")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Could the FDA-approved anti-HIV PR inhibitors be promising anticancer agents? An answer from enhanced docking approach and molecular dynamics analyses
Received 30 April 2015
Accepted for publication 2 June 2015
Published 13 November 2015 Volume 2015:9 Pages 6055—6065
DOI https://doi.org/10.2147/DDDT.S87653
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Olayide A Arodola, Mahmoud ES Soliman
Molecular Modelling and Drug Design Lab, School of Health Sciences, Westville Campus, University of KwaZulu-Natal, Durban, South Africa
Abstract: Based on experimental data, the anticancer activity of nelfinavir (NFV), a US Food and Drug Administration (FDA)-approved HIV-1 protease inhibitor (PI), was reported. Nevertheless, the mechanism of action of NFV is yet to be verified. It was hypothesized that the anticancer activity of NFV is due to its inhibitory effect on heat shock protein 90 (Hsp90), a promising target for anticancer therapy. Such findings prompted us to investigate the potential anticancer activity of all other FDA-approved HIV-1 PIs against human Hsp90. To accomplish this, “loop docking” – an enhanced in-house developed molecular docking approach – followed by molecular dynamic simulations and postdynamic analyses were performed to elaborate on the binding mechanism and relative binding affinities of nine FDA-approved HIV-1 PIs against human Hsp90. Due to the lack of the X-ray crystal structure of human Hsp90, homology modeling was performed to create its 3D structure for subsequent simulations. Results showed that NFV has better binding affinity (ΔG =−9.2 kcal/mol) when compared with other PIs: this is in a reasonable accordance with the experimental data (IC50 3.1 µM). Indinavir, saquinavir, and ritonavir have close binding affinity to NFV (ΔG =−9.0, −8.6, and −8.5 kcal/mol, respectively). Per-residue interaction energy decomposition analysis showed that hydrophobic interaction (most importantly with Val534 and Met602) played the most predominant role in drug binding. To further validate the docking outcome, 5 ns molecular dynamic simulations were performed in order to assess the stability of the docked complexes. To our knowledge, this is the first account of detailed computational investigations aimed to investigate the potential anticancer activity and the binding mechanism of the FDA-approved HIV PIs binding to human Hsp90. Information gained from this study should also provide a route map toward the design, optimization, and further experimental investigation of potential derivatives of PIs to treat HER2+ breast cancer.
Keywords: binding free energy, loop docking, molecular dynamics, HIV-1 protease inhibitors, anticancer
Introduction
Cancer, a heterogeneous disease, is one of the major causes of death worldwide.1 In 2008, GLOBOCAN estimated the occurrence of ~12.7 million cancer cases and 7.6 million cancer deaths (~21,000 cancer deaths a day); of these, 56% (2.8 million) of the cases and 64% (4.8 million) of the deaths occurred in the economically developing countries.1 According to the American Cancer Society, it has been reported that there remains an expected increase in the cancer incidences in the next 20 years, more especially in African regions.2,3
Cancer is an abnormal proliferation of cells that can lead to malignancy and death. These cells have the potential to elude other normal cells through the process called metastasis.3 Causal factors like tobacco, chemicals, radiation, infectious organisms, inherited mutations, and hormones may act together to initiate or accelerate carcinogenesis. Among the numerous types of cancer, breast cancer is the most common cancer diagnosed in women worldwide.4 Approximately two-thirds of breast cancer tumors are hormone-dependent, requiring estrogens to grow, and estrogens are formed in the human body via a multistep route starting from cholesterol.5
Estrogen receptor and human epidermal growth factor 2 (HER2) are familial hormones, which are sensitive receptors of the human epidermal growth factor. HER2 consists of four subtypes (HER1–4),6 which are regulated at the level of expression by hormones; thus, HER2 is of greatest significance as it is a pivotal receptor target for breast cancer treatment. Compared with other types of breast cancer, HER2+ breast cancer tends to be more aggressive and less receptive to hormone treatments. According to published data, it was documented that ~25%–30% of human breast cancers overexpress HER2,7,8 in which heat shock protein has been reported to play a role.9,10 Therefore, to block HER2 overexpression, the subsequent step would be to inhibit the enzyme responsible for producing this estrogen, Hsp90. Hsp90 functions as a molecular chaperone, regulating the proper functionality of other proteins in the body.11
Hsp90 is a homodimer consisting of α and β chains. Each monomer of Hsp90 dimer contains four domains: 1) a highly conserved N-terminal domain (NTD), 2) C-terminal domain (CTD), 3) a middle domain, and 4) a charged linker that connects the NTD and middle domain12 as shown in Figure 1.
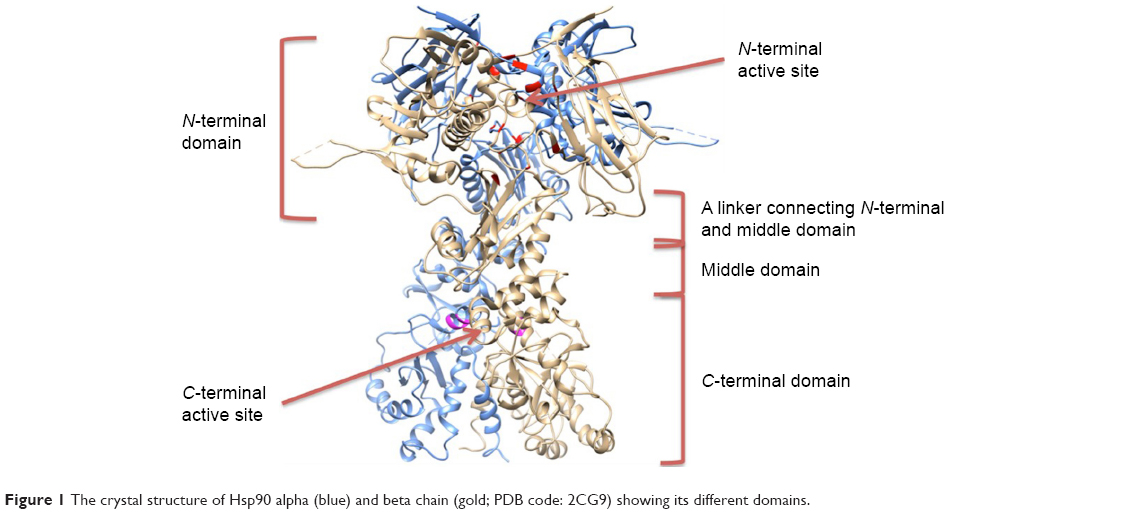 | Figure 1 The crystal structure of Hsp90 alpha (blue) and beta chain (gold; PDB code: 2CG9) showing its different domains. |
Hsp90 contains a highly conserved ATP-binding site near its NTD, as Hsp90 functions are energetically expensive.14 This Hsp90-ATP-binding site has been under intense pharmaceutical investigation, as many designed drugs offer competitive inhibition against ATP for this site.12 The design, development, and discovery of new cancer chemotherapeutic cannot meet the demand and rate at which the disease is manifesting. The devastating effects are evident from the high mortality rate of inflicted patients, due to progression of the disease or inherited infection contracted from having a compromised immunity. Recent studies project that a more efficient procedure for drug development that reduces costs is to find new indications for already approved drugs; this process is referred to as “repositioning”.15–17 “Repositioning” takes advantage of available data on existing drugs, limits risks and costs to pharmaceutical companies, and could advance the evaluation and movement of new cancer therapies to the clinic.
Shim et al18 analyzed the proteolytic profile of yeast Hsp90 full length for both NTD and CTD. This study identified five drugs that exhibited significant inhibitory potential on cell proliferation with the HER2+ breast cancer lines. These include mercaptopurine, nelfinavir mesylate, geftinib, triciribine, and 6-α-methylprednisolone. It was reported that four of the drugs, excluding nelfinavir (NFV) (IC50 =3.1 μM), offered negligible to relatively good inhibitory activity and were thus discarded as viable therapeutics. Shim et al reported the action of NFV in several breast cancer cell lines on a collection of yeast strain and identified Hsp90 as its target. To establish this fact, the effect of NFV on Hsp90 and other cochaperones in rabbit reticulocyte lysates, using a coimmunoprecipitation assay, was determined in order to avoid any chance of changes in protein expression levels by drugs. The possibility that NFV had an anticancer effect on yeast Hsp90 and rabbit reticulocyte lysates raised the possibility that Hsp90 is the target of NFV in mammalian cells. Shim et al further projected that since NFV has no effect on trypsin digestion of NTD and middle domain of the Hsp90, it is likely that NFV binds to the Hsp90 CTD and induces conformational changes in the protein; which is a different mechanism from other Hsp90 inhibitors. The study, however, suggests that although NFV seems to interact with Hsp90 via its CTD such as Novobiocin and induce a conformational change, however, its binding site differs from that of Novobiocin.18 Another study further affirm this assumption that the CTD, which is responsible for maintaining Hsp90 functional homodimeric state and coordinating interactions with several Hsp90 cochaperones, show some promising activities at its binding pocket.12 Khalid et al also hypothesized that Hsp90 CTD binding by different drugs can cause conformational changes in the NTD, which alters its ATPase activity and client protein binding.19
A recent study presented by Srirangam et al reported that another protease inhibitor (PI), ritonavir, partially inhibit the functions of Hsp90 (KD =7.8 μM).20 Based on the findings from the studies performed by Shim et al and Bernstein et al, it can be suggested that the US Food And Drug Administration (FDA)-approved HIV-1 PIs are promising dual inhibitors of cancer and HIV-1 protease.18,21
The study performed by Shim et al, however, has some limitations. Foremost, the precise mode of interaction between NFV and Hsp90 remains a mystery. Second, the objectivity and interpretation of experimental outcome on mammalian cells were not stated. Third, since NFV is the only HIV-1 PI that was investigated in this study, the relative affinities of other HIV PIs are missing. This has prompted us to conduct this extensive computational investigation in order to explore the relative binding affinity, as well as the potential use, of other FDA-approved HIV PIs as anticancer agents.
In this work, a homology model for the human Hsp90 was built, enhanced docking approach was adopted (refer “Methods” section for details), and MD simulations for nine FDA-approved HIV PIs (Figure 2) at the Hsp90 CTD were performed. The 5 ns MD simulations were also performed on the docked complexes to validate the docking results.
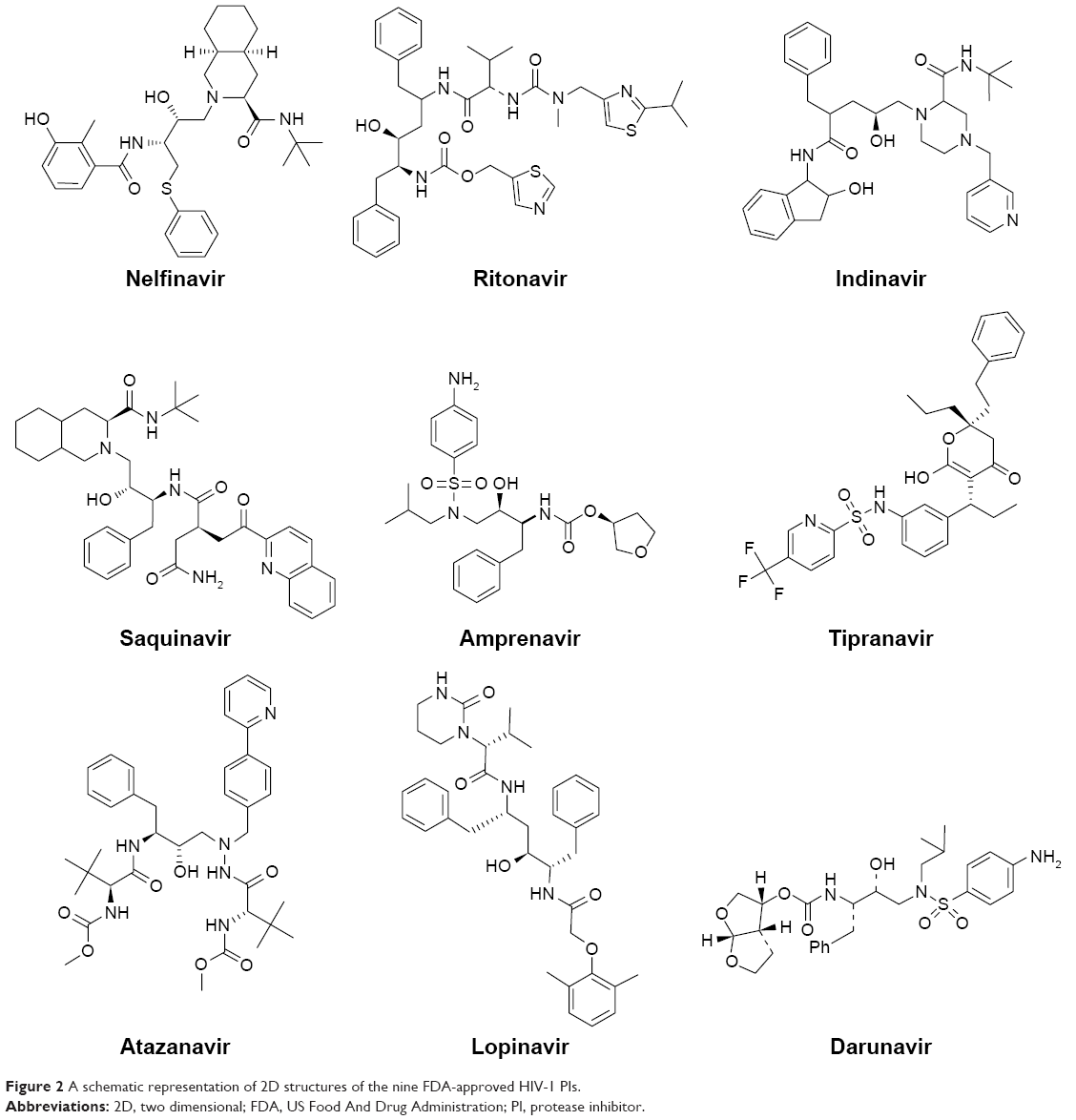 | Figure 2 A schematic representation of 2D structures of the nine FDA-approved HIV-1 PIs. |
We believe that the compilation of the computational and molecular modeling tools presented in this study could serve as powerful tools to answer the question about the repositioning of HIV-1 PIs as anticancer agents. Although this is coming from a computational perspective, this study could open a new gateway to further experimental evaluations of these drugs as anticancer agents.
Computational methodology
Homology modeling of human Hsp90 protein structure
Due to the absence of a full human crystal structure of Hsp90 that comprises of both the NTD and CTD, the crystal structure of the human Hsp90 was modeled using the protein sequence obtained from Uniprot (Uniprot ID: P08238). The full human Hsp90 homolog was modeled by using three crystal structures of Hsp90 as templates: Hsp90 from Saccharomyces cerevisiae (PDB Code: 2CG9), which contained the ATP bound in its active site; Hsp90 MD from Homo sapiens (PDB Code: 3PRY); and Hsp90 CTD from Leishmania major (PDB Code: 3HJC). Homology modeling was performed using the Modeller Software version 9.122 add-on in chimera.23 Multiple sequence alignment was performed on CLUSTALW (Figure S1).24 The active-site residues were determined using Chimera Multi-align Viewer and validated using the SiteHound-web program.25 The homology model of the human Hsp90 was energy minimized and equilibrated via molecular dynamics (MDs) simulations (refer “Molecular Dynamics Simulations” section) and then used for subsequent simulations. The sequence of the target protein was uploaded unto PSIPRED V3.326,27 in order to obtain a predicted 3D secondary structure of the enzyme. Comparing the homolog to the predicted 3D structure and assessment of the bond angles and torsional strain show the validation of the homology model. Ramachandran plot (Figure S2) for the analyses of bond angles and torsional strain was generated using Maestro.28 MolProbity29 results show that 98% of all residues are in the favored regions and >99.8% of all residues are in the allowed regions, which leaves a list of 20 outliers. The list shows that none of the active-site residues are part of these outliers.
Defining the active-site residues in the Hsp90 homology model
Due to the lack of information on the active-site residues for the human Hsp90 CTD, the active-site residues were obtained from the Site-Hound web software.25 Closest active residues to the binding pocket were selected and used for further modeling studies. The identified active site residues for the human Hsp90 homolog CTD were Gln523, Val534, Ser535, Lys538, Thr595, Tyr596, Gly597, Trp598, and Met602. The positions of these active-site residues were mapped in the corresponding human Hsp90β homolog to identify the CTD active-site pockets for further docking and MD simulations.
Building HIV-1 PIs–Hsp90 complexes
Loop docking
“Loop docking” protocol is an automated docking process, which was developed “in house”,30,31 in order to improve reliability and validity of docking results. The main idea of “loop docking” is based on performing consecutive runs of docking calculations starting from pre-docked poses with lower binding energy to ensure that the final docked pose has the lowest binding energy (highest binding affinity) over all runs. The total number of run (n) is defined by user. We found that 20 runs are enough to produce the lowest energy docked structure in most cases. “Loop docking” protocol is depicted in Figure 3. For example, if the initial docking produces a docked conformation with ΔGx =−9.0, the second run will automatically start from this conformation produced from the first run. If the second run produces a conformation with better ΔG value (lower binding energy, for example, ΔGx+1 =−10.3), the third run will select this conformation and leave out the one produced from the previous run and so forth until the total number of run (n) is exhausted.
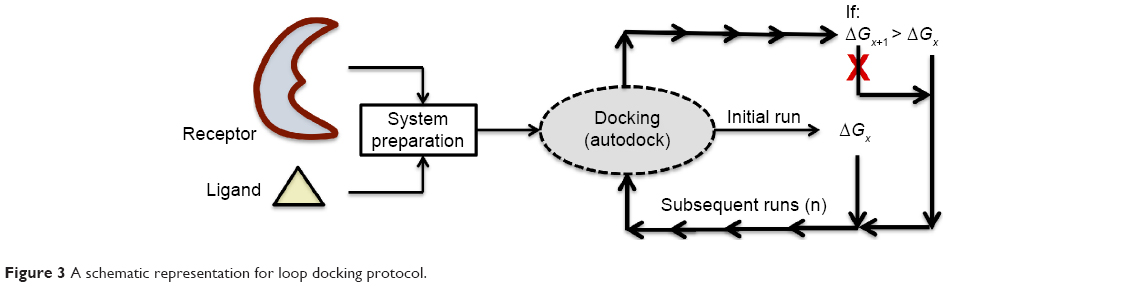 | Figure 3 A schematic representation for loop docking protocol. |
System preparation and docking parameters
The nine HIV-1 PIs were retrieved from ZINC database32 and energy minimized using Avogadro software.33 To ensure thorough conformational sampling of the ligands, the low-energy conformations were generated for each molecule using Omega v1.8b.34 For each compound, the lowest energy conformers were docked into the CTD active sites of Hsp90 homolog using Autodock Vina software.35 The grid box was defined with the grid parameters being X =16, Y =22, and Z =28 for the dimensions and X =−81, Y =−54, and Z =56 for the center grid box at the CTD. All docked human Hsp90–HIV-1 PI complexes were subjected to subsequent MD simulations.
Molecular dynamics simulations
MD simulations were performed using the graphics processor unit (GPU) version of the PMEMD engine provided with the AMBER 12 package.36,37 General amber force field (GAFF) parameters were calculated by antechamber module of AMBER 12 package. Hydrogen atoms of the proteins were added using the LEaP module in AMBER 12.37 The complexes were obtained using Chimera and were solvated in an octahedron box of TIP3PBOX water with buffering distance of 8 Å between the protein surface and the box boundary;38 assuming normal charge states of ionizable groups corresponding to pH 7, sodium (Na+) counter-ions were added to achieve charge neutrality and to mimic biological environment more closely. Cubic periodic boundary conditions were imposed and the long-range electrostatic interactions were treated with the particle-mesh Ewald method implemented in AMBER 12 with a nonbonding cut-off distance of 10 Å. The partial atomic charges for the ligand were obtained using “antechamber”39 module of AMBER 12. Initial energy minimization, with a restraint potential of 500 kcal/mol Å2 applied to the solute, was carried out with the aid of the SANDER module of the AMBER 12 program using the steepest descent method in AMBER 12 for 1,000 steps followed by conjugate gradient protocol for 2,000 steps. Due to the lack of parameters needed for the ligand in the Cornell et al force field,40 the missing parameters were created. Optimization of the ligands were first performed at the HF/6-31G* level with the Gaussian 03 package.41 The standard AMBER force field for bioorganic systems (ff03)42 was used to define the topology and parameter files for the enzyme and PIs using “gaff”43 based on the atom types of the force field model developed by Cornell et al.36 The entire system was freely minimized for 1,000 iterations. Heating was performed for 50 ps from 0 K to 300 K with harmonic restraints of 5 kcal/mol Å2 using a Langevin thermostat with a coupling coefficient of 1/ps. The entire system was then equilibrated at 300 K with a 2 fs time step in the NPT ensemble for 500 ps, and Berendsen temperature coupling44 was used to maintain a constant pressure at 1 bar. The SHAKE algorithm45 was employed on all atoms so as to constrain the bonds of all hydrogen atoms. With no restraints imposed, a production run was performed for 5 ns in an isothermal isobaric (NPT) ensemble using a Berendsen barostat with a target pressure of 1 bar and a pressure-coupling constant of 2 ps for analysis of the energy stabilization and root mean-square deviation (RMSD) values of the complex. The coordinate file was saved every 1 ps, and the trajectory was analyzed every 1 ps using the PTRAJ module implemented in AMBER 12.
Results and discussion
Human Hsp90 homology model
Due to the lack of a human Hsp90 that comprised of the middle domain, CTD, and NTD, a homology model of the human Hsp90β was generated. Using the Hsp90 from S. cerevisiae (PDB Code: 2CG9), crystal structure of Hsp90 MD from H. sapiens (PDB Code: 3PRY), and a crystal structure of Hsp90 CTD from L. major (PDB Code: 3HJC) as structural templates, a human Hsp90 homolog was constructed using software “Modeller” 9.1.22 Structural similarity between the three proteins showed good identity in and around the active site, NTD, middle domain, and CTD, with the majority of active-site residues having nearly identical positions (Figure 4A). The sequences shared a 64.11% (2CG9), 97.39% (3PRY), and 60.14% (3HJC) similarity according to the Multi-align Viewer tool in Chimera, and the model had a zDOPE score of 0.25 after modeling (Figure 4B).
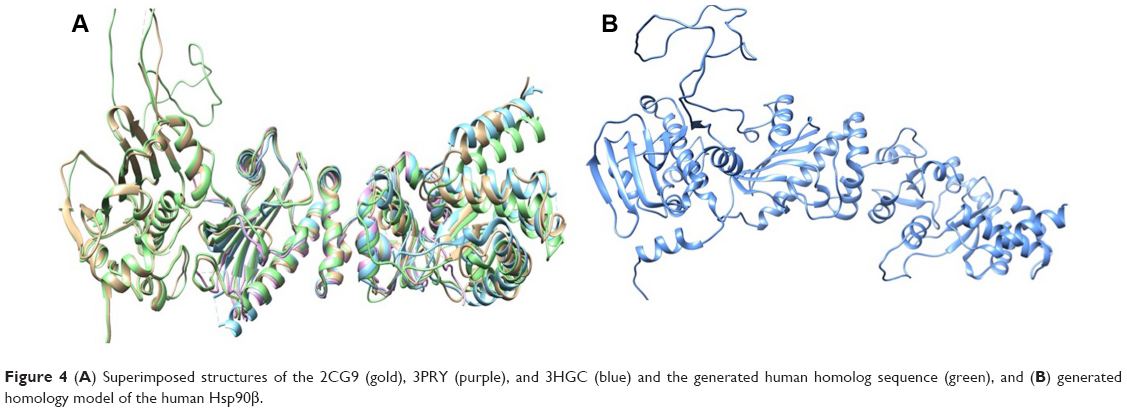 | Figure 4 (A) Superimposed structures of the 2CG9 (gold), 3PRY (purple), and 3HGC (blue) and the generated human homolog sequence (green), and (B) generated homology model of the human Hsp90β. |
Validation of the CTD-binding site
Due to the fact that there is no available crystal structure for the human Hsp90, a homology model was built up using several crystal structures as templates. The active site in the Hsp90 model was primarily identified based on alignment of the active site of the template structures (with known active-site residues; refer “Homology modeling of human Hsp90 protein structure” section). This gives us a rough guide on the approximate positions of the corresponding active-site residues in the homology model. However, this is further validated using SiteHound-web program.25 In fact, we further tested the SiteHound-web program25 with several proteins with known active sites (such as HIV-1 protease and Hsp90 NTD; data presented in Figure S3) to see if the results could be trusted. Surprisingly, we found that SiteHound-web software, to a great extent, precisely predicts these protein active sites. In addition to the earlier validation, we opted to run a relatively long MD simulations (5 ns) in order to allow the ligands to evolve naturally in the enzyme pocket over time; this will ensure that the ligands adjust their position/orientation in the enzyme pocket even if the SiteHound-web and/or docking software were not very accurate in their predictions.
Docking results and validation via MD simulations
From our previous experiences with molecular docking, we found that in most cases, docking results could be artifacts and unreliable, and even the best docked conformation may fly away from the protein’s active site pocket within a few picoseconds on MD simulations. Therefore, to enhance the reliability of docking calculations, we embarked on an enhanced “loop docking” approach (refer “Loop docking” section) followed by relatively long MD simulations to ensure that the docked complexes are quite stable in the protein active site over a reasonable time scale. To this end, all ligand Hsp90 complexes from “loop docking” were further subjected to 5 ns MDs simulation.46–48 Systems stability and overall convergence (ie, RMSD and potential energy) were monitored over the MD trajectories to ensure that the simulated systems are well equilibrated (RMSD and potential energy plots for all simulated systems are provided as Figures S4 and S5, respectively).
Binding affinities and per-residue interaction footprints
Table 1 shows the relative binding affinities of the nine drugs toward Hsp90. NFV showed the highest binding affinity followed by SQV and RTV. As shown in Table 1, all PIs, except Atazanavir (ATV), have better binding affinity compared with Novobiocin, a known human Hsp90 CTD inhibitor. The reason for this could be due to the fact pointed out in Shim et al study that Novobiocin and NFV binds at the CTD, but their binding sites are different. The binding mode of NFV to the Hsp90 CTD is shown in Figure 5. The ligand interaction of Novobiocin to the human Hsp90 homolog is depicted in Figure 6. The 3D coordinates of all docked complexes of the studied systems (nine in total) are provided as Supplementary material.
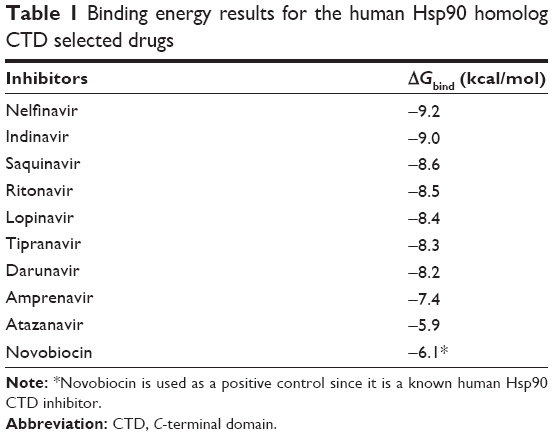 | Table 1 Binding energy results for the human Hsp90 homolog CTD selected drugs |
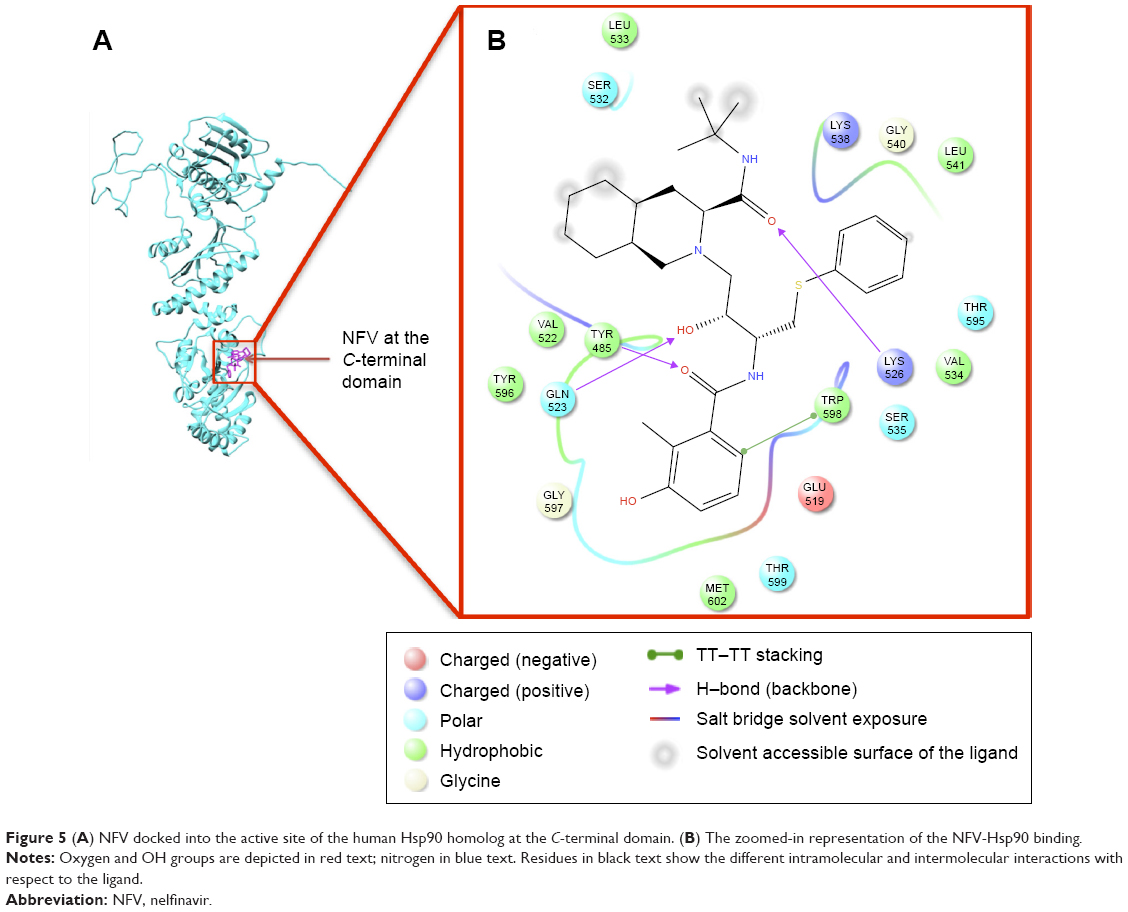 | Figure 5 (A) NFV docked into the active site of the human Hsp90 homolog at the C-terminal domain. (B) The zoomed-in representation of the NFV-Hsp90 binding. |
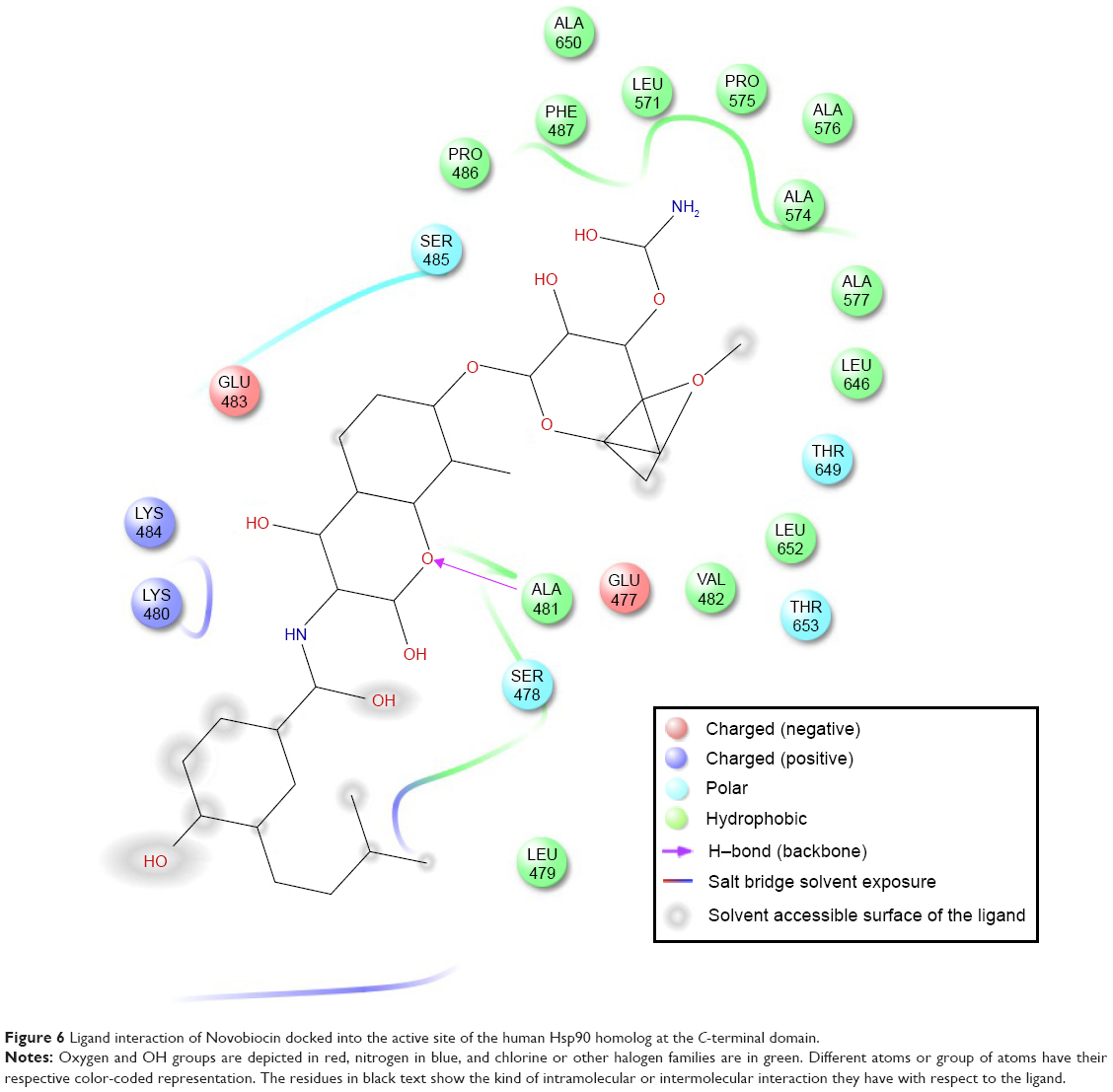 | Figure 6 Ligand interaction of Novobiocin docked into the active site of the human Hsp90 homolog at the C-terminal domain. |
As evident from Figure 5 (refer Figure S6 for the plots of all complexes), all ligands showed a similar interaction pattern with the active-site residues; however, the magnitude of interaction may vary. Generally, hydrophobic interactions with the active site residues Trp598, Met602, Tyr596, Val522, Met620, and Leu533 and hydrogen bonding interaction with the active site residues Gln523 and Tyr596 seem to play a major role in the ligand-enzyme binding.
As shown in Figure 6, the binding site of Novobiocin to the human Hsp90 homolog is distinct from that of NFV. This is a confirmation of the findings in the study carried out by Shim et al18 about the different binding sites of NFV and Novobiocin to the Hsp90 CTD.
To estimate the variation of binding affinity from one ligand to another, per-residue interaction decomposition analysis was performed.
In our previous reports,47,49–53 per-residue interaction energy decomposition analysis using MolDock scoring function54 has proved an efficient and feasible tool to estimate the interaction energy contribution from each amino acid interacting with the ligands.
In the present work, we took one step further to improve the reliability of per-residue interaction energy decomposition results. Instead of calculating the energy contribution using one ligand enzyme complex conformation obtained from docking, which is the conventional approach, we opted to take the average interaction energies over ten conformations extracted evenly from the MD trajectory. The following flowchart (Figure 7) summarizes the docking and per-residue interaction energy decomposition protocols adopted in this work.
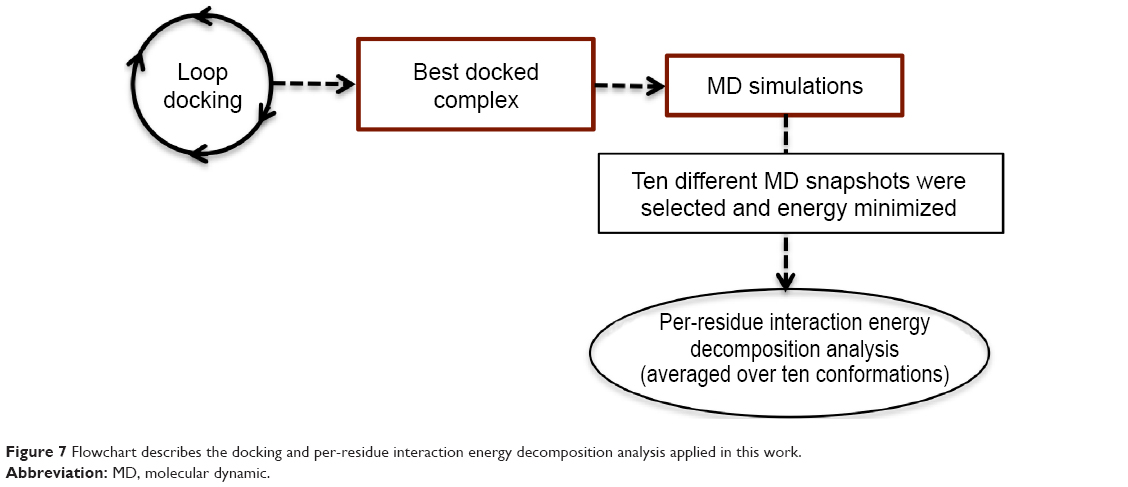 | Figure 7 Flowchart describes the docking and per-residue interaction energy decomposition analysis applied in this work. |
As shown in the interaction energy decomposition plot for NFV (Figure 8; refer Figure S7 for plots of the remaining complexes), the active site amino acids Tyr596, Val534, Lys526, Lys538, Gln523, and Trp598 contributed the best toward NFV binding. Figures 5 and S7 highlight the nature of the binding forces between these amino acids and the ligand.
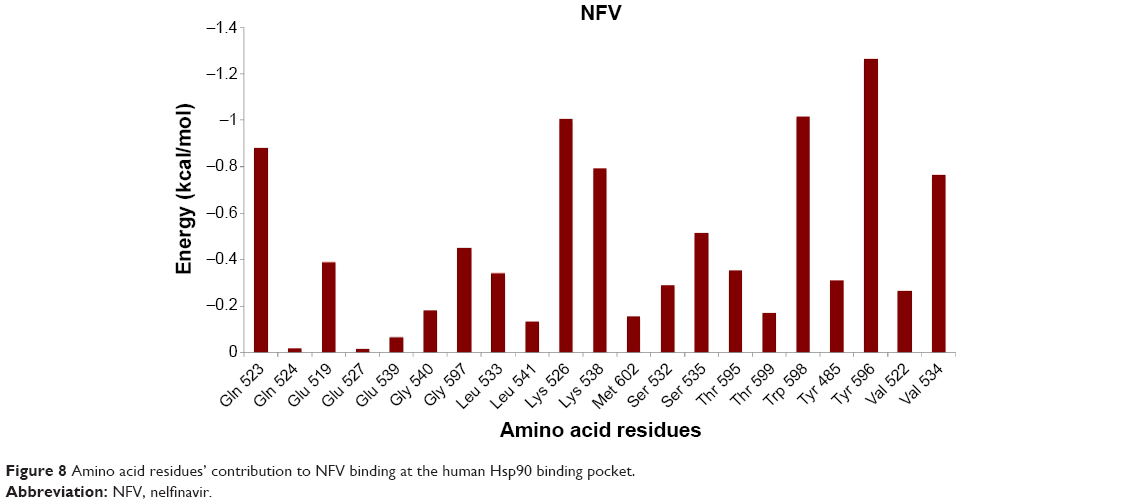 | Figure 8 Amino acid residues’ contribution to NFV binding at the human Hsp90 binding pocket. |
The small difference in the binding affinities of NFV and Indinavir might be due to the fact that Indinavir interacted with the enzyme via hydrogen bond with Gly597, whereas NFV showed hydrogen bond, as well as hydrophobic interaction with the amino acids Tyr485, Gln523, and Lys526. As stated earlier, with exclusion of ATV, all inhibitors showed almost the same binding affinity toward Hsp90. It was interesting to observe that ATV has the lowest binding affinity when compared with all other inhibitors. This could be explained based on two assumptions: 1) the amino acid residues that contributed to ATV binding via hydrophobic interaction are fewer when compared with other PIs and 2) ATV seems to be located slightly out of the active site pocket, when compared with other ligands. This might be due to the presence of ter-butyl and ester groups, which might cause steric conflict with the active-site residue side chains, which leads to a slight displacement of the ligand from the active-site pocket. APV exhibited strong interaction with Gln523 and Tyr596 via hydrogen bonding interaction and hydrophobic interactions, respectively.
We believe that the robust computational tools implemented in this work provide a platform that could assist medicinal chemists in optimizing the activity of studied inhibitors as anticancer as well as designing potential drug candidates against other biological targets.
Conclusion
In this study, the mechanism of binding of nine HIV-1 PIs to the human Hsp90β homolog was investigated in order to gain insight into their potential use as anticancer agents. Enhanced in-house developed molecular docking protocol, “loop docking”, was applied to estimate the relative binding affinities of HIV-1 PIs against Hsp90. MDs simulations were also performed to validate the docking results and ensure the stability of docked inhibitors in the enzyme active site pocket at the CTD. An improved per-residue energy decomposition analysis was performed in order to estimate the contribution of each amino acid toward drug binding. This provides a useful tool in designing and optimizing of more potent inhibitors.
However, the answer for the question we initially imposed, “Could the FDA-approved anti-HIV-1 PR inhibitors be promising anticancer agents?” is based purely on, however validated, silico assumptions and tools. We strongly believe that this report will prompt researchers from different scientific domains to further investigate the potential use of the current FDA-approved HIV-1 PIs as dual anti-HIV-1 and anticancer drugs.
Acknowledgments
The authors acknowledge the School of Health Sciences, UKZN, for financial support and CHPC for technical support. Some parts of this work have been presented at the CHPC National Symposium (National Kruger Park, 2014).
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. The Global Burden of Disease: 2004 Update; 2008. Available from: http://www.who.int/healthinfo/global_burden_disease/GBD_report_2004update_full.pdf. Accessed January 18, 2014. | ||
Jemal A, Bray F, Forman D, et al. Cancer burden in Africa and opportunities for prevention. Cancer. 2012;118:4372–4384. | ||
Unnati S, Ripal S, Sanjeev A, Niyati A. Novel anticancer agents from plant sources. Chin J Nat Med. 2013;11:16–23. | ||
Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. | ||
Cos S, González A, Martínez-Campa C, Mediavilla MD, Alonso-González C, Sánchez-Barceló EJ. Estrogen-signaling pathway: a link between breast cancer and melatonin oncostatic actions. Cancer Detect Prev. 2006;30:118–128. | ||
Stern DF. Tyrosine kinase signalling in breast cancer – ErbB family receptor tyrosine kinases. Breast Cancer Res Treat. 2000;2:176–183. | ||
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human-breast cancer – correlation of relapse and survival with amplification of the HER-2 neu oncogene. Science. 1987;235:177–182. | ||
Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–726. | ||
Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11:263–275. | ||
Kurebayashi J. Biological and clinical significance of HER2 overexpression in breast cancer. Breast Cancer. 2001;8:45–51. | ||
Grover A, Shandilya A, Agrawal V, et al. Hsp90/Cdc37 chaperone/co-chaperone complex, a novel junction anticancer target elucidated by the mode of action of herbal drug Withaferin A. BMC Bioinformatics. 2011;12:1–13. | ||
Perterson LB. Investigation of the Hsp90 C-Terminal Binding Site, Novel Inhibitors and Isoform-Dependent Client Proteins; 2012. Available from: https://kuscholarworks.ku.edu/dspace/bitstream/1808/10218/1/Peterson_ku_0099D_12224_DATA_1.pdf. Accessed August 20, 2013. | ||
Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. | ||
Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. | ||
Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673–683. | ||
Gills J, Lo Piccolo J, Tsurutani J, et al. Nelfinavir, a lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13:5183–5194. | ||
Osborne CK. Steroid hormone receptors in breast cancer management. Breast Cancer Res Treat. 1998;51:227–238. | ||
Shim JS, Rao R, Beebe K, et al. Selective inhibition of HER2-positive breast cancer cells by the HIV protease inhibitor nelfinavir. J Natl Cancer Inst. 2012;104:1576–1590. | ||
Khalid S, Paul S. Identifying a C-terminal ATP binding sites-based novel Hsp90-inhibitor in silico: a plausible therapeutic approach in Alzheimer’s disease. Med Hypotheses. 2014;83:39–46. | ||
Srirangam A, Mitra R, Wang M, et al. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin Cancer Res. 2006;12:1883–1896. | ||
Bernstein WB, Dennis PA. Repositioning HIV protease inhibitors as cancer therapeutics. Curr Opin HIV AIDS. 2008;3:666–675. | ||
Eswar N, Webb B, Marti-Renom MA, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;Chapter 2:Unit 2.9. | ||
Eric F, Pettersen TDG, Conrad C, et al. UCSF chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. | ||
Multiple sequence alignment by CLUSTALW. Kyoto University Bioinformatics Center. Available from: http://www.genome.jp/tools/clustalw/. Accessed October 16, 2013. | ||
SiteHound-web. Available from: http://scbx.mssm.edu/sitehound/sitehound-web/Input.html. Accessed October 15, 2013. | ||
Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. | ||
Buchan DWA, Ward SM, Lobley AE, Nugent TCO, Bryson K, Jones DT. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 2010;38:W563–W568. | ||
Schrodinger. Available from: http://www.schrodinger.com/. Accessed October 20, 2013. | ||
DeepView - Swiss-PdbViewer. The SIB Institute of Bioinformatics. Available from: http://www.expasy.org/spdbv/. Accessed October 21, 2013. | ||
Blake L, Soliman MES. Bifunctional anti-HIV/TB inhibitors: perspective from in-silico design and molecular dynamics simulations. Lett Drug Des Discov. 2013;10:706–712. | ||
Moonsamy S, Soliman MES. Dual acting HIV inhibitors: integrated rational in silico design strategy. Med Chem Res. 2014;23:682–689. | ||
Zinc Database. Available from: http://zinc.docking.org. Accessed September 18, 2013. | ||
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012:4(1):17. | ||
Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model. 2010;50:572–584. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. | ||
Cornell WD, Cieplak P, Bayly CI, et al. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules (vol 117, pg 5179, 1995). J Am Chem Soc. 1996;118:2309–2309. | ||
Case DA, Cheatham TE, Darden T, et al. The amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. | ||
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. | ||
Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J Comput Chem. 2000;21:132–146. | ||
Cornell WD, Cieplak P, Bayly CI, et al. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J Am Chem Soc. 1995;117:5179–5197. | ||
Frisch MJ, Trucks GW, Schlegel HB, et al. Wallingford CT: Gaussian Inc.; 2004. Available from: http://www.gaussian.com/g_misc/g03/citation_g03.htm | ||
Duan Y, Wu C, Chowdhury S, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. | ||
Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. | ||
Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular-dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. | ||
Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-integration of cartesian equations of motion of a system with constraints – molecular-dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. | ||
Durrant J, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol. 2011;9:71. | ||
Blake L, Soliman MS. Identification of irreversible protein splicing inhibitors as potential anti-TB drugs: insight from hybrid non-covalent/covalent docking virtual screening and molecular dynamics simulations. Med Chem Res. 2014;23:2312–2323. | ||
Ahmed SM, Kruger HG, Govender T, et al. Comparison of the molecular dynamics and calculated binding free energies for nine FDA-approved HIV-1 PR drugs against subtype B and C-SA HIV PR. Chem Biol Drug Des. 2013;81:208–218. | ||
Ahmed SM, Maguire GEM, Kruger HG, Govender T. The impact of active site mutations of South African HIV PR on drug resistance: insight from molecular dynamics simulations, binding free energy and per-residue footprints. Chem Biol Drug Des. 2014;83:472–481. | ||
Skelton AA, Maharaj YR, Soliman MES. Target-bound generated pharmacophore model to improve the pharmacophore-based virtual screening: identification of G-protein coupled human CCR2 receptors inhibitors as anti-inflammatory drugs. Cell Mol Bioeng. 2014;7:45–57. | ||
Moonsamy S, Soliman MES. Computer-aided perspective for the design of flexible HIV non-nucleoside reverse transcriptase inhibitors (NNRTIs): de-novo drug design, virtual screening and molecular dynamics simulations. Lett Drug Des Discov. 2014;11:513–524. | ||
Maharaj Y, Soliman MES. Identification of novel gyrase B inhibitors as potential anti-TB drugs: homology modelling, hybrid virtual screening and molecular dynamics simulations. Chem Biol Drug Des. 2013;82:205–215. | ||
Soliman MES. A hybrid structure/pharmacophore-based virtual screening approach to design potential leads: a computer-aided design of South African HIV-1 subtype c protease inhibitors. Drug Dev Res. 2013;74:283–295. | ||
Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem. 2006;49:3315–3321. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.