")
Back to Journals » International Journal of Nanomedicine » Volume 14
Construction and in vivo/in vitro evaluation of a nanoporous ion-responsive targeted drug delivery system for recombinant human interferon α-2b delivery
Authors Liu H, Zhu J , Bao P , Ding Y, Shen Y, Webster TJ , Xu Y
Received 22 March 2019
Accepted for publication 13 June 2019
Published 16 July 2019 Volume 2019:14 Pages 5339—5353
DOI https://doi.org/10.2147/IJN.S209646
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Hongfei Liu, 1 Jie Zhu, 1 Pengyue Bao, 2 Yueping Ding, 3 Yan Shen, 4 Thomas J Webster, 5 Ying Xu 1
1College of Pharmacy, Jiangsu University, Zhenjiang 212013, People’s Republic of China; 2Chia Tai Tianqing Pharmaceutical Group Co., Ltd, Nanjing 210023, People’s Republic of China; 3Jiangsu Sihuan Biopharmaceutical Co., Ltd, Wuxi 214000, People’s Republic of China; 4State Key Laboratory of Natural Medicines, Department of Pharmaceutics, China Pharmaceutical University, Nanjing 210009, People’s Republic of China; 5Department of Chemical Engineering, Northeastern University, Boston, MA 02115, USA
Background: Like most protein macromolecular drugs, the half-life of rhIFNɑ-2b is short, with a low drug utilization rate, and the preparation and release conditions significantly affect its stability.
Methods: A nanoporous ion-responsive targeted drug delivery system (PIRTDDS) was designed to improve drug availability of rhIFNα-2b and target it to the lung passively with sustained release. Chitosan rhIFNα-2b carboxymethyl nanoporous microspheres (CS-rhIFNα-2b-CCPM) were prepared by the column method. Here, an electrostatic self-assembly technique was undertaken to improve and sustain rhIFNα-2b release rate.
Results: The size distribution of the microspheres was 5∼ 15 μm, and the microspheres contained nanopores 300∼ 400 nm in diameter. The in vitro release results showed that rhIFNα-2b and CCPM were mainly bound by ionic bonds. After self-assembling, the release mechanism was transformed into being membrane diffusion. The accumulative release amount for 24 hrs was 83.89%. Results from circular dichrogram and SDS-PAGE electrophoresis showed that there was no significant change in the secondary structure and purity of rhIFNα-2b. Results from inhibition rate experiments for A549 cell proliferation showed that the antitumor activity of CS-rhIFNα-2b-CCPM for 24 hrs retained 91.98% of the stock solution, which proved that the drug-loaded nanoporous microspheres maintained good drug activity. In vivo pharmacokinetic experimental results showed that the drugs in CS-rhIFNα-2b-CCPM can still be detected in vivo after 24 hrs, equivalent to the stock solution at 6 hrs, which indicated that CS-rhIFNα-2b-CCPM had a certain sustained-release effect in vivo. The results of in vivo tissue distribution showed that CS-rhIFNα-2b-CCPM was mainly concentrated in the lungs of mice (1.85 times the stock solution). The pharmacodynamics results showed that CS-rhIFNα-2b-CCPM had an obvious antitumor effect, and the tumor inhibition efficiency was 29.2%.
Conclusion: The results suggested a novel sustained-release formulation with higher drug availability and better lung targeting from CS-rhIFNα-2b-CCPM compared to the reference (the stock solution of rhIFNα-2b), and, thus, should be further studied.
Keywords: nanoporous, ion-responsive, recombinant human interferon α-2b, ion exchange technique, sustained release, lung targeting
Corrigendum for this paper has been published
Introduction
Interferon α-2b (IFNα-2b) is a type of cytokine protein with broad-spectrum antitumor1, antiviral,2 and immune enhancement3 activities. It is one of the most important antitumor biological products in clinical practice today. Recombinant human interferon α-2b (rhIFNα-2b) has the same biological activity as natural human IFNα-2b, which is combined with other drugs to treat small cell or non-small cell lung cancers continuously.4 Like most protein drugs, the half-life of rhIFNɑ-2b is short,5 drug utilization rate is low, and the preparation process and releasing environment significantly influence its stability.6 In brief, the above shortcomings limit it for wider applications. Therefore, a new formulation of this cytokine is desired to improve its encapsulation and sustained release.
The ion-exchange technique has been widely used in many areas, such as ionic chemical drug delivery systems, protein purification, suspension formulations, etc.7,8 It has also been applied in protein polypeptide drug delivery systems. Compared to the crude drug, the drug–resin composite improves the stability and retention rate of enzyme activity. Traditional ion-exchange resins9 (Amberlite GC120 and Amberlite IRP69) are based on polystyrene diethylbenzene, which is nonbiodegradable and difficult to use in the study of protein drug delivery systems for injection. In addition, the protein drug has a molecular weight between 10 and 1000 kDa, so it is difficult to enter the smooth and dense interior of ion-exchange resins in large quantities, resulting in low drug loading. Nanoporous microspheres are suitable for embedding and controlling the release of rhIFNα-2b at a molecular weight of 19 kDa.10
In order to solve the above problems, carboxymethyl chitosan (CC) is proposed in this study as the carrier for ion exchange. CC is a chitosan derivative, and CC is a type of amphoteric polymer with good water solubility, biocompatibility, biodegradability, low toxicity, and pH responsiveness. In recent years, CC has been widely used in biomedical,11–13 tissue engineering,14,15 wound dressing,16 agricultural film, and other health care products. In particular, it has become a new favorite in the field of pharmaceutical research. CC can be prepared into hydrogels,17–19 drug conjugates,20 biodegradable sustained-release carriers, polyelectrolyte complexes,21 etc. In addition, CC can also be used for the delivery of anticancer drugs,22 antibacterial drugs,23 anti-inflammatory drugs,24 and genes.25 Its application in the field of protein polypeptide drug delivery systems has received increasing attention.26
In this article, CC sodium was used as a carrier material to prepare nanoporous microspheres. CC has good biocompatibility and biodegradability27 and is a safe sustained-release carrier. Once crosslinked, carboxymethyl chitosan nano-porous microspheres (CCPM) is insoluble in water, has a certain spatial three-dimensional structure, and retains its carboxyl group for ion exchange. Its carboxyl group is a weak acidic and ionizable group, which can bind the protein drug due to opposite electric charges. In this article, low-toxic ethyl acetate was used as a pore-forming agent to form a nanoporous microsphere. After preparing the blank nanoporous microspheres here, a dynamic drug-loading method was adopted. A dynamic drug-loading method could result in continuous drug concentration in the effluent, which could realize 100% drug loading and greatly reduce the loss of expensive drugs. The mechanism of that CCPM loading and release by ion exchange is shown in Figure 1. When the pH in vitro is less than the rhIFNα-2b isoelectric point, the rhIFNα-2b is positively charged, which can bind CC microsphere ions with negative electricity. The dynamic drug-loading process was stopped immediately when the drug was detected in the effluent, and the drug utilization rate in theory can reach 100%. In the physiological environment of the human body, the positive ions (such as Na+) in the body can release the rhIFNα-2b transposition on the nanoporous microsphere, where the drug plays its biological role.
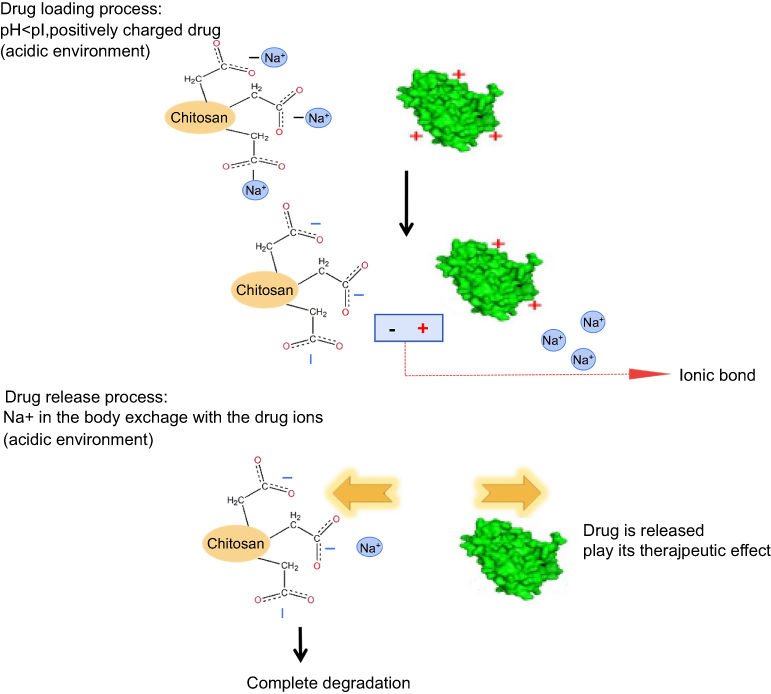 |
Figure 1 The mechanism of CCPM loading and releasing drugs by ion exchange. Abbreviation: CCPM, carboxymethyl chitosan nanoporous microspheres. |
To an extent, the dynamic drug-loading method can solve problems of low encapsulation rate of microspheres and loss of drug activity during drug loading. However, based on existing research,28 we know that ion exchange has a certain slow-release effect, but it is not enough. Therefore, this article adopted an electrostatic self-assembly technique to seal/cover the pores on the surface of the nanoporous microspheres to achieve a further slow-release effect.29 The electrostatic self-assembly technique mainly relies on electrostatic interactions between the assembled molecules to deposit polyelectrolytes alternately on the surface of the substrate and spontaneously form self-assembled films with special structures and functions.30,31 According to the literature,32 the self-assembly of polyelectrolyte layers outside the drug surface can delay the drug-release rate. Therefore, it is feasible that chitosan, with a positive charge, can be used to form a water-insoluble polyelectrolyte complex film on the surface of negatively charged drug-carrying nanoporous microspheres through electrostatic action to achieve slow release.
In general, in this article, a nanoporous ion-responsive targeted drug delivery system (PIRTDDS) was designed to improve drug availability of rhIFNα-2b and target the drug to the lung passively and with sustained release. The drug-loaded nanoporous microspheres were characterized by scanning electron microscopy (SEM), SDS-PAGE, and circular dichrogram (CD). In addition, the drug-release behavior in vitro, anti-lung cancer activity in vitro, pharmacokinetics in vivo, and tissue distribution of the drug-loaded nanoporous microspheres were also evaluated.
Materials
rhIFNα-2b was obtained from Genid Biotechnology Co., Ltd. (Guizhou, China). CC was obtained from Bomei Biotechnology Co., Ltd. (Hefei, China). Coomassie Brilliant Blue G250 and R250 were both purchased from Sinopharm Chemical Reagent Co., Ltd. (Beijing, China). DMEM and thiazolyl blue tetrazolium bromide (MTT) were purchased from Gibco Company (Langley, OK, USA). FBS was purchased from Yuan Ye Biotechnology Co., Ltd. (Shanghai, China). Dimethyl sulfoxide (DMSO) was purchased from Sigma (Shanghai, China). A human lung adenocarcinoma cell line A549 was provided by the Institute of Life Sciences, Jiangsu University, and the use of the cell line was approved by the Ethics Committee of Jiangsu University. rhIFNɑ-2b kit was purchased from Crystal Biological Technique Co., Ltd. (Shanghai, China). CCPM was self-made. All the chemical reagents were obtained from Sinopharm Chemical Reagent Co., Ltd.. Lewis lung carcinoma cells were provided by Shanghai Zibo Biotechnology Co., Ltd.
Animals
Clean Kunming mice (male, 18–24 g) and ICR mice (male, 20–25 g) were obtained from the Experimental Animal Center at Jiangsu University (Zhenjiang, Jiangsu, China). All animal experiments were carried out in accordance with the Guide for Care and Use of Laboratory Animals as adopted and promulgated by the Animal Ethics Committee of Jiangsu University. The experiments were approved by the Animal Ethics Committee of Jiangsu University.
Methods
Preparation of blank CCPM
The blank CCPM was prepared following a previously reported method:33 2 mL of ethyl acetate was used as a pore-forming agent, and 0.1 g CC was dissolved in 5 mL of normal saline at room temperature as the aqueous phase, and then emulsified with 0.1 g tween 80 for 30 mins. The emulsion was poured into polyvinyl alcohol (PVA) (10 mL, 1%, w/v) and again vortexed for 1 min before being placed into 40 mL of liquid paraffin with 0.8 g of span 80. The mixture was stirred at 900 rpm for 1 hr at room temperature. After 1 hr of emulsification, 0.16 mL of a 12.5% glutaraldehyde solution was slowly added at a constant speed. The solution was stirred and solidified for 3 hrs. Then, the nanoporous microspheres were washed by n-hexane three times to make a surface without a floating oil spot, dispersed with anhydrous ethanol, washed with deionized water, and obtained via centrifugation and freeze drying.
Fabrication of rhIFNα-2b-CCPM by ion exchange
The loading process is shown in Figure 2. 450 mg of self-made nanoporous microspheres was soaked with deionized water for 30 mins, then the nanoporous microspheres were packed into a dynamic glass exchange column (Ф 16 mm x 20 cm). 0.75 mg/mL of a rhIFNa-2b solution (0.02 mol/L pH4.6 HAc–NaAc buffer solution) flowed through the column by 0.2 mL/min at 25±0.5°°C for about 44 mins, then deionized water was used to wash the surface of the nanoporous microspheres to remove unbound drugs. The Bradford method was used to determine the total outflow of drugs and to calculate the theoretical drug loading. Last, rhIFNα-2b-CCPM was obtained by freeze-drying.
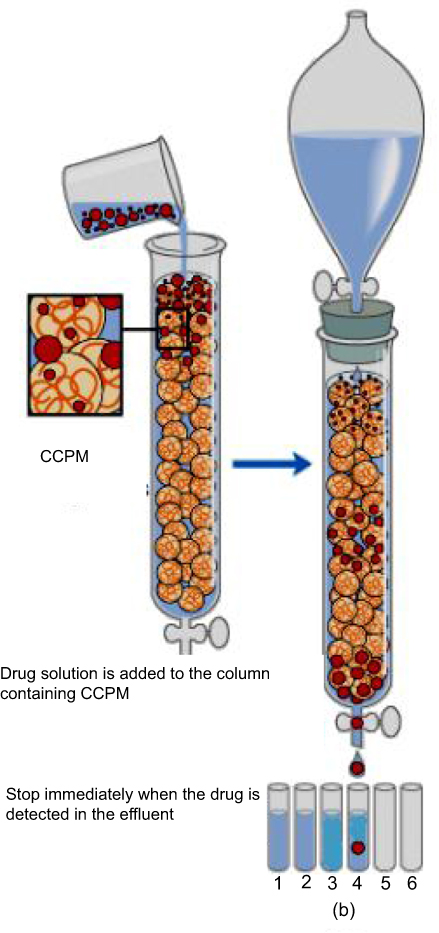 |
Figure 2 The diagram of the dynamic loading drug process.Abbreviation: CCPM, carboxymethyl chitosan nanoporous microspheres. |
Fabrication of CS-rhIFNα-2b-CCPM by self-assembly
5.4 g sodium acetate trihydrate was added to 50 mL of deionized water to prepare 1 mg/mL of a chitosan solution (HAc adjusted to pH 4.6) with ultrasonic treatment for 30 mins (Figure 3). After adding 37.5 mg of wet rhIFNα-2b-CCPM at 25°C, the self-assembly process occurred with magnetic stirring at 100 rpm for 15 mins. After centrifugal depuration, deionized water was added and washed 3 times to freeze and dry the composite nanoporous microsphere.
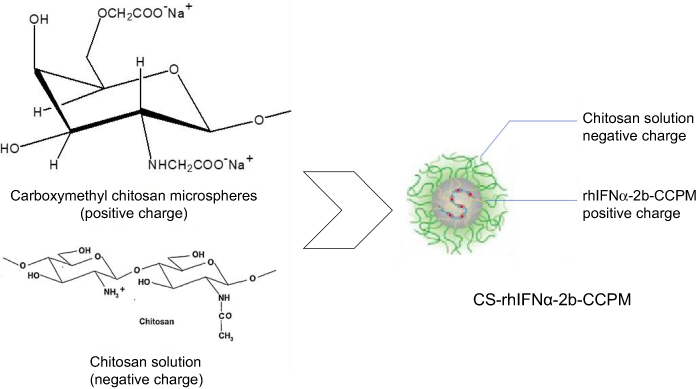 |
Figure 3 Schematic diagram of the electrostatic self-assembly technique.Abbreviations: rhIFNα-2b-CCPM, rhIFNα-2b carboxymethyl chitosan nanoporous microspheres; CS-rhIFNα-2b-CCPM, chitosan rhIFNα-2b carboxymethyl chitosan nanoporous microspheres. |
Characterization
Morphology and particle size distribution of the nanoporous microspheres
The morphological characteristics of CCPM, rhIFNα-2b-CCPM, and CS-rhIFNα-2b-CCPM were determined by SEM (S-4800, Hitachi, Tokyo, Japan). The size of CS-rhIFNα-2b-CCPM was measured by a laser scattering particle size analyzer (LS230, Beckman Coulter, Pasadena, CA, USA).
Drug content determination
25 mg of CS-rhIFNα-2b-CCPM was accurately weighed and added to 2.5 mL of a 1 mol/L HCl solution and sonicated for 1 hr in an ice bath, and then shaken at 50 rpm at 37±0.5°C for 24 hrs. 2.5 mL of 1 mol/L NaOH was added for neutralization. Then, the sample was centrifuged at 10,000 rpm for 5 mins at 4°C. The supernatant was collected and the absorbance was determined by using the Bradford method to calculate the content of rhIFNα-2b. Three parallel tests on each sample were performed, and the actual encapsulation rate (rE) and drug-loading rate (rDL) of the nanoporous microsphere was calculated as indicated in Equations 1 and 2.
(1) (2)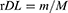
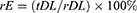
where m is the content of protein mass in the nanoporous microsphere (mg) and M is the content of the nanoporous microsphere mass (mg). The rDL is the actual loading amount (μg/mg), tDL is the theoretical loading amount (μg/mg), and rE is the actual sealing rate.
In vitro release study
A phosphate buffer solution34 (20 mmol/L, 5 mL, pH 7.4), containing 0.02% Tween 80, was added into a 5 mL tube, and then CS-rhIFNα-2b-CCPM (25 mg) was added into the tube and suspended thoroughly. The tube was placed in a 37±0.5°C water bath and shaken at 50 rpm horizontally. After 0.25, 0.5, 1, 2, 3, 4, 6, 12, 16, 24, and 30 hrs, samples were taken out and centrifuged at 10,000 rpm for 5 mins at 4°C. Three replicate tests were performed for the in vitro drug-release studies. The content of rhIFNα-2b was measured by the Bradford method. The cumulated released percent in vitro (Q) of the nanoporous microspheres was calculated as indicated in Equation 3:
(3)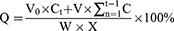
where Ct is the concentration of drug in the medium at the t sampling point, W is the total weight of the nanoporous microspheres in the medium (mg), V0 is the volume of the released medium (mL), V is the volume of each sampling (mL), and X is the amount of drug carried by the nanoporous microsphere (μg/mg).
To determine the drug-release profile, a zero-order release equation (Equation 4), a first-order release equation (Equation 5), the Higuchi equation (Equation 6), and the Hixson and Crowell (Equation 7) and Viswanathan (Equation 8) equations were selected for fitting.
(4) (5) (6) (7) (8)
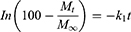
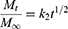
SDS-PAGE
30 μL of different rhIFNα-2b sample solutions, 30 μL of the marker solution plus 10 μL of 4 × SDS gels, and sample buffer were blended and then heated for 10 mins using a 100°C water bath for protein denaturation. The concentration of resolving gel was 12% and stacking gel was 5%. Coomassie brilliant blue R250 was dyed and decolorized.
CD
The rhIFNα-2b-CCPM releasing solution was characterized by a CD and compared with the standard CD of rhIFNα-2b reported in the literature35 to observe the changes of its secondary structure. Test conditions were as follows: the xenon lamp (150 W), room temperature, ultraviolet scanning range area (195~250 nm), the bandwidth of 1 nm, standard sensitivity, response time of 1 s, the scanning speed of 50 nm/min, the data step of 0.5 nm, and an optical path of 1 mm.
Relative biological activity in vitro
Lung adenocarcinoma A549 cells were cultured in DMEM containing 10% FBS and 1% penicillin and streptomycin at 37°C in a humidified atmosphere of 5% CO2.36,37 When the cells entered the logarithmic growth stage, they were seeded into 96-well plates (4x103 cells per well) and cultured in a 200 μL cell suspension. The cells were treated with different concentrations of the rhIFNα-2b stock solution and the rhIFNα-2b-CCPM releasing solution (5, 10, 25, 40, and 50 μg/mL),38 then diluted with DMEM after 24 hrs of incubation. Cell culture medium was added to a negative control group. 96-well plates were incubated in 5% CO2 at 37°C for 72 hrs.
10 μL of the MTT solution39 (5 mg/mL) was added to each well and incubated for 4 hrs. After discarding the cell supernatant and adding 100 μL of DMSO into each well, the 96-well plate was shaken for 10 mins until the purple formazan crystals were solubilized. The OD values of each well were measured with a microplate reader at 490 nm. The cell inhibition rate (IR) from the 24-hr releasing solution was calculated as indicated in Equation 9 and the IR–concentration curve was drawn. Each experiment was repeated three times. The IC50 values were determined using a sigmoidal equilibrium model regression using XLfit version 4.3.2 (ID Business Solutions Ltd., Guildford, Surrey, UK) and are defined as the concentration of rhIFNα-2b required for a 50% reduction in growth/viability:
(9)
where ODc is the absorbance of the control group, ODt is the absorbance of the experimental group, and OD0 is the absorbance of the blank hole.
In vivo analytical methodology
Analysis methods were validated according to the established international guidelines and requirements (Validation of Analytical Methods: Definitions and Terminology, ICH Topic Q2A, and Validation of Analytical Procedure: Methodology, ICH Topic Q2B). Accuracy of the method was assessed on the basis of the coefficient of variation in quality control samples. Three standard kits were used to establish standard curves, and the coefficient of variation between the plates was calculated. For the recovery of plasma and tissue samples, 7.5, 30, and 120 pg/mL of rhIFNα-2b was added to each plasma and tissue homogenate supernatants, respectively. The extraction recovery was calculated with the standard curve.
In vivo pharmacokinetics and tissue distribution study
Eighty Kunming mice40 were randomly divided into three groups: 30 mice in the control group, 45 mice in the experimental group, and 5 mice in the blank group. All of the mice were fasted for 12 hrs before experiments, with free access to water. Before the pharmacokinetic study, the mice were anesthetized with pentobarbital sodium anesthesia (40 mg/kg).
For the control group, the tail vein was administered at 4.645 μg/kg rhIFNα-2b. 200 μL blood samples were withdrawn from the ocular veniplex41–43 and placed in heparinized tubes at predetermined time intervals. Then, the mice were sacrificed, and the tissues were collected.
For the experimental group, 1.5 mg rhIFNα-2b-CCPM was accurately weighed (containing 18.75 μg rhIFNα-2b) and suspended in 20 mL of sterilized water containing 0.02% Tween 80. The sample was injected into the tail vein by 0.005 mL/g. 200 μL blood samples were withdrawn from ocular veniplex and placed in heparinized tubes at predetermined time intervals. Then, the mice were sacrificed, and the tissues were collected. Plasma samples were obtained by centrifugation at 4,000 rpm at 4°C for 15 mins, and then stored in an –80°C refrigerator for analysis.
The procedure followed the NHGRI Animal Care And Use Committee (ACUC) Guideline And Procedures For Retro-orbital.
For the tissue sample pretreatment, the tissue was flushed with PBS (0.01M pH 7.4) to remove residual blood, weighed and cut. Nine times the volume of PBS was mixed with the cut tissue in a homogenizer, fully ground and homogenized by further ultrasonic crushing. Finally, the homogenate was centrifugated at 10,000 rpm for 5~10 mins, and the supernatant was collected and stored at −80°C for measurements. RhIFNα-2b concentrations in the plasma and tissues of mice were determined by the ELISA method.44
The noncompartment model parameters of the plasma concentration–time curves of the stock solution and nanoporous microspheres were calculated by DAS 2.0 software. Taking the injection as a reference, the relative bioavailability of the drug-loaded nanoporous microspheres was calculated. The formula was as follows:
(10)
AUCT is the bioavailability for the drug-loaded nanoporous microspheres and AUCR is the bioavailability of the injection solution.
Pharmacodynamic evaluation
The Lewis cells were rapidly placed in a 37°C water bath to resuscitate, then the cells were cultured in the culture medium in an incubator. When the Lewis cells were grown to logarithmic growth phase and the number of viable cells was greater than 95%, the cells were suspended in PBS to form a 1×107 Lewis cell suspension. The suspension was injected into the subcutaneous of the left forelimb, and each mouse was injected with a 0.2 mL suspension. The tumor growth status of each mouse was observed, and the successfully modeled mice were selected for subsequent experiments.
Thirty-six successfully modeled ICR mice (20–25 g) were randomly divided into three groups: a control group (2 mg/kg, NS), a stock solution group (2 mg/kg, rhIFNα-2b), and a microsphere group (20 mg/kg, CS-rhIFNα-2b-CCPM), administered by the tail vein every day for 15 days.45
The mice weight and tumor volume were measured every other day, and the volume of the tumor was calculated by Equation 11. After the last measurement, all mice were sacrificed, and the tumors were excised, weighed, and photographed. The dissected tumors and major organs, including the heart, liver, spleen, lungs, and kidneys, were gathered and then fixed in a 4% (w/v) paraformaldehyde solution for the following histological examinations that involved H&E staining, which were performed for the evaluation of antitumor effects. The tumor IR was calculated according to the following Equation 12.
(11)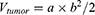
where a is the tumor long diameter (mm) and b is the tumor width (mm).
(12)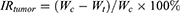
In the formula, Wc is the tumor weight (g) of the control group and Wt is the tumor of the administered group (g).
Statistical analysis
Data were obtained at least in triplicate and expressed as the mean±SD. A t-test (one-sample t-test) was conducted using SPSS statistical software, and the significance was evaluated based on P<0.05.
Results and discussion
Kinetics of drug loading
As can be seen from Figure 4, in the initial stage of dynamic drug loading, the ion-exchange reaction between rhIFNα-2b and the nanoporous microspheres was sufficient, and rhIFNα-2b was rapidly bound to the site on the surface of the nanoporous microspheres. At this time, interferon was not detected in the effluent, and all rhIFNα-2b was loaded into the nanoporous microsphere resin, and the drug utilization rate was 100%. After about 44 mins, the nanoporous microsphere column gradually penetrated, the permeability rapidly increased to 0.1, and the permeability rapidly increased with time, but the drug utilization rate decreased rapidly accordingly. Considering the high price of protein drugs and their drug utilization rate, 44 mins was chosen as the time point to stop drug loading in this study.
 |
Figure 4 The effect of time on permeability and drug utilization rate of the drug-loading process (n=3). |
Characterization
Morphology and particle size distribution of the nanoporous microspheres
As can be seen from Figure 5Ai, the microspheres had a uniform particle size distribution and optimal roundness. Figure 5Aii shows that the microspheres had obvious nanoporous structures of around 300–400 nm in diameter. Figure 5Aiii illustrates that there were still pores remaining on the surface of the nanoporous microspheres after drug loading. Figure 5Aiv shows that the pores on the surface of the nanoporous microspheres were covered, indicating that chitosan was successfully assembled on the surface of the nanoporous microspheres. It is known from the literature46 that the optimal size range required to achieve transient but efficient targeting of pulmonary capillaries after intravenous injection is between 6~10 μm in rats. Particles larger than 25 μm (larger than capillaries) can enter the venous circulation relatively unimpeded via the arteriovenous shunt of the lungs. As shown in Figure 5B, the CS-rhIFNα-2b-CCPM had a uniform distribution of particle sizes between 5 and 15 μm, with an average value of 9.46 μm, suggesting good lung-targeting efficiency.
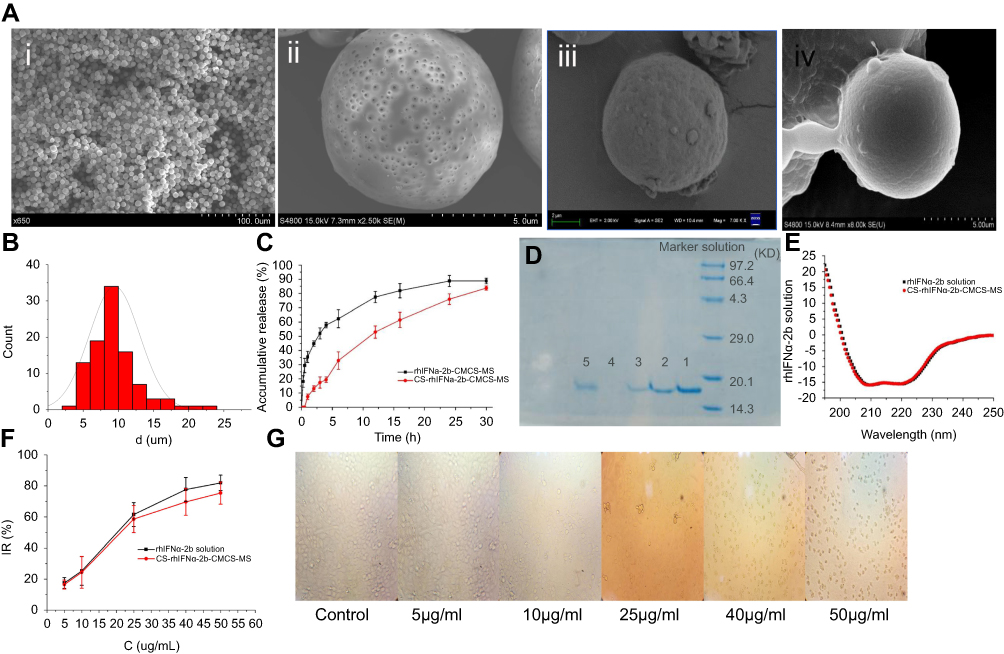 |
Figure 5 Characterization studies. (A) SEM images (ⅰ: CCPM; ⅱ: surface of CCPM; ⅲ: rhIFNα-2b-CCPM; ⅳ: CS-rhIFNα-2b-CCPM); (B) Particle size distribution of CS-rhIFNα-2b-CCPM; (C) Accumulative release from the optimal formulation; (D) Electrophoretogram of different rhIFNα-2b samples dyed by Coomassie brilliant blue (a: rhIFNα-2b solution; b: rhIFNα-2b extracted from nanoporous microspheres; c: rhIFNα-2b release solution for 12 hrs; d: rhIFNα-2b extracted from nanoporous microspheres after in vitro release for 24 hrs; e: rhIFNα-2b in effluent and washing liquid); (E) Circular dichroism spectra of rhIFNα-2b; (F) Inhibition rate of cell proliferation (n=3); and (G) Micrograph of the inhibition effect of the nanoporous microsphere releasing solution with different concentrations on A549 cells. Abbreviations: CCPM, nanoporous microspheres; rhIFNα-2b-CCPM, rhIFNα-2b carboxymethyl chitosan nanoporous microspheres; CS-rhIFNα-2b-CCPM, chitosan rhIFNα-2b carboxymethyl chitosan nanoporous microspheres; IR, inhibition rate. |
Encapsulation efficiency and drug loading
In recent years, much research has been attempted to improve encapsulation efficiency and drug loading. For example, Chen et al47 improved Shirasu porous glass (SPG) membrane emulsification based on traditional preparation methods for protein-loaded composite PLGA microspheres. The drug loading of the composite microspheres can be increased by 8%, and the encapsulation efficiency can be increased by 75.16%. In addition, Varma et al48 prepared chitosan microspheres. The entrapment efficiency of the drug loaded by a passive adsorption method was only 46%. As can be seen from Table 1, the encapsulation efficiency of the nanoporous microspheres significantly increased to 89.91±0.38%, and the actual drug loading was 10.77±2.59%. This was due to the dynamic exchange process where the fresh drug solution underwent an ion-exchange reaction while continuously passing through the resin layer. The dynamic exchange process can effectively separate the exchanged solution from the resin with time and allow for the adequate solution exchange in the whole resin layer. In addition, the nanopores facilitated protein drug penetration and increased the specific surface area of the ion-exchange resins. This not only improved the drug loading of the resin, but also maximized the drug utilization rate.
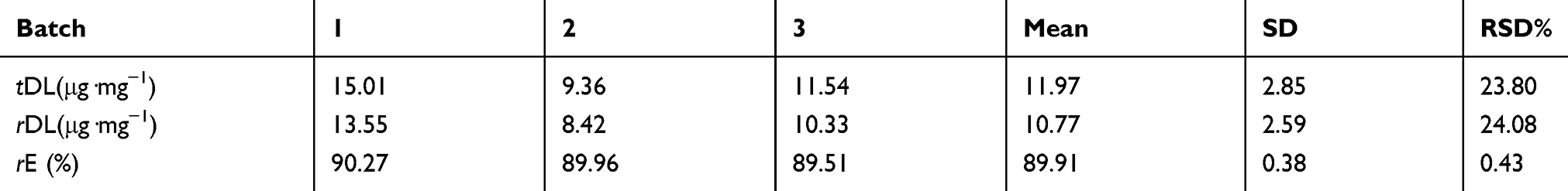 |
Table 1 Drug loading and encapsulation rate of the optimized prescription of CS-rhIFNα-2b-CCPM (n=3) |
In vitro release study
As shown in Figure 5C, compared with the short serum half-life of the bulk drug (2–6 hrs),49 the potential of rhIFN-α-2b-CCPM to prolong the release of rhINF-α-2b was examined in an in vitro release study. The cumulative release rate was 88.78%. Therefore, rhIFN-α-2b-CCPM offered a slow release and was designated as an optimized nanoporous formulation. Next, to induce the sustained-release pattern, the nanopores were coated with chitosan by a self-assembly technique. As shown in Figure 5C, self-assembly did not affect the final release of the protein, but the initial release rate was significantly reduced compared to the self-made drug-loaded microspheres and stealth lipid-coated aquasomes bearing rhIFN-α-2b.49 Only 7.46% was released within 1 hr which effectively slowed down the initial release rate. The cumulative incomplete release may be due to the dynamic balance between drugs and exchangeable ions, and some drugs remained in the nanoporous microspheres and were not released completely. The fitting result from Table 2 showed that the linear relationship between the rhIFN α-2b-CCPM release curve in vitro and Viswanathan model was the best. The results also verified that rhIFNα-2b was bound to the nanoporous microspheres through ionic bonds and released through a particle diffusion mechanism. The in vitro release curve of CS-rhIFN-α-2b-CCPM can be fitted by a first-order release equation, indicating that the release mechanism of the nanoporous microspheres changed to membrane diffusion. The nanoporous microspheres displayed sustained-release characteristics.
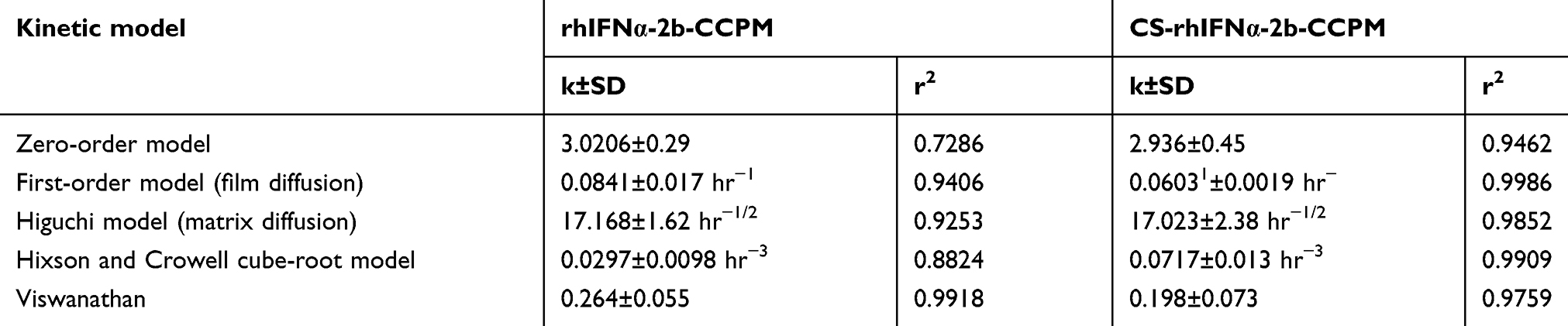 |
Table 2 Results of the drug release behavior fitted by different kinetic models. (mean±SD, n=6) |
SDS-PAGE
The molecular weight of rhIFN α-2b is about 19 kDa. According to the literature,50 rhIFNα2b could be characterized by SDS–PAGE. Figure 5D shows that the positions of the bands for the different samples were identical, and no obvious aggregation and degradation bands were found. It showed that the purity of rhIFN α-2b was higher, and the stability of the protein prepared and released in this article was better. Sample 4 showed that rhIFN α-2b was hardly extracted from the nanoporous microspheres after 24 hrs of release in vitro, which is consistent with the results of the in vitro release. A very shallow band was observed in sample 5, indicating that a small amount of protein penetrated the ion-exchange column.
CD
RhIFNα-2b is a spherical structural protein composed of 5 alpha helices, with negative peaks at 208 nm and 222 nm. Figure 5E shows that the CD spectra of rhIFNα-2b-CCPM released in vitro was similar to the literature.36 Moreover, after the concentration of rhIFNα-2b was determined by the Coomassie brilliant blue method, there was no significant difference in the CD patterns between the same concentrations of the rhIFNα-2b releasing solution and the stock solution. It can be preliminarily concluded that the secondary structure of rhIFNα-2b did not change significantly after dynamic loading and in vitro release.
Relative biological activity at the cellular level in vitro
Figure 5F and G showed that in the range of 5–50 g/ml, the IR of rhIFNα-2b on A549 cells was dose-dependent, and increased with an increase in drug concentration, which had no difference with previous research papers.51 The results indicated that both the stock solution and releasing solution of rhIFNα-2b had an inhibitory effect on A549 proliferation in vitro for 72 hrs. In addition, at the same mass concentration, the relative biological activity of the releasing solution exceeded 91.98%, which indicated preliminarily that the CS-rhIFNα-2b–CCPM still maintained high biological activity against lung cancer cells after release for 24 hrs. The IC50 of rhIFNα-2b-CCPM was 18.915 μg/mL.
In vivo analytical methodology
A linear correlation (r=0.9995) was obtained between concentration and optical density of the rhIFNα-2b standard, and all plasma and tissue samples were between the range 7.5–240 pg/mL. The minimum detectable dose of rhIFNα-2b is 1.0 pg/mL. The maximum variation coefficient between the plates was 13.18%. From the literature,52 it was decided to collect the data for 3 separate experiments. According to the FDA guidelines, the mean value should be within 15% of the nominal value. The average recovery (%) of interferon in plasma and tissue samples is shown in Table 3. All the %RSD were <15%, suggesting that this method was accurate and reliable.
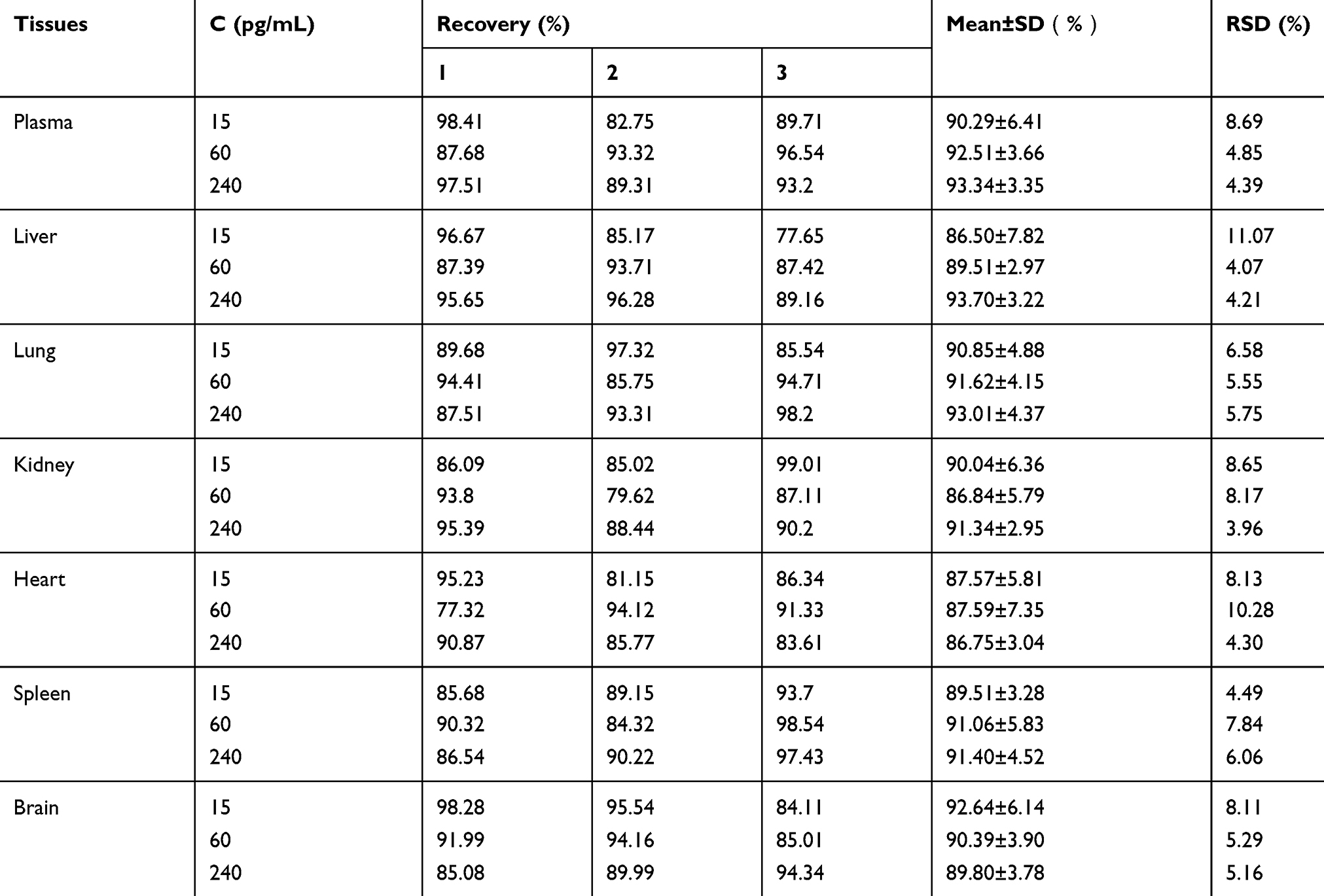 |
Table 3 Results of the recovery test (n=3) |
In vivo pharmacokinetic parameters
Table 4 and Figure 6 show that rhIFNα-2b can be detected in blood from 5 min to 6 hrs after tail vein injection of the rhIFNα-2b solution. Over time, the rhIFNα-2b concentration in blood gradually decreased. After 6 hrs, the concentration of rhIFNα-2b was lower than the quantitative limit (7.5 pg/mL). After tail vein injection in mice, the pharmacokinetic parameters of the rhIFNα-2b solution and rhIFNα-2b nanoporous microspheres were different in vivo. After 1 h, the plasma drug concentration of the rhIFNα-2b nanoporous microspheres was significantly higher than that in the stock solution, and Tmax, t1/2 and the mean residence time (MRT) were longer than those in the stock solution. rhIFNα-2b in the nanoporous microspheres can still be detected at 48 hrs after intravenous injection. It is shown that the nanoporous microsphere group had an obvious sustained-release effect. In summary, these findings were in accordance with the literature.53 The AUC0-∞ of rhIFNα-2b in the nanoporous microsphere group was lower than that in the solution group, which could be due to passive targeting of the nanoporous microspheres to the lung tissue after intravenous injection.
 |
Table 4 Pharmacokinetic parameters in mice plasma (mean±SD, n=3) |
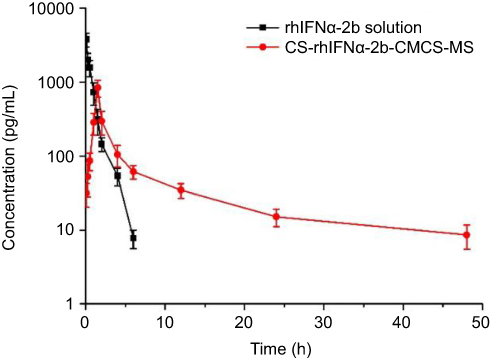 |
Figure 6 Drug concentration–time data following i.v. administration of the rhIFNα-2b solution and nanoporous microspheres in mice (n=3).Abbreviation: CS-rhIFNα-2b-CMCS-MS, chitosan rhIFNα-2b carboxymethyl chitosan nanoporous microspheres. |
Tissue distribution
As can be seen from Figure 7A and B, the rhIFNα-2b solution was mainly distributed in the kidney and lung tissues, suggesting that it may have a good effect on renal and pulmonary tumors. After intravenous injection, the distribution of the nanoporous microspheres in the lung (anticipated target site) significantly increased, which was different from the literature,54 where local retention time was prolonged after the administration of the drug-loaded liposomes. The drugs in the nanoporous microsphere group can still be detected in the tissues at 24 hrs, equivalent to the drugs in the stock solution at 6 hrs, indicating that the nanoporous microspheres had a certain sustained-release effect in vivo.
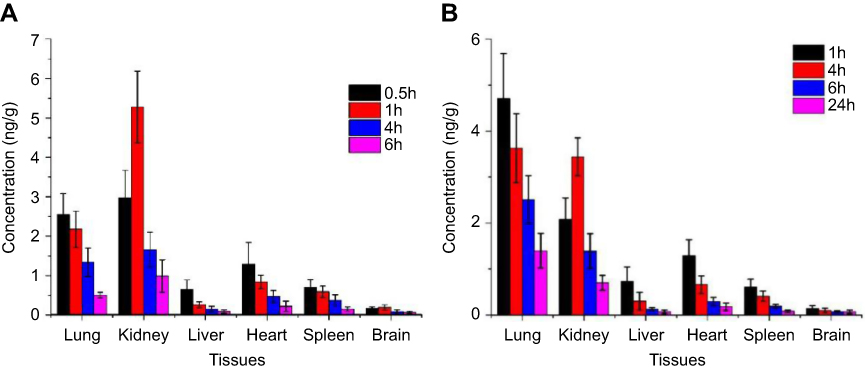 |
Figure 7 Tissue distribution diagram after tail vein injection (ng·g−1) (n=3). (A) histogram of drug concentration in each tissue after 0.5, 1, 4, and 6 hrs after rhIFNα-2b stock solution injection into the tail vein of mice. (B) Histogram of drug concentration in various tissues after the injection of rhIFNα-2b nanoporous microspheres into the tail vein of mice after 1, 4, 6, and 24 hrs. |
As can be seen from Table 5, Ci of the nanoporous microsphere group was 1.85 times that of the solution group, and CS-rhIFNα-2b-CCPM had an obvious lung-targeting effect.
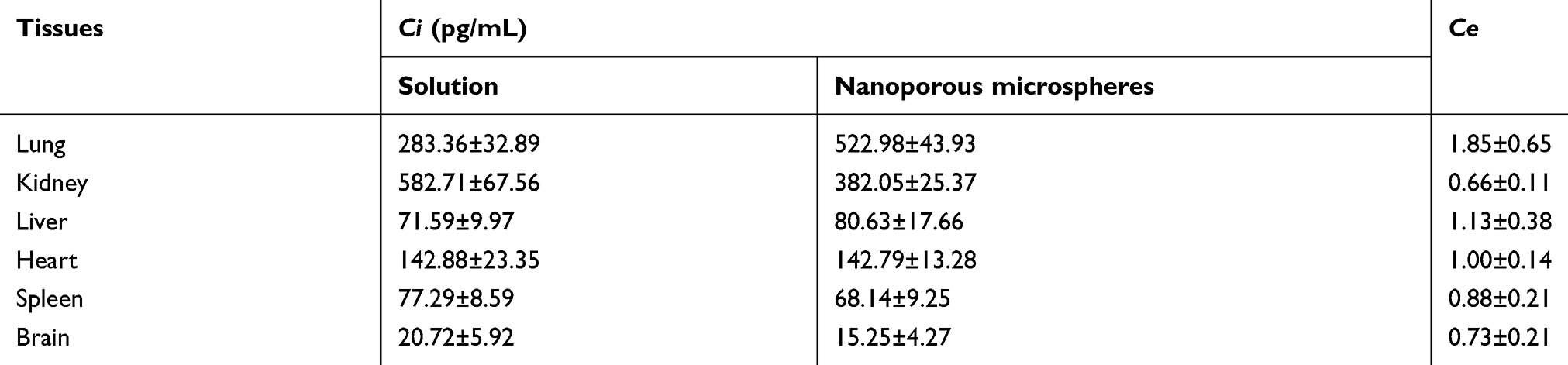 |
Table 5 Target index in mice tissues (mean ± SD, n=3) |
Pharmacodynamic evaluation
As shown in Figure 8A, the growth of tumors significantly slowed down for the stock solution group and the microsphere group compared with the control group. At the end of treatment, the tumor volume of the control group was 1,020.35 mm3, and the tumor weight was 1.37±0.23 g. The tumor volume of the stock solution group was 780.94 mm3 and the tumor weight was 1.05±0.15 g. The tumor volume of the microsphere group was 729.17 mm3, and the tumor weight was 0.97±0.17 g. Tumor inhibition efficiencies of the stock solution group and the microsphere group were 23.4% and 29.2%, respectively. It can be concluded that the tumor volume and weight of the stock solution group and the microsphere group were significantly reduced compared with the control group. Furthermore, the microsphere group showed a better antitumor effect.
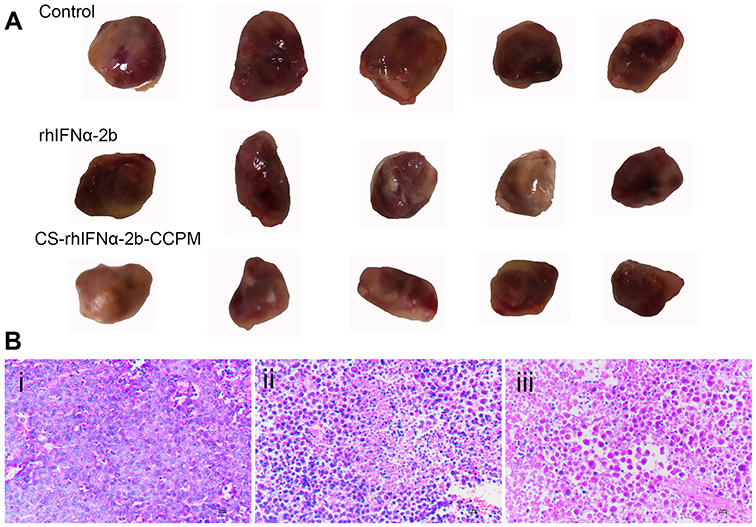 |
Figure 8 The evaluation of the antitumor effect (n=5). (A) The appearance of the tumors. (B) Histopathology slice of tumor (i: the control group; ii: the stock solution group; and iii: the microsphere group).Abbreviation: CS-rhIFNα-2b-CCPM, chitosan rhIFNα-2b carboxymethyl chitosan nanoporous microspheres. |
It can be seen from Figure 8Bi that the tumor cells proliferate actively, with a diffuse distribution, tight arrangement, large cell volume, small nucleus, and dark blue chromatin. Some tumor cells were irregular in shape and size, with pathological mitosis, but there were no typical apoptotic cells. It can be seen from Figure 8Bii that there were obvious apoptotic cells in the tumor cells (the nucleus became smaller or almost disappeared). The uniformity of cell arrangement and distribution was worse, the morphological differentiation intensified, the tumor cell density decreased, the nuclear fragmentation increased, and there were some scattered empty bubbles. Similarly, it can be seen from Figure 8Biii that there were more apoptotic tumor cells (the nucleus became smaller or almost disappeared) while the number of active proliferating cancer cells reduced. Thus, apoptotic tumor cells were not evenly and uniformly distributed with intensified morphological differentiation, nuclear fragmentation and significantly increased scattered vacuoles. The results showed that the microsphere group (CS-rhIFNα-2b-CCPM) had a stronger antitumor effect.
Conclusion
In view of the large molecular weight and poor physical and chemical stability of protein macromolecules, CS-rhIFNα-2b-CCPM was prepared for the first time to achieve a slow drug release in the lungs and enhance the specificity of pharmacological activity. This method has high encapsulation efficiency and high drug activity retention rate. CS-rhIFNα-2b-CCPM had an obvious sustained-release effect and a good targeting ability in vitro and in vivo. The in vitro cell experiments and anti-tumor effects in vivo proved that the prepared microspheres have a better antitumor effect, providing a new direction to study protein-based drug sustained-release preparations.
Acknowledgments
This study was sponsored by The Zhenjiang Social Development Program (project number SH2017003), The Jiangyin Industry Prospective Technique Research and Development Program (project number JY0602A 010101180020PB), The Postgraduate Research & Practice Innovation Program of Jiangsu Province (project number KYCX18_2290), The Research Foundation for Advanced Scholars of Jiangsu University (project number 11JDG122), The China Postdoctoral Science Foundation (project number 2017M610309), and The Training Project of Jiangsu University for Young Key Teachers (project number 5521290003).
Disclosure
There is no interest dispute between the project and the company that the authors were employed by. The authors report no other conflicts of interest in this work.
References
1. Pagán Carrasco S, Arranz Maestro D. Topical interferon alpha-2B topic as the first therapeutic option in a clinical case of conjunctival intraepithelial neoplasia. Archivos De La Sociedad Española De Oftalmología (English Edition). 2017;92(9):442–446. doi:10.1016/j.oftale.2017.03.019
2. Zhang N, Liu ZW, Han QY, Chen J, Lv Y. Xanthohumol enhances antiviral effect of interferon α-2b against bovine viral diarrhea virus, a surrogate of hepatitis C virus. Phytomedicine. 2010;17(5):310–316. doi:10.1016/j.phymed.2009.08.005
3. Coulombe F, Jaworska J, Verway M, et al. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity. 2014;40(4):554–568. doi:10.1016/j.immuni.2014.02.013
4. Hong WS, Jett JR, Sasaki Y, et al. Colony inhibitory effect of recombinant human tumor necrosis factor (rH-TNF) and/or recombinant human interferon (rH-IFN)-alpha, -beta and -gamma on human lung cancer cell lines. Jpn J Clin Oncol. 1987;17(1):49–55.
5. Xu J, Tan L, Goodrum KJ, Kieliszewski MJ. High-yields and extended serum half-life of human interferon alpha2b expressed in tobacco cells as arabinogalactan-protein fusions. Biotechnol Bioeng. 2010;97(5):997–1008. doi:10.1002/(ISSN)1097-0290
6. Yang L, Yang W, Bi D, ZENG Q. A novel method to prepare highly encapsulated interferon-α-2b containing liposomes for intramuscular sustained release. Eur J Pharm Biopharm. 2006;64(1):0–15. doi:10.1016/j.ejpb.2006.03.003
7. Gu JF. Application of ion-exchange resin drug carrier in drug delivery system. Ion Exch Adsorpt. 2010;26(1):89–96.
8. Liu HF, Su XY, Li X, et al. Development of prolonged release microspheres of metformin hydrochloride using ion exchange resins. J Chin Pharm Sci. 2006;15(3):155–161.
9. Liu HF, Zhang S, Nie S, et al. Preparation and characterization of a novel pH-sensitive ion exchange resin. Chem Pharm Bull. 2005;53(6):631–633.
10. Gull I, Samra ZQ, Aslam MS, Athar MA. Heterologous expression, immunochemical and computational analysis of recombinant human interferon alpha 2b. Springerplus. 2013;2(1):264. doi:10.1186/2193-1801-2-264
11. Liang X, Wang H, Tian H, LUO H, CHANG J. Synthesis, structure and properties of novel quaternized carboxymethyl chitosan with drug loading capacity. Acta Physico-Chimica Sinica. 2008;24(2):223–229. doi:10.1016/S1872-1508(08)60011-X
12. Wang J, Chen JS, Zong JY, et al. Calcium carbonate/carboxymethyl chitosan hybrid microspheres and nanospheres for drug delivery. J Phys Chem C. 2010;114(44):18940–18945. doi:10.1021/jp105906p
13. Luo Y, Teng Z, Wang Q. Development of zein nanoparticles coated with carboxymethyl chitosan for encapsulation and controlled release of vitamin D3. J Agric Food Chem. 2012;60(3):836–843. doi:10.1021/jf204194z
14. Mattioli-Belmonte DM, Gigante A, Muzzarelli RAA, et al. N,N-dicarboxymethyl chitosan as delivery agent for bone morphogenetic protein in the repair of articular cartilage. Med Biol Eng Comput. 1999;37(1):130. doi:10.1007/BF02513279
15. Budiraharjo R, Neoh KG, Kang ET. Hydroxyapatite-coated carboxymethyl chitosan scaffolds for promoting osteoblast and stem cell differentiation. J Colloid Interface Sci. 2012;366(1):224–232. doi:10.1016/j.jcis.2011.09.072
16. Chen XG, Wang Z, Liu WS, Park H-J. The effect of carboxymethyl-chitosan on proliferation and collagen secretion of normal and keloid skin fibroblasts. Biomaterials. 2002;23(23):4609–4614.
17. Hiroki A, Tran HT, Nagasawa N, Yagi T, Tamada M. Metal adsorption of carboxymethyl cellulose/carboxymethyl chitosan blend hydrogels prepared by Gamma irradiation. Radiat Phys Chem. 2009;78(12):1076–1080. doi:10.1016/j.radphyschem.2009.05.003
18. Guo BL, Gao QY. Preparation and properties of a pH/temperature-responsive carboxymethyl chitosan/poly(N-isopropylacrylamide) semi-IPN hydrogel for oral delivery of drugs. Carbohydr Res. 2007;342(16):2416–2422. doi:10.1016/j.carres.2007.07.007
19. Yang C, Xu L, Zhou Y, et al. A green fabrication approach of gelatin/CM-chitosan hybrid hydrogel for wound healing. Carbohydr Polym. 2010;82(4):1297–1305. doi:10.1016/j.carbpol.2010.07.013
20. Hu L, Sun Y, Wu Y. Advances in chitosan-based drug delivery vehicles. Nanoscale. 2013;5(8):3103–3111. doi:10.1039/c3nr00338h
21. Gujarathi NA, Rane BR, Patel JK. PH sensitive polyelectrolyte complex of O-carboxymethyl chitosan and poly (acrylic acid) cross-linked with calcium for sustained delivery of acid susceptible drugs. Int J Pharm. 2012;436(1–2):418–425. doi:10.1016/j.ijpharm.2012.07.016
22. Feng C, Wang Z, Jiang C, et al. Chitosan/o-carboxymethyl chitosan nanoparticles for efficient and safe oral anticancer drug delivery: in vitro and in vivo evaluation. Int J Pharm. 2013;457(1):158–167. doi:10.1016/j.ijpharm.2013.07.079
23. Liu Y, Zhao D, Wang JT. Preparation of o-carboxymethyl chitosan by schiff base and antibacterial activity. Adv Mater Res. 2013;647:794–797. doi:10.4028/www.scientific.net/AMR.647.794
24. Narayanan D, Ninan N, Jayakumar R, et al. O-carboxymethyl chitosan nanoparticles for controlled release of non-steroidal anti-inflammatory drugs. Adv Sci. 2014;6(5):
25. Mccarthy JD, Agnone J, Johnson EW. Preparation and characterization of o-carboxymethyl chitosan ion-complexed poly(l-lysine) for drug and gene delivery system. J Korean Ind Eng Chem. 2010;21(6):2267–2292.
26. Liu Z, Jiao Y, Zhang Z. Calcium-carboxymethyl chitosan hydrogel beads for protein drug delivery system. J Appl Polym Sci. 2007;103(5):3164–3168. doi:10.1002/(ISSN)1097-4628
27. Lu G, Sheng B, Wei Y, et al. Collagen nanofiber-covered porous biodegradable carboxymethyl chitosan microcarriers for tissue engineering cartilage. Eur Polym J. 2008;44(9):2820–2829. doi:10.1016/j.eurpolymj.2008.06.021
28. Raghunathan Y, Amsel L, Hinsvark O, Bryant W. Sustained-release drug delivery system I: coated ion-exchange resin system for phenylpropanolamine and other drugs. J Pharm Sci. 2010;70(4):379–384. doi:10.1002/jps.2600700409
29. Chen M. Electrostatic self-assembly of multilayer copolymeric membranes on the surface of porous tantalum implants for sustained release of doxorubicin. Int J Nanomed. 2011;6:3057–3064. doi:10.2147/IJN.S25918
30. Mendes AC, Strohmenger T, Goycoolea F, Chronakis IS. Electrostatic self-assembly of polysaccharides into nanofibers. Colloids Surf A. 2017;531:182–188. doi:10.1016/j.colsurfa.2017.07.044
31. Fu J, Zhang M, Liu L, Xiao L, Li M, Ao Y. Layer-by-Layer electrostatic self-assembly silica/graphene oxide onto carbon fiber surface for enhance interfacial strength of epoxy composites. Mater Lett. 2018;236:69–72. doi:10.1016/j.matlet.2018.10.077
32. Ai H, Jones SA, Villiers MMD, Lvov YM. Nano-encapsulation of furosemide microcrystals for controlled drug release. J Controlled Release. 2003;86(1):59–68. doi:10.1016/S0168-3659(02)00322-X
33. Li B, Zhen XC, Chen LB, et al. Preparation and characterization of porous chitosan microspheres. J Nanjing Univ. 2010;46(2):186–191.
34. Li X, Yuan H, Zhang C, et al. Preparation and in‐vitro/in‐vivo evaluation of curcumin nanosuspension with solubility enhancement. J Pharm Pharmacol. 2016;68(8):980–988. doi:10.1111/jphp.12575
35. Shi XC, Rao CM, Pei DN, et al. Studies on circular dichroism (CD) spectra of recombinant human interferon alpha 2a. Chin J Pharm Anal. 2005;25(10):1169–1172.
36. Nigro E, Stiuso P, Matera MG, et al. The anti-proliferative effects of adiponectin on human lung adenocarcinoma A549 cells and oxidative stress involvement. Pulm Pharmacol Ther. 2019;55:25–30. doi:10.1016/j.pupt.2019.01.004
37. Milligan G, Alvarezcurto E, Watterson KR, et al. Short-chain fatty acids increase expression and secretion of stromal cell-derived factor-1 in mouse and human pre-adipocytes. Hormones. 2014;13(4):532–542. doi:10.14310/horm.2002.1519
38. Zhou Q, Gui S, Zhou Q, Wang Y, Samant R. Melatonin inhibits the migration of human lung adenocarcinoma A549 cell lines involving JNK/MAPK pathway. PLoS One. 2014;9(7):e101132. doi:10.1371/journal.pone.0101132
39. Grela E, Kozłowska J, Grabowiecka A. Current methodology of MTT assay in bacteria – A review. Acta Histochem. 2018;120(4):303–311. doi:10.1016/j.acthis.2018.03.007
40. Zhang X, Xia J, Zhang W, Luo Y, Sun W, Zhou W. Study on pharmacokinetics and tissue distribution of single dose oral tryptanthrin in Kunming mice by validated reversed-phase high-performance liquid chromatography with ultraviolet detection. Integr Med Res. 2017;6(3):269–279. doi:10.1016/j.imr.2017.05.001
41. Shukla DJ, Vyas HA, Vyas MK, Ashok BK, Ravishankar B. A comparative study on chronic administration of Go Ghrita (cow ghee) and Avika Ghrita (ewe ghee) in albino rats. Ayu. 2012;33(3):435. doi:10.4103/0974-8520.108862
42. Bougie DW, Nayak D, Boylan B, Newman PJ, Aster RH. Drug-dependent clearance of human platelets in the NOD/SCID mouse by antibodies from patients with drug-induced immune thrombocytopenia. Blood. 2010;116(16):3033–3038. doi:10.1182/blood-2010-03-277764
43. Van Herck H, Baumans V, Brandt C, et al. Blood sampling from the retro-orbital plexus, the saphenous vein and the tail vein in rats: comparative effects on selected behavioral and blood variables. Lab Anim. 2001;35(2):131–139. doi:10.1258/0023677011911499
44. Lequin RM. Enzyme Immunoassay (EIA)/Enzyme-Linked Immunosorbent Assay (ELISA). Clin Chem. 2005;51(12):2415–2418. doi:10.1373/clinchem.2005.051532
45. Lu L, Lu Y, Zou H, et al. Pharmacokinetics Study of Intereron α-2b PLGA Microspheres in Rats by ELISA. Chin Pharm J. 2008;23.
46. Kutscher HL, Chao P, Deshmukh M, et al. Threshold size for optimal passive pulmonary targeting and retention of rigid microparticles in rats. J Controlled Release. 2010;143(1):31–37. doi:10.1016/j.jconrel.2009.12.019
47. Chen Z, Luo YY, Guo ZF. CA-gel/PLGA composite microspheres loaded protein by SPG membrane emulsification. Acta Scientiarum Naturalium Universitatis Sunyatseni. 2016;55(5):96–102. doi:10.13471/j.cnki.acta.snus.2016.05.017
48. Varma S, Sadasivan C. A long acting biodegradable-controlled delivery of chitosan microspheres loaded with tetanus toxoide as model antigen. Biomed Pharmacother. 2014;68(2):225–230. doi:10.1016/j.biopha.2013.08.009
49. Kaur K, Kush P, Pandey RS, Madan J, Jain UK, Katare OP. Stealth lipid coated aquasomes bearing recombinant human interferon-α-2b offered prolonged release and enhanced cytotoxicity in ovarian cancer cells. Biomed Pharmacother. 2015;69:267–276. doi:10.1016/j.biopha.2014.12.007
50. Hermeling S, Schellekens H, Maas C, et al. Antibody response to aggregated human interferon alpha2b in wild-type and transgenic immune tolerant mice depends on type and level of aggregation. J Pharm Sci. 2010;95(5):1084–1096. doi:10.1002/jps.20599
51. Martyre MC, Beaupain R, Falcoff E. Potentiation of antiproliferative activity by mixtures of human recombinant IFN-α2 and−γ on growth of human cancer nodules maintained in continuous organotypic culture. Eur J Cancer Clin Oncol. 1987;23(7):917–922.
52. Ezzat HM, Elnaggar YSR, Abdallah OY. Improved oral bioavailability of the anticancer drug catechin using chitosomes: design, in-vitro appraisal and in-vivo studies. Int J Pharm. 2019. doi:10.1016/j.ijpharm.2019.05.034
53. Yang F, Shu Y, Yang Y, et al. The pharmacokinetics of recombinant human interferon-alpha-2b poly (lactic-co-glycolic acid) microspheres in rats. J Microencapsulation. 2011;28(6):483–489. doi:10.3109/02652048.2011.586065
54. Yang L, Yang W, Bi D, Zeng Q. A novel method to prepare highly encapsulated interferon-α-2b containing liposomes for intramuscular sustained release. Eur J Pharm Biopharm. 2006;64(1):9–15. doi:10.1016/j.ejpb.2006.03.003
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.