")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Computer-aided vaccine designing approach against fish pathogens Edwardsiella tarda and Flavobacterium columnare using bioinformatics softwares
Authors Mahendran R, Jeyabaskar S, Sitharaman G, Michael D, Vincent Paul A
Received 3 September 2015
Accepted for publication 4 February 2016
Published 23 May 2016 Volume 2016:10 Pages 1703—1714
DOI https://doi.org/10.2147/DDDT.S95691
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Radha Mahendran,1 Suganya Jeyabaskar,1 Gayathri Sitharaman,1 Rajamani Dinakaran Michael,2 Agnal Vincent Paul1
1Department of Bioinformatics, 2Centre for Fish Immunology, School of Life Sciences, Vels University, Pallavaram, Chennai, Tamil Nadu, India
Abstract: Edwardsiella tarda and Flavobacterium columnare are two important intracellular pathogenic bacteria that cause the infectious diseases edwardsiellosis and columnaris in wild and cultured fish. Prediction of major histocompatibility complex (MHC) binding is an important issue in T-cell epitope prediction. In a healthy immune system, the T-cells must recognize epitopes and induce the immune response. In this study, T-cell epitopes were predicted by using in silico immunoinformatics approach with the help of bioinformatics tools that are less expensive and are not time consuming. Such identification of binding interaction between peptides and MHC alleles aids in the discovery of new peptide vaccines. We have reported the potential peptides chosen from the outer membrane proteins (OMPs) of E. tarda and F. columnare, which interact well with MHC class I alleles. OMPs from E. tarda and F. columnare were selected and analyzed based on their antigenic and immunogenic properties. The OMPs of the genes TolC and FCOL_04620, respectively, from E. tarda and F. columnare were taken for study. Finally, two epitopes from the OMP of E. tarda exhibited excellent protein–peptide interaction when docked with MHC class I alleles. Five epitopes from the OMP of F. columnare had good protein–peptide interaction when docked with MHC class I alleles. Further in vitro studies can aid in the development of potential peptide vaccines using the predicted peptides.
Keywords: E. tarda, F. columnare, edwardsiellosis, columnaris, T-cell epitopes, MHC class I, peptide vaccine, outer membrane proteins
Introduction
Most fish are susceptible to various bacterial infections. Fish are in intimate contact with higher number of environmental pathogens. Hence, these pathogens easily attach to and degrade the tissues of fish.1 Degradation of fish tissue and the diseases caused by pathogens constitute the main reason for huge economic losses in aquaculture. Aquaculture is the fastest growing food production industry in the world. Hence, there is a high demand for identifying ways to combat fish diseases. Existing pathogens’ host range and virulence are found to be increasing and this scenario has created a challenge to fish health researchers. Aquaculture researchers are trying to identify more efficacious vaccines and potent prophylactic and therapeutic drugs against bacterial fish diseases.2
Edwardsiella tarda and Flavobacterium columnare are two virulent intracellular bacterial pathogens responsible for causing edwardsiellosis and columnaris in most fish species.
Features of E. tarda and F. columnare
E. tarda belongs to the Enterobacteriaceae family. E. tarda are Gram-negative facultative anaerobic rods that are motile. They possess peritrichous flagella and they have a length of 2–3 μm and diameter of 1 μm.3–5 The organism has been isolated from warmwater fish and it is found worldwide. The mode of transmission of disease is by means of exposure to aquatic environments and animals and by means of ingestion of contaminated fish.6 Most strains of E. tarda are resistant to drugs such as penicillin, polymyxin B, oxacillin, rifampin, and colistin.7,8 When there is an imbalance in environmental conditions, such as the presence of high organic content, high water temperature, and poor water quality, edwardsiellosis occurs in fish.9 The symptoms of infected fish include spiral movement, abnormal swimming, and floating near the surface of water. The mortality rate of the disease is very high among different populations of fish of various age groups. Hence, edwardsiellosis causes serious threats to catfish and flounder farming. This emphasizes the need for the development of new vaccines. Vaccination with an ideal immunogenic protein would be the best way of preventing the diseases rather than using antibiotics.
F. columnare is a highly prevalent Gram-negative rod that is motile in nature. It leads to columnaris disease among freshwater fish throughout the world.10 Several species of fish that are commercially very important are severely affected by columnaris. The mortality rate is very high, and this leads to a drastic economic loss.11,12 Columnaris mostly affects species of salmonids, carp, peach, eel, tilapia, channel catfish and goldfish. The fish affected by columnaris exhibit yellowish brown lesions on their fin, skin, and gills. The bacteria grow in individual gill filaments.13 These considerations lead to the increased need for the development of new vaccines against columnaris.
Immunoinformatic approach for vaccine design
The use of antibiotics to treat diseases has led to antibiotic resistance among pathogens. Currently, vaccines to treat columnaris are available. However, there is a danger of reversion in case of live attenuated vaccines.14 In this study, we have identified potential T-cell epitopes that can be used as effective vaccine candidates by means of using immunoinformatic tools, software, and databases. T-cells are reported to be mediators exhibiting competent immune responses elicited by the target vaccine.15 Such reports have catalyzed research into and development of in silico immunoinformatics methods for further exploring T-cell epitopes in peptide sequences.16 The first step in vaccine designing to identify the T-cell epitope is to determine which peptide of the proteome of the virulent bacterial pathogen will bind to the major histocompatibility complex (MHC) molecules. The strength of MHC molecule binding to epitopes is an important parameter in determining T-cell immunogenicity.17 In this study, we have modeled peptide–MHC complexes and studied their interactions using appropriate immunoinformatics tools.
Outer membrane proteins and cytotoxic T-cells
We analyzed the sequences of the outer membrane proteins (OMPs) of the pathogens based on their inherent ability to elicit immune responses during the time of infection. Cytotoxic T-lymphocyte (CTL) epitopes were predicted from the OMPs of the virulent pathogens. CTLs are responsible for killing infected cells. The infected antigen-presenting cell activates the T-cell to become an effector cell and further, the T-cell can kill any target cell infected by the pathogen. CTLs attack the target cells and make the target undergo apoptosis or self-destruction.
MHC protein–peptide complex
The MHC molecule binds to the peptide fragments of the pathogens and, later, the peptides are displayed on the cell surface. The CTLs recognize the peptide–protein complex and kill the infected cell. The underlying mechanism behind the antigen processing and its presentation to the T-cell is as follows. Peptides are processed by the proteosome in the cytosol and transported to the endoplasmic reticulum, the site of MHC synthesis. MHC proteins bind to the peptides at the endoplasmic reticulum. A protein complex called T cell activating protein (TAP) moves the peptide into the MHC I molecule and later it is transported to the cell surface. The terminal amino group of the peptide binds to the invariant pocket present at one end of the class I MHC groove. The terminal carboxyl group of the peptide binds to the invariant pocket present at the other end of the groove of the class I MHC molecule. Both the N- and C-termini of the peptide are secured by hydrogen bond interactions and networks. Anchor amino acids of the peptide bind to the specificity pockets of the groove. The T-cell receptor recognizes the side chains of the other amino acids pointing outward. Diverse sequences of peptides bind to each allelic form of a class I MHC protein. The specificity pockets that differ along the groove bind to the specific amino acids of the side chain of the peptide. Later, each allelic form binds and presents a specific characteristic of the peptides.18 The MHC molecule promiscuously binds peptides tightly because the virulent pathogens will try to mutate the epitope of the MHC molecule. Thus, MHC molecule binds to a variety of peptides with high affinity.19
Methods
Protein sequence retrieval
Ethical approval was not required from the Institutional Animal Ethics Committee of Vels University as the data is available in the public domain, and therefore the data is anonymized. OMPs were chosen for this study owing to their significant antigenic and high immunogenic properties. OMPs were retrieved from the UniProt database, which is a highly annotated protein sequence database that contains accurate structural and functional information of proteins. The amino acid sequences of the OMPs were retrieved in FASTA format from UniProt.20,21
OMP of E. tarda: TolC
TolC (Swiss-Prot: Q4ACC9) is an OMP responsible for the transport of toxic compounds and virulent proteins. It has the capacity to induce resistance in bacteria against different antibiotics.22,23
OMP of F. columnare: OMPA FCOL_04620
OMPA FCOL_04620 (Swiss-Prot: G8X7P2) is a protein in the outermost lipid bilayer of the cell envelope, which contains protein and lipopolysaccharide structures that are highly toxic.24
Prediction of antigenic sites
Prediction of antigenic sites was performed using Kolaskar and Tongaonkar antigenicity tool (http://tools.immuneepitope.org/tools/bcell/iedb_input). A semiempirical approach is used in the prediction of peptides, which depends on various physical and chemical properties of the amino acids and is also based on the frequency of occurrence of residues in experimentally known epitopes. It has the efficiency to predict antigens with ~75% accuracy, and the threshold value is 1.000.25
Prediction of CTL epitopes
CTL epitopes were predicted using the NetCTL.1.2 server (http://www.cbs.dtu.dk/services/NetCTL). CTLs destroy the infected cells, and their activation normally takes place on the surface of antigen-presenting MHC molecules.26 The NetCTL 1.2 server integrates prediction of TAP transport efficiency, MHC class I binding, and proteasomal C-terminal cleavage. The FASTA sequence of the organism was given as input. Peptide length and human leukocyte antigen (HLA) alleles were selected and submitted. T-cell epitopes were generated as output. Artificial neural network is used to predict MHC class I binding and proteasomal C-terminal cleavage. Weight matrix is used to predict TAP transport efficiency.
Modeling and docking of protein–peptide complex
MODPROPEP tool was used for the modeling of protein–peptide complexes.27 Peptide sequence was given as input, and the crystal structure of MHC alleles (HLA-A0201 and HLA-B2705) taken from Protein Data Bank (PDB) was used as a template for modeling the protein–peptide complex.
Rosetta FlexPepDock tool was used for high-resolution peptide docking (http://flexpepdock.furmanlab.cs.huji.ac.il). FlexPepDock uses the Rosetta fragment library and a representation of the receptor and peptide structures for simultaneous docking and de novo folding of peptides. The space of the backbone conformation of the peptide and orientations of a rigid body on the receptor surface of a binding site are sampled simultaneously and efficiently. The final results are reported as high-resolution models.28 The peptide structure is accurately refined by FlexPepDock, starting from up to 5.5 Å root mean square deviation (RMSD) of its native conformation. It also allows side chain flexibility of receptor and complete flexibility of the peptide.29 The overall conformation of the peptide in a centroid mode is detected by using fragment based sampling.30
Input model
The modeled complex from MODPROPEP was given as input PDB structure for docking analysis.
Selection of models
Top-scoring models were selected as models for interaction study. The Rosetta cluster application results in clusters of 500 top-scoring models with cluster radius cutoff of 2 Å peptide backbone atom RMSD. A representative model is selected from each cluster according to the top energy score and, finally, all clusters are ranked based on the energy.31
Energy function
The energy functions and energy calculations are well calibrated in the Rosetta protocol used by FlexPepDock. The Rosetta library consists of fragments of proteins derived from solved protein structures. The Dunbrack Rotamer library and the Rosetta side chain repacking protocol were used.32,33
Interaction study
The hydrogen bond interactions between the peptide and the MHC protein complex were studied using Argus Lab software.
Docked peptide–protein complexes obtained from FlexPepDock were visualized in Argus Lab. The peptide residues bound in the groove of the MHC complex were selected and grouped separately using the option “Make a group from the selected residues”. A group consisting of the peptide residues was obtained. Hydrogen bond interaction between the peptide and the MHC protein was retrieved using the option “Show hydrogen bonds”. Hydrogen bonds, along with their atomic distances, are displayed in the peptide–MHC complex. The stability of the peptide–protein complex was evaluated using the number of hydrogen bonds formed between the complexes.
Results
Predicting antigenic sites in OMPs is of major importance for synthesis of peptide vaccines. We predicted the antigenic sites of the OMPs of E. tarda and F. columnare using Kolaskar and Tongaonkar antigenicity tool.
Antigenic sites in OMP of E. tarda
There were six antigenic sites predicted in the OMP of E. tarda (Table 1). Proline at the eighth position, found in the antigenic peptide from positions 4 to >24 (KKLLPLLIGLSLSGFSALSQA), was identified as having the maximum residual score of 1.164, well over the threshold score of 1.000. It should be noted that the amino acids Gln (Q) and Ala (A) from this antigenic site were also predicted as comprising a CTL epitope, thus significantly enhancing the scope of amino acids Q and A to be treated as potential peptide vaccine candidates.
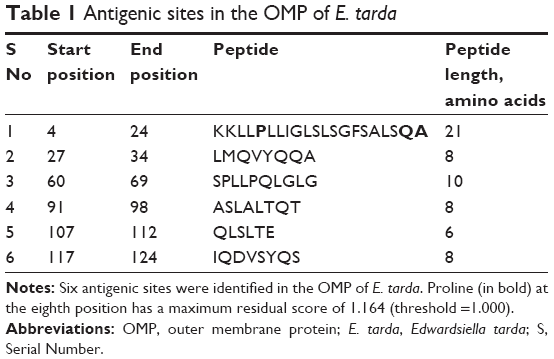 | Table 1 Antigenic sites in the OMP of E. tarda |
Antigenic sites in OMP of F. columnare
We identified 31 antigenic sites from OMPA FCOL_04620 of F. columnare (Table 2). Phenylalanine at the seventh position found at the antigenic site IILFTVLGLSLHGG had a maximum residual score of 1.170. The longest peptide was a 26-residue peptide GDSYYYNAKLVDAHKWYAELFALGET from the 61st position to >86th position. We found that the amino acids Glu (E) and Thr (T) at positions 85 and 86 also exhibited the immunogenic properties of CTL epitopes. Furthermore, the amino acids Phe (F) and Ala (A) at positions 288 and 289, as well as the amino acids from positions 574–586, namely, His (H), Thr (T), Asp (D), Cys (C), Arg (R), Gln (Q), Thr (T), Val (V), and Ala (A), were also predicted to be CTL epitopes.
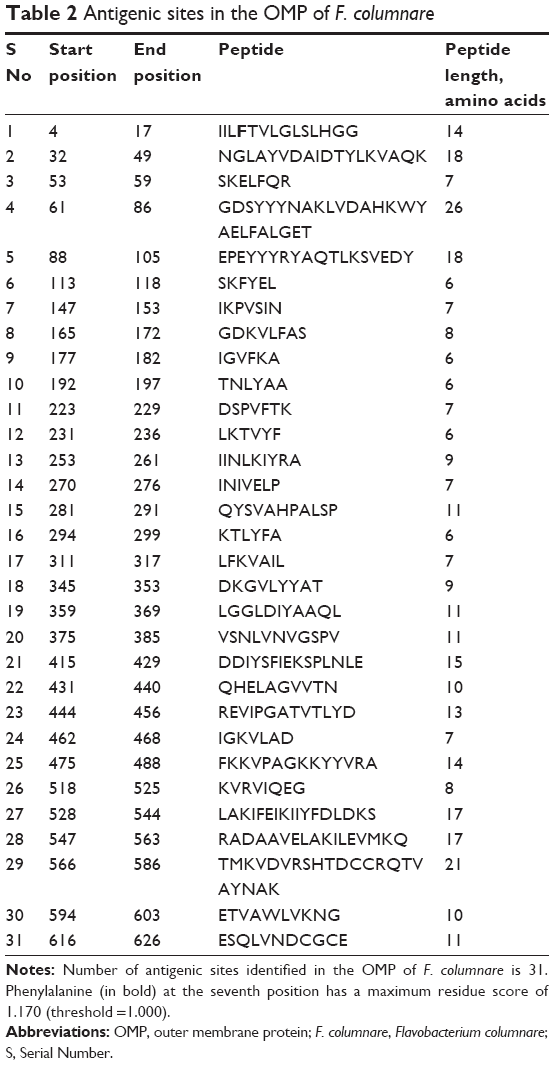 | Table 2 Antigenic sites in the OMP of F. columnare |
CTL epitope prediction
CTL epitopes are potential candidates for peptide vaccine design for various diseases. We predicted CTL epitopes using NetServer 1.2 prediction tool. Peptides were predicted based on their MHC binding affinity, proteasomal C-terminal cleavage, and transport affinity. Prediction was carried out using MHC supertype A1.
We identified four peptide sequences whose prediction scores were >0.75000 as CTL epitopes (Table 3). As stated earlier, the amino acids Gln and Ala were also predicted as antigenic sites. Hence, QAENLMQVY, one of the CTL epitopes predicted from E. tarda, can be considered a potential vaccine candidate.
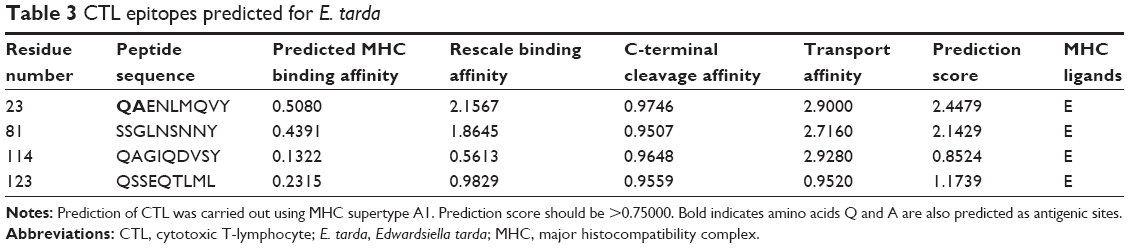 | Table 3 CTL epitopes predicted for E. tarda |
Eighteen peptide sequences from the OMP of F. columnare were predicted as CTL epitopes (Table 4). We found that most of the peptide sequences had more than one amino acid residues that were antigenic in nature. Moreover, we also found a lot of mismatch between the predicted antigenic sites and CTL epitope peptides. For example, YVDAIDTYL and QTLKSVEDY, epitopes with residue numbers 36 and 97, respectively, were also predicted as antigenic sites. But the docking results did not throw up the peptides as vaccine candidates. On the other hand, the predicted epitopes FAKEINSKY and NSDQYSVAH, with residue numbers 212 and 278, respectively, had no antigenic sites predicted but were found to be bound well with MHC class I when docked, showing significant hydrogen bonding, indicating that the peptides might be worthy candidates for peptide vaccine design. Similar was the case for the peptide sequences from residue numbers 151, 227, 289, 465, and 485, wherein we identified the amino acids (in bold) to be antigenic in nature, but not desirable as candidates for vaccine design. One interesting finding that can be considered significant is the peptide sequence HTDCRQTVA from the residue number 574. All the amino acids from the CTL epitope were also predicted as antigenic sites in our study, and hence, we can confidently say that the herein-mentioned epitope can elicit immune responses against these pathogens very effectively.
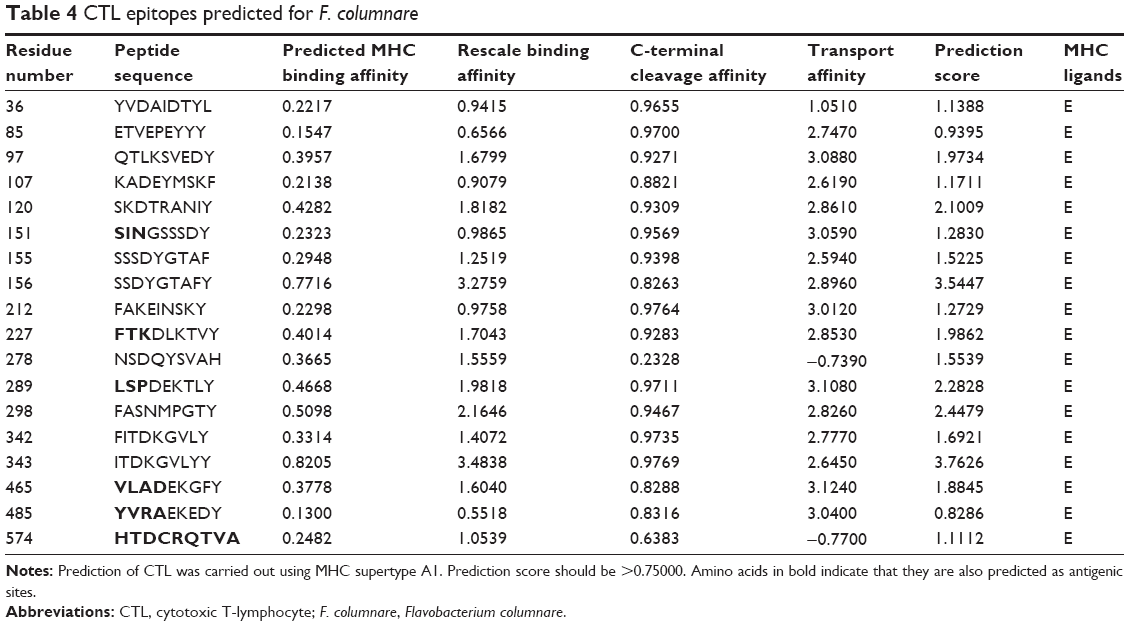 | Table 4 CTL epitopes predicted for F. columnare |
Docking results
The predicted CTL epitopes of the OMPs of E. tarda and F. columnare were docked with MHC class I proteins using FlexPepDock server. It is a high-resolution docking server implemented in the Rosetta framework. The docking server provided well-calibrated energy functions and efficient energy calculations. The stability of docked structures was studied using Argus Lab. Argus Lab software helped us in the hydrogen bond interaction studies between the peptide and the protein complex, thus enabling in-depth understanding of the docked structure.
Docking results for TolC of E. tarda
Four CTL epitopes predicted from TolC of E. tarda were docked with MHC class I HLA-A0201. We identified two peptides QAENLMQVY and SSGLNSNNY, which bound well with HLA-A0201. Figure 1A shows the docked structure of QAENLMQVY–MHC bound complex. We identified 12 hydrogen bond formations with interatomic distances of ~2.8–3.0 Å, thus giving stability to the docked structure (Table 5). Significant H-bonding interactions were seen between the residues Asn, Gln, Tyr, Val, Glu, and Ala of the peptide and Thr, Asp, Trp, Tyr, Gln, and Glu of the MHC protein. The docked structure of SSGLNSNNY is shown in Figure 1B. We identified four hydrogen bond formations between the peptide and the protein (Table 6). Residues Ser, Gly, and Tyr had significant hydrogen-bonding interactions with Glu, Tyr, Thr, and Asp of the MHC protein.
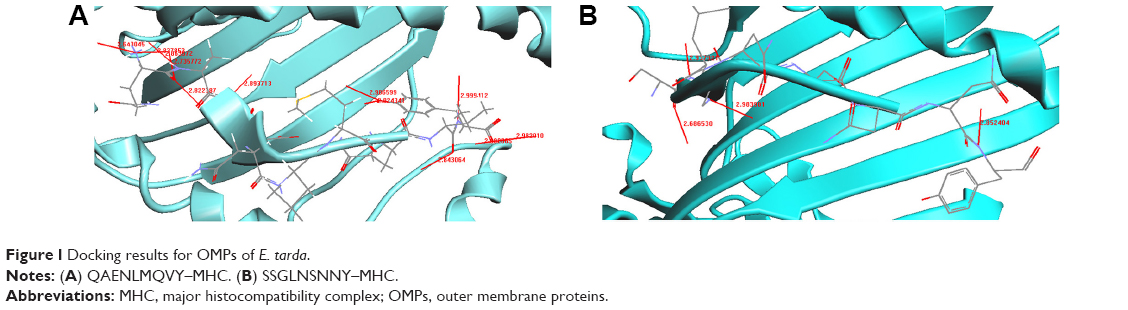 | Figure 1 Docking results for OMPs of E. tarda. |
 | Table 5 Peptide QAENLMQVY–MHC HLA-A0201 interaction |
 | Table 6 Peptide SSGLNSNNY-MHC HLA-A0201 interaction |
Docking results for OMPA FCOL_04620 of F. columnare
Out of 18 epitopes predicted, five epitopes, namely, ETVEPEYYY, FAKEINSKY, FASNMPGTY, HTDCRQTVA, and NSDQYSVAH, bound well with MHC-HLA-B2705 and exhibited excellent protein–peptide interactions.
The docked structure of the peptide ETVEPEYYY with the MHC protein is shown in Figure 2A. It can be seen from Table 7 that the overall stability of the structure is well preserved by the formation of eight H-bonds. The amino acid residues that participated in the bonding were Tyr, Glu, and Thr from the MHC and Asn, Tyr, Thr, and Trp from the peptide. The docked structure of the FAKEINSKY–MHC complex is shown in Figure 2B. We identified 15 H-bonds between the peptides (Phe, Ile, and Ala) and the MHC protein (Tyr, Asn, Thr, and Trp) (Table 8). It should once again be reiterated that although this peptide is considered to be one of the peptide vaccine candidates, none of the amino acid residues are predicted to be antigenic. Figure 2C represents the docked structure of the FASNMPGTY–MHC complex. The structure is well stabilized by the formation of 12 H-bonds (Table 9). By analyzing this peptide, we found that the amino acid residues Phe and Ala are capable of eliciting good immune responses because they are also predicted to be antigenic sites. The interaction between the peptide HTDCRQTVA and the MHC complex is shown in Figure 2D. The docked structure is well stabilized because of the formation of 16 H-bonds (Table 10). The structure is well conserved because of the presence of a greater number of polar residues such as His, Thr, and Arg in the peptide. This peptide should be noted as being a significant finding because all amino acid residues are predicted to be antigenic in nature and hence the chance of eliciting better immune responses is more compared to other peptides. The docked structure of NSDQYSVAH–MHC is shown in Figure 2E and their respective hydrogen bond interactions are shown in Table 11. We identified many polar and hydrophobic residues that take part in hydrogen bonding. Though this peptide has been predicted as a worthy vaccine candidate by means of docking studies, we are skeptical regarding the finding because none of the amino acids in the peptide sequence exhibit the property of antigenicity and immunogenicity.
 | Table 7 Peptide ETVEPEYYY–MHC HLA-B2705 interaction |
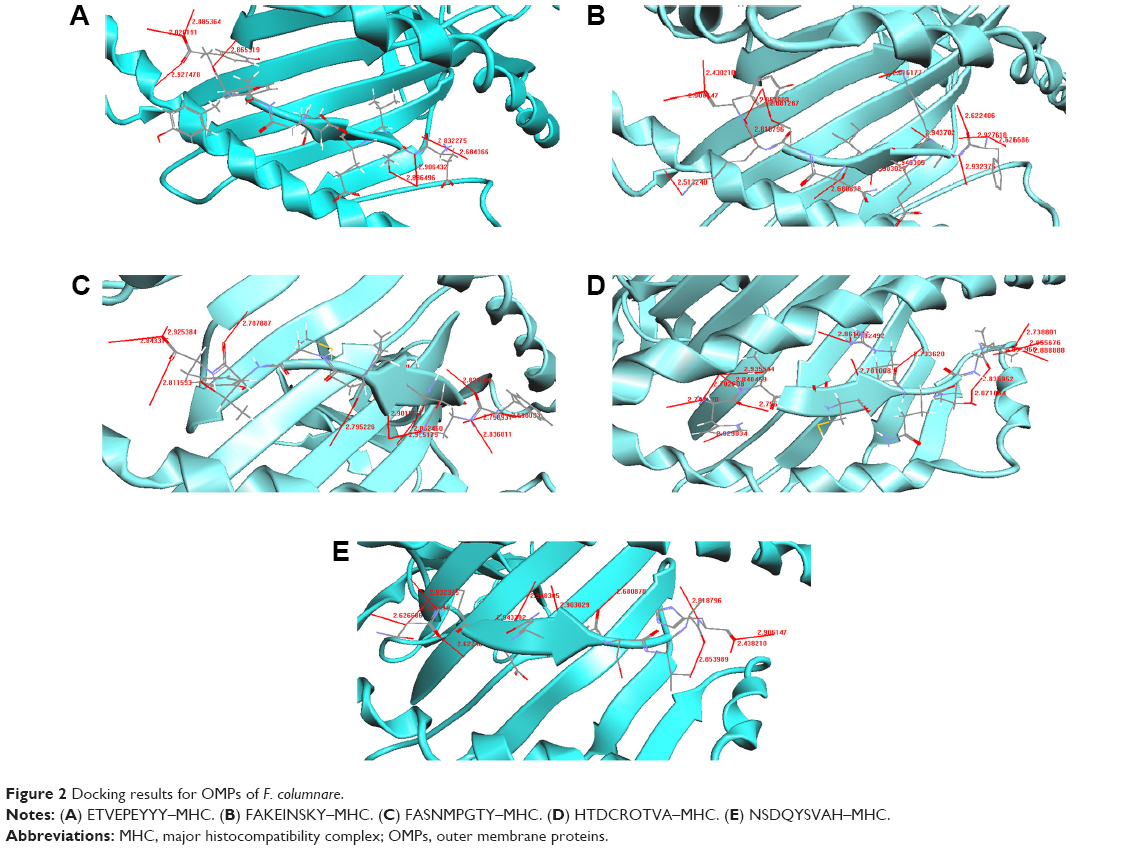 | Figure 2 Docking results for OMPs of F. columnare. |
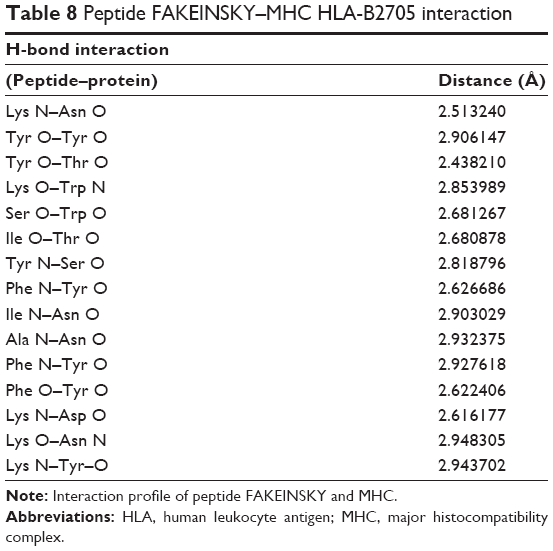 | Table 8 Peptide FAKEINSKY–MHC HLA-B2705 interaction |
 | Table 9 Peptide FASNMPGTY–MHC HLA-B2705 interaction |
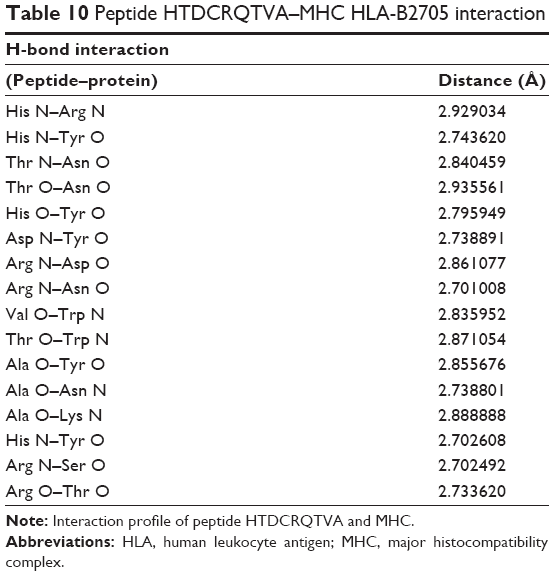 | Table 10 Peptide HTDCRQTVA–MHC HLA-B2705 interaction |
 | Table 11 Peptide NSDQYSVAH–MHC HLA-B2705 interaction |
Discussion
The main aim of this study was to identify potential T-cell epitopes for designing peptide vaccines. An immunoinformatics approach was carried out to perform this study. In silico immunoinformatics tools were used to perform prediction analysis and docking between the peptide and the protein.
Analysis of pathogenomics on a genome-wide scale with the help of in silico immunoinformatics tools aids in the identification of new vaccines.34 Conventional experimental approaches have several limitations. The complete spectrum of potential antigens is analyzed by using in silico immunoinformatics study. The antigens that cannot be expressed in vitro due to the nonfeasibility of pathogen culturing can also be analyzed in silico. Several immunoresearch groups have reported in silico vaccine candidates, which have produced promising preclinical results.35,36
We have identified potential T-cell epitopes that bind with MHC protein from the OMPs of the infectious pathogens E. tarda and F. columnare. The two potential E. tarda epitopes from that can be treated as candidates for peptide vaccine production are QAENLMQVY and SSGLNSNNY. Five F. columnare epitopes that can be used as potential targets for peptide vaccine design were identified. The peptides are ETVEPEYYY, FAKEINSKY, FASNMPGTY, HTDCRQTVA, and NSDQYSVAH.
The prediction of antigenic sites helps in identifying potential residues from the OMPs. There are six antigenic peptides predicted in the OMP of E. tarda and the OMP of F. columnare has 31 predicted peptides. These residues are antigenic in nature and can be treated as potential targets for vaccine design.
T-cell epitope prediction is a milestone in the field of in silico vaccine designing.37 Several research groups that have been involved in development of different vaccines against various infectious diseases, including autoimmune disease and cancer, have reported excellent results by using in silico epitope prediction methods.38 Because the in silico process reduces the number of in vitro experimental techniques, CTL epitope prediction by this method serves as an important tool in the vaccine designing approach.
Four peptide sequences were identified as epitopes in the OMP of E. tarda. The amino acids Gln and Ala serve also as potential antigenic sites, as can be noted from Table 1. Eighteen peptide sequences were identified as epitopes in the OMP of F. columnare. It must be noted that most of the peptide sequences had more than one amino acid residue that was antigenic in nature.
Among the four peptide sequences identified as epitopes in E. tarda, two epitopes – QAENLMQVY and SSGLNSNNY – were identified as potential candidates for peptide vaccine synthesis. When docked with MHC–HLA alleles, these peptides bound well with the MHC protein and had excellent hydrogen bond interactions with them. Similarly, the five epitopes from the OMP of F. columnare – ETVEPEYYY, FAKEINSKY, FASNMPGTY, HTDCRQTVA, and NSDQYSVAH – were selected as excellent candidates for peptide vaccine synthesis. This analysis was performed first on the basis of the docking between peptide and protein and, later, on the basis of the stability of the docked complexes. For understanding protein folding, protein movement, and protein structure, hydrogen bonds and their relative strengths are very important.
The peptide binding site is located in a groove between the α1 and α2 domains, bound by two α-helices on top of the eight-strand antiparallel β-sheets as the binding floor. In peptide–MHC bound complexes, the N- and C-termini of the peptide are nestled into the MHC protein, with extensive hydrogen bond networks and salt bridges. The central portion of the peptide bulges out and is more exposed to the surface.
Most of the epitope prediction methods depend on structural information and use algorithms that predict potentially promiscuous binders. A stable complex of ligands and receptors is formed when there is proper geometric and electrostatic complementarity between them; only then can the MHC molecules recognize the antigens. Several computational studies use a protocol that aligns the known sequences of the MHC molecule and identifies residues that favor the binding of peptides.39,40 This information, along with crystallographic knowledge, serves as a guide to understanding the principle behind peptide–MHC recognition.39,40
Docking is a widely used important methodology in bioinformatics for studying interactions between molecules.41 Earlier, docking studies were only used for analyzing peptides that bind only with class I MHC molecules.42,43 Later, a research group investigated residues with different physical properties and evaluated the properties of acceptable residues.44 Furthermore, a study45 designed peptides with high binding affinity by using a genetic algorithm and later their binding energy values were reported.
After the in vitro experimental identification of the binding affinity of peptides, there was a good correlation observed between the in silico predicted and experimentally identified affinity values. Later on, docking was used to investigate peptides binding to class II MHC molecules.46 Thus, docking serves as an important tool in investigating the interaction between a T-cell receptor and an MHC–ligand complex.47,48
Interaction profile of TolC from E. tarda
The docking results for OMP TolC are shown in Figure 1A and B. Peptides QAENLMQVY and SSGLNSNNY are nestled in between the grooves of the MHC. The residues Asn, Gln, Tyr, Val, Glu, and Ala of the peptide QAENLMQVY show significant H-bonding with the MHC protein residues Thr, Asp, Trp, Tyr, Gln, and Glu (Table 5). There are 12 hydrogen bonds between in the peptide–protein complex. Typically, hydrogen bonds in peptides are ~2.8–3 Å in length between N and O atoms. Accordingly, from the table, it can be noted that the distances between N and O atoms for the residues Tyr N–Asp O, Val N–Trp O, Glu N–Tyr O, Asn N–Gln O, and Ala N–Glu O are ~2.8–3 Å, ensuring the stability of the protein–peptide complex.
In the SSGLNSNNY–MHC bound complex, residues Ser, Gly, and Tyr of the peptide had significant H-bonding interactions with the MHC residues Glu, Tyr, Thr, and Asp (Table 6). Four hydrogen bonds formed between the peptide and the MHC protein ensure the needed stability. The interatomic distances between the atoms are ~2.8–3 Å, thereby holding the complex rigidly.
Interaction profile of OMPA FCOL_04620 of F. columnare
Peptides ETVEPEYYY, FAKEINSKY, FASNMPGTY, HTDCRQTVA, and NSDQYSVAH bound well with MHC and exhibit excellent protein–peptide interactions.
The interaction between ETVEPEYYY and MHC is shown in Figure 2A. Residues Tyr, Glu, and Thr form hydrogen bonds with the MHC protein residues Asn, Tyr, Trp, and Asn (Table 7). Tyr and Asn are polar residues that participate in hydrogen bonding. They cover the surface of the molecule and are in contact with the solvent due to their ability to form hydrogen bonds. The overall stability of the complex is well preserved because of the formation of eight hydrogen bonds.
The FAKEINSKY–MHC complex is shown in Figure 2B. Peptides use more hydrogen bonds than proteins in binding to their protein partner. The residues Lys, Tyr, Ser, Ile, Ala, and Phe of the peptide FAKEINSKY show significant H-bonding with the MHC protein residues Asn, Tyr, Trp, Thr, Asn, and Tyr. In this complex, there are as many as 15 hydrogen bonds formed between the peptide and the MHC complex (Table 8). Many of the hydrogen bonds involve the peptide backbone. Residues Phe, Ile, and Ala form the core of the complex because of their strong hydrophobicity. Polar residues such as Tyr, Asn, Thr, and Trp of the protein complex participate in hydrogen bonding with the peptide, thus providing structural rigidity to the complex.
The FASNMPGTY–MHC bound complex is shown in Figure 2C. It must be noted that almost all residues of the peptide, namely, Thr, Tyr, Met, and Ser, are polar amino acids. The residues Thr, Tyr, Met, Ser, Ala, and Phe of the peptide FASNMPGTY show significant H-bonding with the MHC protein residues Trp, Tyr, Thr, Asn, Asn, and Tyr. They are in contact with the solvent due to their ability to form hydrogen bonds. The interactions between the polar residues and their respective protein residues are often crucial for the stabilization of the complex. Hydrophobic residues Phe and Ala form the core of the complex (Table 9). The FASNMPGTY–MHC complex is stabilized by the formation of 12 hydrogen bonds.
Peptide HTDCRQTVA was docked with MHC (Figure 2D). The hydrogen bonds between the peptide and MHC protein are shown in Table 10. The residues His, Thr, Arg, Val, Ala, and Asp of the peptide HTDCRQTVA show significant H-bonding with the MHC protein residues Arg, Asn, Asn, Trp, Tyr, and Tyr. The structure is stabilized by the formation of 16 hydrogen bonds. The structure is conserved because of the presence of greater number of polar residues such as His, Thr, and Arg in the peptide. Most of the complexes have a hydrophobic core that is not accessible to the solvent, and hydrophobic amino acids build up the hydrophobic core. In this complex, the hydrophobic core contains Val and Ala residues. This peptide is a potential candidate for vaccine synthesis also because of the fact that the residues of this peptide are predicted to be antigenic sites (Table 2, residue number 574).
The potential epitope NSDQYSVAH and its MHC-bound complex are shown in Figure 2E. Their respective interactions can be studied from Table 11. The residues His, Ala, Asp, Tyr, Ser, Asn, Gln, Val, and Glu of the peptide NSDQYSVAH show significant H-bonding with the MHC protein residues Tyr, Trp, Thr, Asn, Asn, and Tyr. The interaction study shows that there are 12 hydrogen bonds formed between the peptide and the MHC protein, and the complex’s stability is enhanced because of the presence of large number of hydrogen bonds. There are numerous polar and hydrophobic residues that take part in the bonding between the peptide and the protein. Although this peptide is identified as a potential T-cell epitope because of its binding and interaction with the MHC complex, it must be noted that none of the residues from the peptide are predicted to be an antigenic site (Table 2). Similar is the case for two other predicted epitopes, SSGLNSNNY and FAKEINSKY. Hence, it is a wait-and-watch scenario until the epitopes are taken forward for in vitro peptide vaccine synthesis.
A recent study reported that in silico analysis enhances the precision and identification of functional CTL epitopes.49 The authors predicted the CTL epitopes of conserved hepatitis C virus (HCV) proteins and measured their binding affinity with MHC class I molecules in vitro. They also determined the immunogenicity of the identified epitopes in vivo to verify the protective HCV CTL epitopes. They validated in silico approaches and concluded that potential CTL epitope candidates for therapeutic vaccine development can be identified and narrowed down. They have also suggested combining several algorithms for MHC class I and proteasomal cleavage site prediction for more accurate CTL epitope prediction. These findings show the significance of the in silico approach for peptide vaccine synthesis and its practical application in vitro.49 We hope that the predicted CTL epitopes from our study, when taken for in vitro experiments, will produce peptide vaccines with therapeutic usages.
Conclusion
The development of vast genome sequence databases along with structural and genomic data, coupled together with computational tools, aids in the design and discovery of novel vaccine candidates. Recently, many refined assays that measure the T-cell responses against various vaccine candidates have been developed, which have facilitated the progress in vaccine-designing research.49 This study identified potential peptides from the OMPs of fish pathogens, which bound well with MHC class I alleles. It can be noted from the results that most of the peptides bound well between the α1 and α2 grooves of the MHC class I complex. There were significant hydrogen bonding interactions between the residues of the peptides and the MHC. The peptides QAENLMQVY and SSGLNSNNY from the OMP of E. tarda and the peptides ETVEPEYYY, FAKEINSKY, FASNMPGTY, HTDCRQTVA, and NSDQYSVAH from F. columnare showed excellent peptide–protein interactions.
The designing of peptide vaccines in silico is a very novel approach in computer-aided vaccine design. By means of different in silico approaches, an attempt has been made in identifying potential peptides that can be used for designing a peptide vaccine. We need to validate the predicted epitopes by proceeding with in vitro experiments. Much of the research and most of the studies in recent times have concentrated on the successful transition of in silico peptide vaccines to in vitro peptide vaccines that have enormous therapeutic use. The therapeutic uses of peptide-based vaccines range from the treatment of allergic diseases, autoimmune diseases, and asthma to combating cancers such as lung cancer, pancreatic cancer, gastric cancer, prostate cancer, and breast cancer. Peptide vaccines are widely used because of their low molecular weight, specificity toward target tumor cells, and low toxicity toward normal tissues. The first standard vaccine for the treatment of prostate tumor, accepted by the US Food and Drug Administration is sipuleucel-T.50 Clinical trials now focus on administering a combination of peptides and nanomaterials using an improved delivery system because clinical trials using peptide-alone therapy showed limited efficacy.50 We are moving forward to conduct further in vitro research on the CTL epitopes predicted in this study at the Centre for Fish Immunology, School of Life Sciences, Vels University, based on the positive and encouraging results obtained from the previous in vitro experiments. We conclude that the predicted CTL epitopes possess huge therapeutic potential, with higher scope in the future, and they can be considered for testing in clinical trials by various immunologists involved in vaccine research.
Acknowledgment
We acknowledge Vels Institute of Science, Technology, and Advanced Studies (VISTAS) for providing us with the required infrastructure and support system needed.
Disclosure
The authors report no conflicts of interest in this work.
References
Ellis E. Innate host defense mechanisms of fish against viruses and bacteria. Dev Comp Immunol. 2001;25(8–9):827–839. | ||
Sudheesh PS, Al-Ghabshi A, Al-Mazooei N, Al-Habsi S. Comparative pathogenomics of bacteria causing infectious disease in fish. Int J Evol Biol. 2012;2012:457264. | ||
Abbott SL. Klebsiella, Enterobacter, Citrobacter, Serratia, Plesiomonas, and other Enterobacteriaceae. Man Clin Microbiol. 2007;9:698–715. | ||
Plumb JA. Edwardsiella septicaemias. Fish Dis Disord. 1999;3:479–521. | ||
Mohanty BR, Sahoo PK. Edwardsiellosis in fish: a brief review. J Biosci. 2007;32:1331–1344. | ||
Stock I, Wiedemann B. Natural antibiotic susceptibilities of Edwardsiella tarda, E. ictaluri, and E. hoshinae. Antimicrob Agents Chemother. 2001;45(8):2245–2255. | ||
Farmer JJ, Boatwright KD, Janda JM. Enterobacteriaceae: introduction and identification. Man Clin Microbiol. 2007;9:649–669. | ||
Park SB, Aoki T, Jung TS. Pathogenesis of and strategies for preventing Edwardsiella tarda infection in fish. Vet Res. 2012;43:67. | ||
Woo PTK, Bruno DW. Viral, bacterial and fungal infections in Edwardsiella septicaemias. Fish Dis Disord. 2011;3(2):570–605. | ||
Tekedar HC, Karsi A, Gillaspy AF, et al. Genome sequence of fish pathogen Flavobacterium columnare ATCC 49512. J Bacteriol. 2012;194(10):2763–2764. | ||
Declercq AM, Haesebrouck F, Van den Broeck W, Bossier P, Decostere A. Columnaris disease in fish: a review with emphasis on bacterium-host interactions. Vet Res. 2013;44:27. | ||
Hawke JP, Thune RL. Systemic isolation and antimicrobial susceptibility of Cytophaga columnaris from commercially reared channel catfish. J Aquat Anim Health. 1992;4(2):109–113. | ||
Zhu W, Yang G, Zhang Y, Yuan J, An L. Generation of biotechnology-derived Flavobacterium columnare ghosts by PhiX174 gene E-mediated inactivation and the potential as vaccine candidates against infection in grass carp. J Biomed Biotechnol. 2012;2012:760730. | ||
Gianfrani C, Oseroff C, Sidney J, Chesnut RW, Sette A. Human memory CTL response specific for influenza A virus is broad and multispecific. Hum Immunol. 2000;61(5):438–452. | ||
Cohen T, Moise L, Martin W, De Groot AS. Chapter 11, Immunoinformatics: the next step in vaccine design. In: Sintchenko V, editor. Infectious Disease Informatics. Berlin: Springer Publications; 2006:223–244. | ||
Gillespie GM, Wills MR, Appay V, et al. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8(+) T lymphocytes in healthy seropositive donors. J Virol. 2000;74(17):8140–8150. | ||
Lazarski CA, Chaves FA, Jenks SA, et al. The kinetic stability of MHC class II. Peptide complexes is a key parameter that dictates immunodominance. Immunology. 2005;23(1):29–40. | ||
Benjamin JM, Beeson C. Binding interactions between peptides and proteins of the class II major histocompatibility complex. Med Res Rev. 2002;22(2):168–203. | ||
Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 4th edition. New York: Garland Science; 2002. T Cells and MHC Proteins. | ||
UniProtKB - Q4ACC9 (Q4ACC9_EDWTA) [webpage on the Internet]. UniProt. Available from: http://www.uniprot.org/uniprot/Q4ACC9. Accessed March 5, 2016. | ||
UniProtKB - G8X7P2 (G8X7P2_FLACA) [webpage on the Internet]. UniProt. Available from: http://www.uniprot.org/uniprot/G8X7P2. Accessed March 5, 2016. | ||
Koronakis V, Li J, Koronakis E, Stauffer K. Structure of TolC, the outer membrane component of the bacterial type I efflux system, derived from two-dimensional crystals. Mol Microbiol. 1997;23(3):617–626. | ||
Feriancikova L, Bardy SL, Wang L, Li J, Xu S. Effects of outer membrane protein TolC on the transport of Escherichia coli within saturated quartz sands. Environ Sci Technol. 2013;47(11):5720–5728. | ||
UniProt [webpage on the Internet]. UniProtKB – G8X7P2 (G8X7P2_FLACA). Available from: http://www.uniprot.org/uniprot/G8X7P2. Accessed September 6, 2013. | ||
Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276(1–2):172–174. | ||
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 2007;8:424. | ||
Kumar N, Mohanty D. MODPROPEP: a program for knowledge based modeling of protein-peptide complexes. Nucleic Acids Res. 2007;35:W549–W555. | ||
Raveh B, London N, Schueler-Furman O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins. 2010;78:2029–2040. | ||
Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2006;383:66–93. | ||
Li Z, Scheraga HA. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc Natl Acad Sci. 1987;84:6611–6615. | ||
Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci. 2000;97(19):10383–10388. | ||
Dunbrack RL, Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6(8):1661–1681. | ||
Yeh T. Structural Analysis of Peptide Binding to Class I MHC Protein. Accelyrs.com. 2011; case study: Discovery Studio. | ||
Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2000;3:445–450. | ||
Groot DAS, Rappuoli R. Genome-derived vaccines. Expert Rev Vaccines. 2004;3(1):59–76. | ||
Davies MN, Flower DR. Harnessing bioinformatics to discover new vaccines. Drug Discov Today. 2007;12:389–395. | ||
Florea L, Halldórsson B, Kohlbacher O, Schwartz R, Hoffman S, Istrail S. Epitope prediction algorithms for peptide-based vaccine design. Proc IEEE. 2003;2:17–26. | ||
Brusic V, Bajic VB, Petrovsky N. Computational methods for prediction of T-cell epitopes – a framework for modelling, testing, and applications. Elsevier. 2004;34(4):436–443. | ||
Kellam P, Holzerlandt R, Gramoustianou E, Jenner R, Kwan A. Immunoinformatics viral bioinformatics: computational views of host and pathogen. In: Immunoinformatics: Bioinformatic Strategies for Better Understanding of Immune Function: Novartis Foundation Symposium. Vol. 254. Chichester: John Wiley & Sons, Ltd; 2008:234–249. | ||
Schonbach C, Ranganathan S, Brusic V, editors. Immunoinformatics. Immunomics Reviews Series. New York, NY: Springer; 2008:19–49. | ||
Vajda S, Camacho CJ. Protein-protein docking: is the glass half-full or half-empty? Trends Biotechnol. 2004;22:110–116. | ||
Sezerman U, Vajda S, Cornette J, DeLisi C. Toward computational determination of peptide–receptor structure. Protein Sci. 1993;2:1827–1843. | ||
Rosenfeld R, Zheng Q, Vajda S, DeLisi C. Flexible docking of peptides to class I majorhistocompatibility-complex receptors. Genet Anal. 1995;12:1–21. | ||
Zeng J, Treutlein HR, Rudy GB. Predicting sequences and structures of MHC-binding peptides: a computational combinatorial approach. J Comput Aided Mol Des. 2001;15:573–586. | ||
Del Carpio CA, Hennig T, Fickel S, Yoshimori A. A combined bioinformatic approach oriented to the analysis and design of peptides with high affinity to MHC class I molecules. Immunol Cell Biol. 2002;80:286–299. | ||
Tzakos AG, Fuchs P, van Nuland NA, et al. NMR and molecular dynamics studies of an autoimmune myelin basic protein peptide and its antagonist: structural implications for the MHC II (IAu)–peptide complex from docking calculations. Eur J Biochem. 2004;271:3399–3413. | ||
Wu LC, Tuot DS, Lyons DS, Garcia KC, Davis MM. Two-step binding mechanism for T-cell receptor recognition of peptide MHC. Nature. 2002;418:552–556. | ||
Buslepp J, Wang H, Biddison WE, Appella E, Collins EJ. A correlation between TCR Valpha docking on MHC and CD8 dependence: implications for T cell selection. Immunity. 2003;19:595–606. | ||
Ip PP, Nijman HW, Daemen T. Epitope prediction assays combined with validation assays strongly narrows down putative cytotoxic T lymphocyte epitopes. Vaccines. 2015;3:203–220. | ||
Xiao Y-F, Jie M-M, Li B-S, et al. Peptide-based treatment: a promising cancer therapy. J Immunol Res. 2015;2015:761820. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.