r B-ALL, although their long-term study was limited by bridging to allo-HSCT after CAR-T cells for a majority of patients.69 Finally, Fred Hutchinson’s team has observed persistence of CAR-T cells in a defined CD4:CD8 ratio beyond 6 months, especially in patients receiving lymphodepleting regimen containing CY/Flu.37
Many explanations can be hypothesized from the results above. First, lymphodepletion clearly enhances CARs efficacy as demonstrated in murine and clinical studies, principally by eradication of regulatory T cells.49,70 Therefore, CY/Flu compared with CY alone was demonstrated superior by Turtle et al from FRCHC.37 In the CTL019 pivotal trial recently published by Maude et al, almost all patients received a lymphodepletion with the combination of CY/Flu.67
Then, the long-term persistence of CAR-T cells seems to correlate with TCM and TSCM concentration in the infused products.71,72 Some authors have demonstrated that CD8+ TCM have a superior long-term persistence in primate models.73 As stated above, recent work on murine model demonstrates that TCR activation in CARs leads to premature exhaustion and clearance of CD8 CARs.65
Regarding co-stimulatory domain, 41BB vs CD28 is clearly a component to explore as many confounding factors between clinical studies complicate their interpretation. In vivo, CD28 drives more rapid expansion with early tumor control, whereas 41BB drives more memory T-cell functions with a slower kinetic.74 Moreover, the question of third-generation CAR-T cells in order to combine this efficacy and persistence is currently under study.
The infused dose is another aspect that should be clarified as wide range of doses has been employed in studies (Table 1). Either a dose correlated to the burden of the disease or a fixed dose remains to be explored, taking into account toxicity. Recent data on CTL019 demonstrate no correlation between dose and expansion.67
To measure the persistence of CAR-T, flow cytometry and qPCR are available. There is a strong correlation between the circulating levels of CD3+ CAR-T cells measured by flow cytometry and qPCR. However, flow cytometry measures CAR-T cells by their expressed surface protein and qPCR measures the integration of the transgene into T-cell. Therefore, qPCR may not necessarily reflect functional CAR-T cells.66 Of note, qPCR was used for CTL019 studies. Duration of B-cell aplasia is indeed a surrogate to the CAR-T cells persistence and functionality.
Efficacy studies of CTL019 in r/r ALL
Since the initial clinical report by Grupp et al16 in 2013, CTL019 continue to show promising results in patients with r/r ALL. The latter reported the use of CAR-T cells in two children with r/r ALL, including one after hematopoietic stem cell transplantation and blinatumomab administration. They received 1.4×106 and 1.2×107 CTL019 cells/kg with etoposide and CY as lymphodepletion for one of the patients. The other patient had no lymphodepletion before as a highly cytotoxic therapy was administered 6 weeks before. Both experienced CR at 1 month post-infusion. One patient had sustained remission 11 months later at the time of publication and the other relapsed 2 months later with a CD19 negative leukemia escape.16
In 2014, Maude et al published the clinical results of their pilot study of CTL019 at CHOP for r/r ALL.48 The pediatric cohort (22 years old and younger) included 25 patients, and 5 adult patients from the population studied. Among them, two were blinatumomab-resistant and 15 had a previous HSCT. Lymphodepleting regimen was variable, including 50% Flu/CY. Some patients received no lymphodepletion. One month after infusion, 90% (27/30) of them were in complete morphologic remission and 22/27 were MRD negative by flow cytometry. At 6 months, EFS was 67% and OS rate was 78%, with a 68% probability of CTL019 persistence. There was no death related to the product.
The recent CTL019 study published (ELIANA trial) has confirmed the previous results. This was a phase II multicenter trial in pediatric and young adults that enrolled 92 patients. Of note, among those 92 patients enrolled, 17 were not infused: seven because of manufacturer issues, seven died before infusion, and three had severe adverse events. They received between 0.2 and 5.4×106 CTL019 cells/kg (median: 3.1×106 CTL019 cells/kg) and 96% had lymphodepleting chemotherapy before infusion, mostly CY/Flu. At 12 months, EFS rate was 50% and OS was 76%. Moreover, as stated before, CTL019 was noted to persist for 20+ months, with a median duration of 168 days. All responding patients had B-cell aplasia.67
Given those impressive results, CAR-T cells studies are currently running internationally. At the time of publication, three clinical trials are ongoing for CTL019. A phase II open-label multicenter trial for pediatric and young adults with r/r B-ALL is recruiting patients. This trial is available in centers in Canada and Europe and is sponsored by Novartis (NCT03123939). There is also a phase 1 with a humanized CTL019 (huCTL019) for pediatric and young adults with ALL or Diffuse large B-cell lymphoma (DLBCL) open in CHOP at Pennsylvania (NCT02374333). Finally, a phase II adult trial of CTL019 is actually recruiting ALL patients with MRD positive disease for use of CAR-T cells in upfront therapy (NCT02935543).
Safety and tolerability
Besides their remarkable results, CAR-T cells can cause toxicity by several mechanisms such as damage in normal tissues if the tumor-associated antigen is expressed in those tissues;75 acute anaphylaxis76 and tumor lysis syndrome (TLS).48 Two common and potential severe toxicities have been described with CAR-T therapies: CRS, an entity caused by cytokines released from activated CAR-T cells, and neurologic toxicity that can occur, associated or not with CRS.77 Both toxicities will be detailed in this section. Other most common adverse effects affecting more than 20% of patients receiving tisagenlecleucel are: hypogammaglobulinemia, infections–pathogen unspecified, fever, viral infectious disorders, loss of appetite, nausea, diarrhea, vomiting, headache, encephalopathy and delirium, hypotension, bleeding episodes, tachycardia, hypoxia, fatigue, and acute kidney injury. Of note, grade 3 and 4 events were experienced by 88% of patients.67
In the CTL019 pilot study, no death was linked to the treatment and seven patients died after 30 days, all from relapse or progression of B-ALL.48 In ELIANA trial, 19 deaths occurred, mostly from ALL relapse but one from CRS complication and two from infection, within 30 days of infusion.67 Because of those serious adverse reactions, including some fatal complications in patients receiving tisagenlecleucel, the FDA approved this product with a Risk Evaluation and Mitigation plan.78
For its effective application, clinicians must learn how to manage these associated toxicities. The underlying goal is a fair balance between preventing life-threatening toxicities and keeping the best chance for a beneficial antitumor effect. The CARTOX (CAR-T-cell-therapy-associated TOXicities) Working Group was created with the goal of providing general guidelines to manage those patients. The group has recently published a framework for a proactive management strategy of different and frequent complications after CAR-T treatment for adult patients.79 The final proposition is a three-step approach to assessment and management of acute toxicities associated with CAR-T-cell therapy.
Step 1: Systematic monitoring of patient’s clinical and biological parameters to determine the nature of the CAR-T cell-related toxicity, in order to diagnose CRS, CAR-T cell-related encephalopathy syndrome (CRES), and hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS).
Step 2: Grading the severity of CRS, CRES, and HLH/MAS using the criteria adapted from Lee et al and the Common Terminology Criteria for Adverse Events, version 4.0.77,80
Step 3: Treating the toxicities according to the suggested management algorithms for CRS, CRES, and HLH/MAS.
The use of the CTCAE80 to grade CRS is debatable as it was developed before the emergence of cellular therapy and can underestimate this phenomenon.81 The University of Pennsylvania (Penn) grading system for CRS, a more clinical approach that has some differences compared with the Lee grading system, has been developed and used in the different CTL019 trials, including the ELIANA trial.77,81
Cytokine release syndrome (CRS)
The CRS is a systematic inflammation response that is produced by elevated levels of cytokines, associated with T-cell in vivo activation and proliferation.82 CRS is not unique to anti-CD19 therapies because it is related to T-cell engagement with a tumor antigen and symptoms may be delayed, depending on the kinetics of T-cell activation, the main difference between cytokine storm (eg, after transplant) and CRS.81 The clinical features include high fever, malaise, fatigue, myalgia, nausea, anorexia, tachycardia/hypotension, capillary leak, cardiac dysfunction, renal impairment, hepatic failure, and disseminated intravascular coagulation.77 Most patients experience CRS 1–14 days after CAR-T cell infusion.48,81 The median time was 4 days in all recent trials, with more severe presentation if symptoms appear after 24 hours of CAR-T infusion.38,48,83 It is quite unusual for CRS to start after 2 weeks of CAR-T infusion.14,48 Accurate predictors for severe CRS have not yet a defined place in clinical practice, because of technical difficulties to monitor serum cytokines and its cost. Currently, under investigation is the use of well-known C-reactive protein, which is made by hepatocytes in response to IL-6, as a laboratory marker of CRS onset and severity.84 As stated above, a high leukemia burden seems to be associated with a higher risk of CRS.48 One of the key mediators of CRS after CAR-T-cell therapy is IL-6, but a dramatic increase in other cytokines, such as interferon-gamma, granulocyte macrophage-colony stimulation factor (GM-CSF), IL-10, and IL-5, is also present.82,84–86
In the ELIANA trial, CRS occurred in 77% of cases.67 Notably, 35/75 patients were admitted to the intensive care unit for CRS management with 19 of them needing vasopressor support, 10 needing mechanical ventilation, 7 dialysis, and 28 received tocilizumab.
Tocilizumab (Atlizumab [Chugai Pharmaceutical Co., Tokyo, Japan], Actemra [Hoffmann-La Roche, Basel, Switzerland]) is a humanized monoclonal antibody against IL-6 receptor (IL-6R).87 It blocks IL-6-induced signal transduction pathways through competitive inhibition of IL-6 binding to its receptors and has been currently used to treat patients with rheumatoid arthritis.88 Tocilizumab has become a mainstay in the control of complications from T-cell-engaging therapies for several groups after being reported by CHOP that CRS can be successfully ameliorated with this IL-6R inhibitor.16,48 Concomitant to the approval of tisagenlecleucel, the FDA has approved it as a treatment for CRS after CAR-T-cell therapy. The recommended dose of tocilizumab for CRS is 12 mg/kg for children under 30 kg and 8 mg/kg for patients more than 30 kg (pediatric and adults).78
Siltuximab (CNTO 328, Sylvant; Janssen Pharmaceutica, Beerse, Belgium) is a human murine chimeric monoclonal antibody against IL-6.89 It is FDA-approved since 2014 for the treatment of multicentric Castleman’s Disease.90 It is used off-label for management of CRS and, as tocilizumab, can induce rapid reversal of symptoms in most patients.16,48,91 Even if tocilizumab is the only approved drug to treat CRS, its advantage, compared with siltuximab, remains to be proved. A theoretical disadvantage of tocilizumab is the fact that its administration is associated with an increase of the IL-6 level in the blood (as it binds the IL-6 receptor) and possible passive diffusion of IL-6 into the central nervous system, which could increase the risk of neurotoxicity with tocilizumab.79,87
Corticosteroids can also control inflammatory responses and are effective in the management of CRS, CRES, and HLH/MAS associated with cellular therapies. However, as corticosteroids suppress T-cell function and can induce T-cell apoptosis, their use can impair the function and the durability of the cells infused.79,92,93 So, corticosteroids should be reserved for CRS that is refractory to anti IL-6 therapy.
No unified grading system has been validated prospectively for CRS, but it is a first step to managing complications. The guidelines will evolve with our better knowledge of cellular therapies. However, since CAR-T-cell therapy is going beyond the research context, it is imperative that a grading system that can be applied globally be developed in the near future. In Table 2, we summarize the two grading systems available and guidelines for management of CRS published so far.
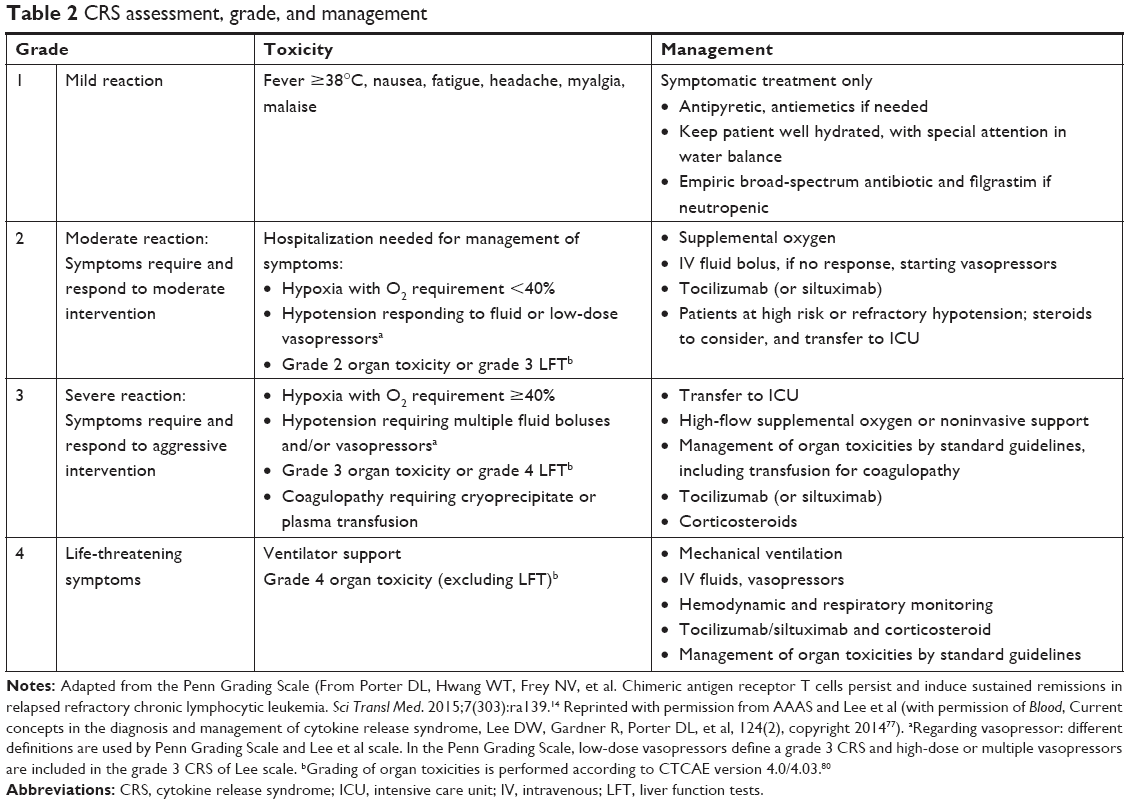 | Table 2 CRS assessment, grade, and management |
CAR-T-cell-related encephalopathy syndrome (CRES)
The pathophysiology of neurological toxicity is still unclear and the neurological symptoms do not follow the same time course as systemic CRS. Two explanations are considered: first, passive diffusion of cytokines into the brain,83,93 and second, trafficking of T-cells to the brain.16,48,83,94 Both hypotheses are supported by findings in the serum and in the CSF, respectively. The clinical presentation is a toxic encephalopathy, presenting as decreased attention, dysphasia, and impaired handwriting. Patients can experience other symptoms including confusion, disorientation, agitation, aphasia, somnolence, and tremors.37,95,96 In severe cases (grade 2 or more), seizures, increased intracranial pressure, papilledema, and cerebral edema also occur.79
In the ELIANA trial, neurologic effects were present in 40% of patients within 8 weeks after infusion and 13% were grade 3.67 In the pilot study, 13/25 patients had cerebral toxicity with self-limiting symptoms and complete resolution without apparent sequelae.48 No cerebral edema or grade 4 toxicity was reported in those two studies.
CRES has been reported as a biphasic manifestation: the first phase occurs with high fever and other CRS symptoms in the first 5 days after infusion and a second phase that occurs after CRS subsides, usually after 5 days of infusion. In about 10% of patients, however, a delayed neurotoxicity can occur 3–4 weeks after CAR-T-cell therapy.79
For its treatment, anti IL-6 can reverse the CRES concomitant with CRS, but it is not effective when CRS is not present. In this situation, corticosteroids remain the treatment of choice. CRES usually lasts for 2–4 days, but it can vary from a few hours to weeks. CRES occurring post-CRS is usually more severe.79,81 It is generally reversible; however, some rare fatal cases have been reported.37,82,97
HLH/MAS
HLH is a rare condition characterized by inappropriate immune activation and cytokine release that typically presents with fever and splenomegaly in association with hyperferritinemia, coagulopathy, hypertriglyceridemia, and cytopenias.98 These traditional criteria are not specific and are frequently present even in patients with low-grade CRS, which can complicate the diagnosis of HLH/MAS in the context of CAR-T-cell therapy. In CTL019 trials, this condition was not described thus far. However, severe CRS can evolve into HLH and clinicians must be aware about this complication in order to avoid fatal outcomes.
CARTOX has proposed a definition for HLH/MAS after CAR-T-cell therapy: a peak serum ferritin level of >10,000 ng/mL with any two of the following: grade ≥3 increase in serum bilirubin, aspartate aminotransferase, or alanine aminotransferase levels; grade ≥3 oliguria or increase in serum creatinine levels; grade ≥3 pulmonary edema; presence of hemophagocytosis in bone marrow or organs based on histopathological assessment of cell morphology and/or CD68 immunohistochemistry.79
These patients should be treated with anti-IL6 therapy as for CRS (and corticosteroids if clinically indicated), which has been able to reverse this clinical situation. If the patient does not improve in 48 hours, additional therapy with etoposide should be considered and intrathecal cytarabine as well for patients with neurotoxicity-associated HLH. Even if there is no evidence to support the latter practice in patients with CAR-T-cell-associated HLH, this recommendation is supported by the preference for those drugs in refractory HLH.79
It is critical that the community continue to investigate the pathophysiology and patterns of these unique toxicities and find ways to modify its administration or the CAR-T constructs to increase safety.99 In addition to medications, preparation of clinical staff to recognize toxicity and effectively manage these patients is key to improving overall safety.
B-cell aplasia
Therapies targeting B-cells such as anti-CD20 antibodies like rituximab, kinase inhibitors such as ibrutinib, and CAR-T cells can lead to absolute and functional hypogammaglobulinemia and/or the inability to respond to vaccines.100
CAR-T cells can probably be associated with “on target/off tumor” because of their direct attack on normal tissues that have the shared expression of the targeted antigen. This situation is well illustrated by the extended B-cell aplasia and hypogammaglobulinemia presented after anti-CD19 CAR-T-cell therapies.75,93,95 In the ELIANA trial, the probability of B cell aplasia at 6 months was 83%, and was present in all patients responding to CAR-T cells.79 Fortunately, for CTL019 and other anti-CD19 CAR-T therapies, this situation can be easily managed with intravenous or subcutaneous immunoglobulin replacement therapy without compromising the patient’s quality of life (QoL). Otherwise, if the target is present in the vital organs, the on target/off tumor effect can result in severe or fatal toxicity, perhaps one of the greatest obstacles for CAR-T-cell therapy in other diseases such as solid tumors.101,102
The immunoglobulin replacement therapy remains controversial and needs to be individualized, considering the disease, therapy, and serum antibody titers. The recommendation resulting from the last review by Ueda et al is a starting dose of 500 mg/kg/monthly intravenously or 200 mg/kg/weekly subcutaneously with a target serum IgG level over 400–500 mg/dL.100 Serum IgG level and/or specific antibody titers and infections should be monitored and used to individualize therapy.
Patient-focused perspective
Since more than 85% of children will survive 5 years or longer after a diagnosis of ALL in developed countries, the diminished QoL of the survivors obviously becomes a concern.2,103 On the other hand, it means that approximately 15% of children diagnosed with childhood ALL will experience a relapse.104 Indeed, for adults with ALL, it is only 20%–40% who will achieve long-term remission.6,7
For most relapsed patients, the treatment of choice remains an allo-HSCT,105 with a success rate of about 40%.106,107 However, morbidity after HSCT remains elevated for long-term survivors and includes endocrinology complications such as impairment growth, hormone deficiency, thyroid and sex hormone deficiencies, infertility, and second malignancies.103,108–110 In this context, CAR-T-cell therapy has shown remission rates and OS that seem to considerably exceed other therapies. However, some crucial points should also be taken into account with this innovative therapy, such as its unique toxicity profile, and its development and costs. Indeed, the cost of $475,000 announced by Novartis for a single infusion, compared with the estimate of allo-HSCT of about $200,000, has been viewed with a lot of criticism.111,112 Furthermore, this number does not include collateral costs during therapy, such as apheresis, cell infusion, and management of complications, including intensive care unit stays and use of tocilizumab.78
Eligible patients for this new therapy need to face more obstacles: geographic barriers, with less than 40 centers in the US authorized to administer the therapy and the time required to manufacture these cells, which can prevent patients with a more aggressive disease from receiving the therapy. In the ELIANA trial, 18% of patients were unable to receive the infusion, leading to the death of six patients, while seven patients failed to pass the manufacturing stage.67 All these points can overestimate the potential of cure of anti-CD19 CAR-T therapies. Of note, Novartis foresees that the actual time of manufacture for tisagenlecleucel will be shorter compared with the manufacturing time required during the last clinical trials.
Quality of life (QoL)
A French prospective study published in 2017 by Michel et al showed the results of a late-effect program (the LEA program) comparing transplanted vs non-transplanted children in a long-term follow-up.113 Despite major physical complications in the HSCT group, clinical consequences on the QoL were relatively mild. Those findings demonstrate acceptance and adaptation of a handicap, allowing him/her to have a better QoL. Also, an important point highlighted in this study was that some events adversely affecting QoL of transplant patients have been detected in adults but not in children, suggesting impairment in the adulthood of these younger survivors.
If we look at the specific population of patients who have received an infusion of CTL019, we see such an important difference in instantaneous QoL, starting from day 28, and continued improvement after 3 and 6 months as shown in ELIANA’s QoL study presented at ASH Meeting in 2017.114 Studies described above used different QoL scales, limiting the comparison between them. Moreover, it is essential to analyze this QoL in two different ways: short-term follow-up comparing with relapsed and refractory leukemia and long-term follow-up comparing with a normal population and children after HSCT, for example.
The timing of the CAR-T-cell treatment should also be considered to assess QoL. If CAR-T is given after two or three lines of treatment, patients might present less cumulative toxicity vs if CAR-T is given after being refractory to several lines of treatment or after a first transplant. It is likely that there would be significant differences regarding morbidities burden between these two groups of patients.
Although it is early to show the superiority of CAR-T-cell therapy to deliver a better QoL, the first results are promising once the adverse effects period ends without permanent morbidities. As stated above, those results need to be compared in a prospective long-term follow-up scenario with other patients who received chemotherapy with or without allo-HSCT instead of CAR-T cells, and with normal population control.
Conclusion
CTL019 and other CAR-T-cell therapies represent an impressive new approach of anti-cancer therapy and have a strong potential to transform lives of patients with fatal diseases. Optimal structure, T-cells selection and enrichment, doses, and lymphodepleting regimen remain relevant and hot topics in the field of cellular therapy. In spite of those questions, tisagenlecleucel has proven its efficacy leading to its approval by the FDA. However, its peculiar and severe toxicities must be improved in future trials.
Its recent approval has opened singular discussion about the need for a new policy and improved infrastructure to achieve the best clinical benefit, and, moreover, raised the concern about better access for patients worldwide. The price of a single infusion is undoubtedly an obstacle to this commercialization.112 Actually, tisagenlecleucel has the promise of meaningful benefits but at a considerable social cost and with toxicities.
Disclosure
Henrique Bittencourt and Pierre Teira are consulting for Novartis Oncology. The other authors report no conflicts of interest in this work.
References
Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review 1975–2014. Bethesda, MD: National Cancer Institute; 2017; based on November 2016 SEER data submission. Available from: https://seer.cancer.gov/csr/1975_2014/. Accessed May 2018. | ||
Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol. 2012;30(14):1663–1669. | ||
Tasian SK, Hunger SP. Genomic characterization of paediatric acute lymphoblastic leukaemia: an opportunity for precision medicine therapeutics. Br J Haematol. 2017;176(6):867–882. | ||
Nguyen K, Devidas M, Cheng SC, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia. 2008;22(12):2142–2150. | ||
Wolach O, Amitai I, Deangelo DJ. Current challenges and opportunities in treating adult patients with Philadelphia-negative acute lymphoblastic leukaemia. Br J Haematol. 2017;179(5):705–723. | ||
Kantarjian H, Thomas D, O’Brien S, et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer. 2004;101(12):2788–2801. | ||
Sive JI, Buck G, Fielding A, et al. Outcomes in older adults with acute lymphoblastic leukaemia (ALL): results from the international MRC UKALL XII/ECOG2993 trial. Br J Haematol. 2012;157(4):463–471. | ||
Roberts KG, Gu Z, Payne-Turner D, et al. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J Clin Oncol. 2017;35(4):394–401. | ||
Wetzler M, Dodge RK, Mrózek K, et al. Prospective karyotype analysis in adult acute lymphoblastic leukemia: the cancer and leukemia Group B experience. Blood. 1999;93(11):3983–3993. | ||
Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–724. | ||
Weissman AM, Baniyash M, Hou D, Samelson LE, Burgess WH, Klausner RD. Molecular cloning of the zeta chain of the T cell antigen receptor. Science. 1988;239(4843):1018–1021. | ||
van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499–509. | ||
Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–4102. | ||
Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):ra139. | ||
Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):ra73. | ||
Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. | ||
Tedder TF, Zhou LJ, Engel P. The CD19/CD21 signal transduction complex of B lymphocytes. Immunol Today. 1994;15(9):437–442. | ||
Matsuo Y, Drexler HG. Establishment and characterization of human B cell precursor-leukemia cell lines. Leuk Res. 1998;22(7):567–579. | ||
Rosenberg SA, Lotze MT, Muul LM, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313(23):1485–1492. | ||
Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. | ||
Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013;10(5):267–276. | ||
Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCR zeta/CD28 receptor. Nat Biotechnol. 2002;20(1):70–75. | ||
Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–684. | ||
Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. | ||
Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125(26):4017–4023. | ||
Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. | ||
Levine BL, Miskin J, Wonnacott K, Keir C. Global Manufacturing of CAR T Cell Therapy. Mol Ther Methods Clin Dev. 2017;4:92–101. | ||
Senzon R. Determining Patient Readiness for Leukapheresis. In: Collection L, editor. Manual Leukapheresis Reference Manual. V4 ed. Novartis; 2017:20. | ||
Karakukcu M, Unal E. Stem cell mobilization and collection from pediatric patients and healthy children. Transfus Apher Sci. 2015;53(1):17–22. | ||
McGuirk J, Waller EK, Qayed M, et al. Building blocks for institutional preparation of CTL019 delivery. Cytotherapy. 2017;19(9):1015–1024. | ||
Somerville RP, Devillier L, Parkhurst MR, Rosenberg SA, Dudley ME. Clinical scale rapid expansion of lymphocytes for adoptive cell transfer therapy in the WAVE® bioreactor. J Transl Med. 2012;10(1):69. | ||
FDA. BLA 125646 Tisagenlecleucel Novartis Pharmaceuticals Corporation. 2017. Available from: https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM566166.pdf. Accessed May 2018. | ||
Mullinax JE, Hall M, Prabhakaran S, et al. Combination of Ipilimumab and Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes for Patients with Metastatic Melanoma. Front Oncol. 2018;8:44. | ||
Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12(10):671–684. | ||
Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. | ||
Sommermeyer D, Hudecek M, Kosasih PL, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30(2):492–500. | ||
Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–2138. | ||
Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–3331. | ||
Wang X, Popplewell LL, Wagner JR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127(24):2980–2990. | ||
Cruz CR, Micklethwaite KP, Savoldo B, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122(17):2965–2973. | ||
Vile RG, Russell SJ. Retroviruses as vectors. Br Med Bull. 1995;51(1):12–30. | ||
Aronovich EL, Mcivor RS, Hackett PB. The Sleeping Beauty transposon system: a non-viral vector for gene therapy. Hum Mol Genet. 2011;20(R1):R14–R20. | ||
Kebriaei P, Singh H, Huls MH, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest. 2016;126(9):3363–3376. | ||
Varmus HE, Quintrell N, Ortiz S. Retroviruses as mutagens: insertion and excision of a nontransforming provirus alter expression of a resident transforming provirus. Cell. 1981;25(1):23–36. | ||
Vannucci L, Lai M, Chiuppesi F, Ceccherini-Nelli L, Pistello M. Viral vectors: a look back and ahead on gene transfer technology. New Microbiol. 2013;36(1):1–22. | ||
Scholler J, Brady TL, Binder-Scholl G. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4132(132):ra153. | ||
Lamers CH, van Elzakker P, Luider BA, et al. Retroviral vectors for clinical immunogene therapy are stable for up to 9 years. Cancer Gene Ther. 2008;15(4):268–274. | ||
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. | ||
Brentjens RJ, Rivière I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. | ||
Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med. 1995;181(5):1653–1659. | ||
Brentjens RJ, Latouche JB, Santos E, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9(3):279–286. | ||
Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012;119(3):696–706. | ||
Pulè MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12(5):933–941. | ||
Guedan S, Chen X, Madar A, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014;124(7):1070–1080. | ||
Norelli M, Casucci M, Bonini C, Bondanza A. Clinical pharmacology of CAR-T cells: Linking cellular pharmacodynamics to pharmacokinetics and antitumor effects. Biochim Biophys Acta. 2016;1865(1):90–100. | ||
Cannons JL, Choi Y, Watts TH. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J Immunol. 2000;165(11):6193–6204. | ||
Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9(4):271–285. | ||
Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J Immunol. 2002;169(9):4882–4888. | ||
Nam KO, Kang H, Shin SM, et al. Cross-linking of 4-1BB activates TCR-signaling pathways in CD8+ T lymphocytes. J Immunol. 2005;174(4):1898–1905. | ||
Daniel-Meshulam I, Horovitz-Fried M, Cohen CJ. Enhanced antitumor activity mediated by human 4-1BB-engineered T cells. Int J Cancer. 2013;133(12):2903–2913. | ||
Wen T, Bukczynski J, Watts TH. 4-1BB ligand-mediated costimulation of human T cells induces CD4 and CD8 T cell expansion, cytokine production, and the development of cytolytic effector function. J Immunol. 2002;168(10):4897–4906. | ||
Lee HW, Nam KO, Park SJ, Kwon BS. 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur J Immunol. 2003;33(8):2133–2141. | ||
Kersh EN, Shaw AS, Allen PM. Fidelity of T cell activation through multistep T cell receptor zeta phosphorylation. Science. 1998;281(5376):572–575. | ||
Baer A, Colon-Moran W, Xiang J, Stapleton JT, Bhattarai N. Src-family kinases negatively regulate NFAT signaling in resting human T cells. PLoS One. 2017;12(10):e0187123. | ||
Yang Y, Kohler ME, Chien CD, et al. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci Transl Med. 2017;9(417):eaag1209. | ||
Mueller KT, Maude SL, Porter DL, et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130(21):2317–2325. | ||
Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med Overseas Ed. 2018;378(5):439–448. | ||
Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. | ||
Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. | ||
Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116(19):3875–3886. | ||
Xu Y, Zhang M, Ramos CA, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR. CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123(24):3750–3759. | ||
Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118(23):6050–6056. | ||
Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118(1):294–305. | ||
Zhao Z, Condomines M, van der Stegen SJC, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28(4):415–428. | ||
Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. | ||
Maus MV, Haas AR, Beatty GL, et al. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol Res. 2013;1(1):26–31. | ||
Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. | ||
FDA. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125276s114lbl.pdf. Accessed May 2018. | ||
Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy – assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. | ||
US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE); version 4.0; 2010. Available from: https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/Archive/CTCAE_4.0_2009-05-29_QuickReference_8.5x11.pdf. Accessed May, 2018. | ||
Porter D, Frey N, Wood PA, Weng Y, Grupp SA. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol. 2018;11(1):35. | ||
Teachey DT, Lacey SF, Shaw PA, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;6(6):664–679. | ||
Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. | ||
Davila ML, Riviere I, Wang X. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Transl Med. 2014;6224(224):ra225. | ||
Hay KA, Hanafi LA, Li D, Kevin A, L-Ah H, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130(21):2295–2306. | ||
Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20(2):119–122. | ||
Mea M. Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int Immunopharmacol. 2015;5:1731–1740. | ||
Singh JA, Beg S, Lopez-Olivo MA. Tocilizumab for rheumatoid arthritis: a Cochrane systematic review. J Rheumatol. 2011;38(1):10–20. | ||
Zaki MH, Nemeth JA, Trikha M. CNTO 328, a monoclonal antibody to IL-6, inhibits human tumor-induced cachexia in nude mice. Int J Cancer. 2004;111(4):592–595. | ||
Sitenga J, Aird G, Ahmed A, Silberstein PT. Impact of siltuximab on patient-related outcomes in multicentric Castleman’s disease. Patient Relat Outcome Meas. 2018;9:35–41. | ||
Chen F, Teachey DT, Pequignot E, et al. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR T cell therapy. J Immunol Methods. 2016;434:1–8. | ||
Sea G. Analysis of a Global registration trial of the efficacy and safety of CTL019 in pediatric and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL) – Abstract. Blood. 2016;128:221. | ||
Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol Ther. 2017;25(1):285–295. | ||
Hu Y, Sun J, Wu Z, et al. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J Hematol Oncol. 2016;9(1):70. | ||
Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–549. | ||
Brudno JN, Somerville RP, Shi V, Jnea B, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol. 2016;34(10):1112–1121. | ||
Bhoj VG, Arhontoulis D, Wertheim G, et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. 2016;128(3):360–370. | ||
Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr Opin Pediatr. 2012;24(1):9–15. | ||
Perales MA, Kebriaei P, Kean LS, Sadelain M. Reprint of: Building a Safer and Faster CAR: Seatbelts, Airbags, and CRISPR. Biol Blood Marrow Transplant. 2018;24(3S):S15–S19. | ||
Ueda M, Berger M, Gale RP, Lazarus HM. Immunoglobulin therapy in hematologic neoplasms and after hematopoietic cell transplantation. Blood Rev. 2018;32(2):106–115. | ||
Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011. | ||
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. | ||
Speechley KN, Barrera M, Shaw AK, Morrison HI, Maunsell E. Health-related quality of life among child and adolescent survivors of childhood cancer. J Clin Oncol. 2006;24(16):2536–2543. | ||
Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14(6):e205–e217. | ||
Oliansky DM, Camitta B, Gaynon P, et al. Role of cytotoxic therapy with hematopoietic stem cell transplantation in the treatment of pediatric acute lymphoblastic leukemia: update of the 2005 evidence-based review. Biol Blood Marrow Transplant. 2012;18(4):505–522. | ||
Gökbuget N, Stanze D, Beck J, et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120(10):2032–2041. | ||
Moussalem M, Esperou Bourdeau H, Devergie A, et al. Allogeneic bone marrow transplantation for childhood acute lymphoblastic leukemia in second remission: factors predictive of survival, relapse and graft-versus-host disease. Bone Marrow Transplant. 1995;15(6):943–947. | ||
Socié G, Clift RA, Blaise D, et al. Busulfan plus cyclophosphamide compared with total-body irradiation plus cyclophosphamide before marrow transplantation for myeloid leukemia: long-term follow-up of 4 randomized studies. Blood. 2001;98(13):3569–3574. | ||
Bernard F, Auquier P, Herrmann I, et al. Health status of childhood leukemia survivors who received hematopoietic cell transplantation after BU or TBI: an LEA study. Bone Marrow Transplant. 2014;49(5):709–716. | ||
Khera N, Storer B, Flowers ME, et al. Nonmalignant late effects and compromised functional status in survivors of hematopoietic cell transplantation. J Clin Oncol. 2012;30(1):71–77. | ||
Majhail NS, Mau LW, Denzen EM, Arneson TJ. Costs of autologous and allogeneic hematopoietic cell transplantation in the United States: a study using a large national private claims database. Bone Marrow Transplant. 2013;48(2):294–300. | ||
Prasad V. Immunotherapy: Tisagenlecleucel – the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol. 2018;15(1):11–12. | ||
Michel G, Bordigoni P, Simeoni MC, et al. Health status and quality of life in long-term survivors of childhood leukaemia: the impact of haematopoietic stem cell transplantation. Bone Marrow Transplant. 2007;40(9):897–904. | ||
Dietz A, Grupp SA, Laetsch TW, Theodore W, Stefansky H. Patient-Reported Quality of Life Following CTL019 in Pediatric and Young Adult Patients with Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia. Journal of Clinical Oncology. 2017;15_suppl:10523–10523. | ||
Park JH, Rivière I, Gonen M, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378(5):449–459. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.