")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Comparison of efficacy and safety of oral agents for the treatment of relapsing–remitting multiple sclerosis
Authors Guarnera C, Bramanti P , Mazzon E
Received 20 March 2017
Accepted for publication 15 June 2017
Published 28 July 2017 Volume 2017:11 Pages 2193—2207
DOI https://doi.org/10.2147/DDDT.S137572
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Cristina Guarnera, Placido Bramanti, Emanuela Mazzon
IRCCS Centro Neurolesi “Bonino-Pulejo”, Messina, Italy
Abstract: In the therapeutic scenario of disease-modifying therapies for relapsing–remitting multiple sclerosis, the introduction of oral agents, starting in 2010 with fingolimod, has been a huge step forward in therapeutic options due to the easier administration route. Three oral drugs fingolimod, teriflunomide, and dimethyl fumarate, which are clinically approved for the treatment of relapsing–remitting multiple sclerosis, are reviewed in this work. Results of Phase III clinical trials and their extension studies showed that the three oral agents significantly reduced the annualized relapse rate – a superior efficacy compared to placebo. Fingolimod 0.5 mg consistently reduced clinical relapses and brain volume loss. In all Phase III studies, teriflunomide 14 mg dose showed a reduction in the risk of disability accumulation. Regarding safety profile, fingolimod had more safety issues than the other two agents. For this reason, it should be strictly monitored for risks of infections, cancers, and certain transitory effects such as irregular cardiac function, decreased lymphocyte count, and a higher level of liver enzymes. Adverse effects of teriflunomide are well characterized and can be considered manageable. The main risks marked with dimethyl fumarate were flushing and gastrointestinal events.
Keywords: oral agents, comparison, efficacy, safety, relapsing-remitting multiple sclerosis
Introduction
Before fingolimod was used in the treatment of RRMS, the disease was managed by injectable drugs. Subsequently, the other two oral agents, teriflunomide and dimethyl fumarate, were approved for the treatment of RRMS. The introduction of these new oral therapies has been a huge step forward in the treatment of RRMS, firstly for ease of their administration. However, several parameters such as clinical efficacy, ability to reduce lesions on MRI, safety, and tolerability should be evaluated. In order to evaluate the benefit–risk profile of fingolimod, teriflunomide, and dimethyl fumarate, the efficacy and safety profiles are reviewed here. In this review, we have analyzed randomized Phase III clinical trials and their extensions studies. Furthermore, we have included post hoc analyses and safety data regarding monitoring programs and real-world data. The endpoints of efficacy were relapse rate, disability accumulation, and MRI measures. The endpoints of safety were AEs.
Mechanisms of action
The mechanism of action by which fingolimod, teriflunomide, and dimethyl fumarate, respectively, exert their effect in RRMS has not yet been completely defined. Fingolimod is a S1P receptor modulator present on the surface of lymphocytes and CNS cells. It binds to the S1P1 receptor after getting phosphorylated (by sphingosine kinase) to its active form fingolimod phosphate. Subsequently, it inhibits the egress of lymphocytes by lymph nodes, resulting in redistribution of lymphocytes. This redistribution potentially reduces the infiltration of pathogenic lymphocytes into the CNS and consequently abnormal autoimmune processes.1 Moreover, it can cross the blood–brain barrier and can bind to the S1P1 receptors on CNS cells, with possible direct actions on these cells. As a result of lymphocyte retention, during fingolimod treatment, the number of peripheral blood lymphocytes gets reduced. After discontinuation of treatment, the lymphocyte counts become normal within 1 or 2 months. This shows that this effect is reversible and reflects the redistribution of lymphocytes into lymphoid tissues (the mechanism of action is shown in Table 1). Teriflunomide inhibits pyrimidine biosynthesis in activated lymphocytes by selectively and reversibly blocking the mitochondrial enzyme DHODH (expressed on proliferating lymphocytes).2,3 Consequently, in the periphery, it reduces the proliferation of stimulated T and B lymphocytes (thought to be responsible for damaging inflammatory processes involved in MS) and diminishes the number of activated cells available to migrate into the CNS.4 Teriflunomide inhibits DHODH in activated and proliferating lymphocytes, whereas resting lymphocytes are not affected and are preserved from normal immune surveillance (Table 1).5 Dimethyl fumarate is an ester of fumaric acid with cytoprotective and neuroprotective effects. Dimethyl fumarate and its main active metabolite, mono-methyl fumarate, have been shown to increase the levels of Nrf2, involved in the antioxidant response. Dimethyl fumarate and mono-methyl fumarate increase cellular redox potential, glutathione and ATP levels, and mitochondrial membrane potential. The upregulation of an Nrf2-dependent antioxidant response may explain the cytoprotective function of neurons (Table 1).6
 | Table 1 Mechanism of action of three oral agents |
Approved doses and metabolic pathways
Fingolimod
Fingolimod has been approved by the US FDA and the EMA for the treatment of RRMS at 0.5 mg dose once daily. Since food does not change its pharmacokinetics parameters – Cmax or area under the curve – fingolimod can be taken independently. Fingolimod is slowly absorbed (with a Tmax of 12–16 hours) and its apparent absolute bioavailability is very high (93%). Fingolimod and its active metabolite can extensively bind to plasma proteins (99.7%). In red blood cells, free form of fingolimod is found in high concentrations (86%) whereas phosphate-bound form (fingolimod phosphate) is found in low concentration (<17%). Reversible phosphorylation to fingolimod phosphate, oxidative biotransformation mainly through CYP4F2 and subsequent fatty acid–like degradation to inactive metabolites, and formation of inactive ceramide metabolites are the primary biotransformation pathways of fingolimod. Fingolimod is majorly metabolized by CYP4F2, and a minor part of it is metabolized by CYP2D6, 2E1, 3A4, and 4F12.7,8 The inducers or inhibitors of these isoenzymes may alter the concentrations of fingolimod and its primary metabolite. Since multiple CYP isoenzymes are involved in the oxidation of fingolimod, in the presence of a single CYP-specific inhibitor, fingolimod metabolism probably does not undergo significant inhibition. After fingolimod administration, fingolimod (23.3%), fingolimod phosphate (10.3%), and inactive metabolites (M3 carboxylic acid metabolite 8.3%, M29 ceramide metabolite 8.9%, M30 ceramide metabolite 7.3%) are the major circulatory components detected in plasma. Fingolimod phosphate is a pharmacologically active metabolite. Fingolimod has a half-life (t1/2) of 6–9 days. Approximately 81% of the administered dose is eliminated via urine as inactive metabolites, whereas ~2.5% of fingolimod and same percent of fingolimod phosphate are excreted via feces. Pharmacokinetics parameters were calculated by data obtained from healthy volunteers and MS patients.
Teriflunomide
Teriflunomide has been approved by FDA and EMA for the treatment of RRMS at 14 mg dose once daily. It is absorbed almost completely (~100%) within 1–4 hours (time median of maximum plasma concentrations). Food has no effect on its pharmacokinetics. Teriflunomide is >99% protein-bound (albumin) and mainly distributed in plasma. It undergoes partial metabolism and is the major component detected in plasma. Hydrolysis and oxidation are the primary routes of biotransformation, whereas N-acetylation and sulfate conjugation are the secondary routes. It is excreted mainly through bile as unmodified drug. Teriflunomide is very slowly eliminated from plasma. Approximately 60% of the administered dose is excreted via feces (37.5%) and urine (22.6%) over 21 days. After repeated doses, median t1/2 is ~19 days.9,10 Elimination may be accelerated by administration of cholestyramine. Pharmacokinetics parameters were calculated by data obtained from healthy volunteers and MS patients.
Dimethyl fumarate
Dimethyl fumarate was the last oral agent approved by the FDA and EMA for the treatment of RRMS. The treatment comprises a 120 mg starting dose twice a day for 7 days. After 7 days, the dose should be increased to a maintenance dose of 240 mg twice a day.11,12 Immediately after administration, dimethyl fumarate is converted (by hydrolysis) to its active metabolite, MMF. Since dimethyl fumarate is not quantifiable in the blood, pharmacokinetics analyses refer to MMF, which is absorbed (median Tmax) within 2–5.5 hours of administration. Food does not have any significant effects on its pharmacokinetics. The plasma protein binding capacity of dimethyl fumarate ranges from 27% to 40% and is extensively metabolized. Less than 0.1% of the dose is eliminated in urine as unmodified drug. Before it reaches the systemic circulation, dimethyl fumarate is metabolized by esterases in the GI tract, blood, and tissues. Further metabolism of MMF occurs through the tricarboxylic acid cycle (CYPP450 system is not involved). MMF, glucose, fumaric acid, and citric acid are the major metabolites found in plasma. A large proportion (60%) of dimethyl fumarate is excreted mainly by CO2 exhalation and a small proportion by renal and fecal elimination (15.5% and 0.9%, respectively).11,12 The terminal t1/2 of MMF is very short (about 1 hour), and in most of the conditions, no circulating MMF is found after 24 hours of administration. Pharmacokinetic data were obtained from patients with MS and healthy volunteers.
Efficacy data: relapse disability and MRI outcomes
Fingolimod
Fingolimod was the first oral agent used in the treatment of RRMS.8 Efficacy and safety of this drug compared to placebo and IFN β 1a have been investigated in three large Phase III clinical trials: FREEDOMS, TRANSFORMS, and FREEDOMS II. Results of these studies strongly showed that fingolimod 0.5 mg/day consistently reduced clinical relapses and MRI parameters, mainly brain volume loss. FREEDOMS was the first randomized, double-blind, placebo-controlled Phase III trial, by Novartis Pharma AG, Basel, Switzerland, that compared oral fingolimod 0.5 mg and 1.25 mg daily with placebo. Patients with RRMS were evaluated for 24 months.13 In that study, the following inclusion parameters were used: a relapsing course, 18–55 years of age, an EDSS score up to 5.5, and one or more documented relapses in the preceding year and two or more in the previous 2 years. Exclusion parameters were relapse or use of corticosteroid within 30 days prior to randomization, discontinuation of interferon beta or GA from more than 3 months, drugs or disease-induced immunosuppression, active infection, macular edema, and diabetes mellitus or relevant systemic diseases. Compared with placebo, fingolimod 0.5 mg and 1.25 mg significantly reduced ARR at 54% and 60%, respectively. Reduction in the risk of disability progression was 17.7% for the 0.5 mg dose, 16.6% for 1.25 mg dose, and 24.1% for placebo. EDSS scores remained stable or slightly improved with fingolimod and worsened with placebo.13 Also the results of MRI parameters were positive with fingolimod. Brain volume loss had a 30% reduction with fingolimod. At 6, 12, and 24 months, gadolinium-enhancing and new or enlarged lesions on T2-weighted MRI were consistently less than those with placebo. These clinical and MRI findings were in line with those of the 6-month Phase II randomized study tested with placebo and its extension study at 36 months. In the Phase II study, fingolimod (at 5.0/1.25 mg doses daily compared to 0.5/1.25 mg doses in the Phase III trials) compared to placebo significantly reduced ARR (50%, P≤0.01) and gadolinium-enhanced lesions (fingolimod 77%–82% and placebo 47%).14,15 These reductions in relapse rate and MRI activity seen at 6 months were maintained in the extension study. In fact, ARR in patients who continued fingolimod treatment was 0.20/0.21 at 36 months. A high proportion of patients treated with fingolimod (70%–78%) did not show new T2 lesions in the 36 month period.15 Reductions in ARR (0.2 relapses/year), MRI activity, and disability progression (modest) with fingolimod were confirmed in the 5-year Phase II study.16 Treatment with fingolimod up to 5 years confirmed sustained and low clinical and MRI activity. Post hoc analysis of FREEDOMS study showed that 33% of patients treated with fingolimod 0.5 mg showed NEDA versus 13% of patients treated with placebo. Therefore, the approved dose of fingolimod confirmed an improvement in disease activity.17 TRANSFORMS was a randomized, multicentered, double-blind Phase III study by the drug manufacturer that compared oral fingolimod 0.5 mg and 1.25 mg once a day versus intramuscular IFN β 1a 30 μg weekly for 12 months. That study included RRMS patients with the following criteria: a relapsing course, 18–55 years of age, an EDSS score up to 5.5, and at least a documented relapse in the previous year or two in the previous 2 years. Patients with a documented relapse and use of corticosteroid therapy in the 30 days before randomization, active infection, drugs- or disease-induced immunosuppression, macular edema, and relevant systemic diseases were excluded.18 Clinical and MRI results of this trial confirmed those of previous studies. Fingolimod significantly reduced ARR compared with IFN β 1a in both doses: 0.16 at 0.5 mg dose, 0.20 at 1.25 mg, 0.33 with IFN β 1a (consisting of a relative reduction of 38%–58%). Instead no differences were observed in time to the disability progression (this was expected in a 1-year study).18 At 12 months, hyperintense T2-weighted lesions, gadolinium-enhanced lesions, and reduction in brain volume on MRI were significantly lower in patients treated with fingolimod compared with those treated with IFN β 1a. At the end of the study, 46% of patients treated with the 0.5 mg dose had NEDA, and 38% with the 1.25 mg dose versus 34% with IFN β 1a had NEDA.19 FREEDOMS II was another randomized, double-blind, Phase III study of 24 months carried out mainly in the USA by the drug manufacturer Novartis Pharma AG, because the US FDA requested additional measures relating to cardiac parameters, which were not included in the FREEDOMS study.20 FREEDOMS II reconfirmed the results of FREEDOMS in reducing relapse rates, lesion activity, and brain volume loss on MRI. Study participants were selected according to the following criteria: RRMS, 18–55 years of age, an EDSS score up to 5.5, one or more documented relapses in the previous year or two or more relapses in the previous 2 years, relapse or steroid treatment within 30 days prior to randomization, naïve-patients and previously treated patients who had interrupted the interferon β GA for at least 30 days and natalizumab for at least 60 days prior to randomization. Exclusion criteria were drugs- or disease-induced immunosuppression, active infection, significant systemic diseases, macular edema, diabetes mellitus, specific cardiac, pulmonary, and hepatic disorders, and history of a cancer with the exception of basal or squamous carcinoma removed. As in FREEDOMS, in this study, two doses of fingolimod 0.5 mg and 1.25 mg were compared with placebo. Subsequently patients receiving fingolimod 1.25 mg were switched in blinded manner to the 0.5 mg dose (but have been assessed as the group with 1.25 mg dose).20 FREEDOMS II reaffirmed some results of FREEDOMS. Compared with placebo, fingolimod showed a 48% relative reduction in relapse rates with the 0.5 mg dose and a 50% with the 1.25 mg dose. Instead, no relevant effect has been observed in 3-month or 6-month confirmed disability progression. Median MSFC score improved with fingolimod compared with placebo in 24 months. The MRI outcomes based on the new or enlarged T2 lesions and gadolinium-enhancing T1 lesions (at 6, 12, and 24 months) and brain volume loss (at month 24) were significantly lower with fingolimod treatment (P<0.0001) than placebo. Reduction in brain volume loss was maintained throughout the study (at 6, 12, and 24 months). No differences between the fingolimod-treated group and placebo were found regarding the percentage change of T1 hypointense lesions. FREEDOMS II, confirming the results of the previous controlled Phase III study, showed that fingolimod 0.5 mg improves relapse rate and MRI outcomes. The extension studies of FREEDOMS and TRANSFORMS confirmed the previous findings and efficacy of fingolimod in the long term. In the extension study, the patients who continued fingolimod 0.5 mg had a 48% reduction in ARR and those who continued fingolimod 1.25 mg had a 54% reduction. ARR also improved in the groups that switched from placebo to fingolimod 0.5 or 1.25 mg (P<0.001 for both groups), mainly in the group that switched to fingolimod 0.5 mg.21 In the continuous-fingolimod groups, more patients were free from 3-month confirmed disability progression (P<0.05) compared with the groups of all patients who switched from placebo to fingolimod. Also reduced brain volume loss (P<0.05) was observed in the continuous-fingolimod groups. In the placebo-fingolimod groups, brain volume loss improved after switching (placebo–fingolimod 0.5 mg group: P<0.01). In the TRANSFORMS extension study, patients re-treated with fingolimod 0.5 mg showed a 0.17 reduction in ARR versus 0.27 of the patients who switched from IFN β 1a to fingolimod 0.5 or 1.25 mg. The data of these two extensions showed a sustained effect on the level of disease activity with continuous fingolimod therapy during the extensions from the core studies. Since continuous-fingolimod patients from the pivotal studies had greater improvements than those who received fingolimod in the extensions, fingolimod proved effective if started earlier. Moreover, in TRANSFORMS extension study, patients who switched from IFN β 1a had a 50% improvement in the ARR (from 0.40 to 0.20), confirming the validity of the switch from a first-line treatment to fingolimod.22 These patients also experienced a lower brain volume loss and MRI activity (percent brain volume loss observed in the core study and at the end of the study was −1.01% for patients re-treated with fingolimod continuously and −0.96% for those who switched from IFN β 1a to fingolimod). A post hoc analysis showed that patients who switched from IFN β 1a to fingolimod in the first year showed a 50% increase in NEDA. NEDA is a generic definition of no evidence of disease activity. NEDA-3 is correctly defined as absence of relapses, new or enlarged T2 and gadolinium lesions, and 3-month confirmed disability progression. Achievement of NEDA status has been proposed as the therapeutic goal in RRMS. A study analyzed the individual parameters of NEDA-3 (31% patients treated with fingolimod 0.5 mg achieved NEDA-3 status compared with 9.9% of placebo at 2 years, P<0.0001) and the impact of adding brain volume loss to NEDA-3 (NEDA-4), using data from the two Phase III trials of fingolimod.23 In a 2 year period, 19.7% of patients treated with fingolimod 0.5 mg achieved NEDA-4 status compared with 5.3% of placebo (P<0.0001). Therefore, fingolimod has been favored for NEDA-4 (odds ratio 4.01–4.41, P<0.0001).23 Results from a retrospective, real-world study named MS-MRIUS confirmed the importance of including four key measures of disease activity: relapses, MRI lesions, disability progression, and brain volume loss. In a 16-month period, 59.6% of patients achieved NEDA-3 and 37.5% NEDA-4. Among these patients in NEDA-4 status, 86.5% treated with fingolimod 0.5 mg were free from relapses, 91.1% had no disability progression, and 79.7% had no new or enlarged MRI lesions. These data confirmed that fingolimod is highly effective long-term treatment option for MS.24 In order to estimate the indirect relative risk of achieving NEDA status, fingolimod has been compared with teriflunomide and dimethyl fumarate by analyzing data from their Phase III trials. Results of that study showed that fingolimod treatment had a higher probability of achieving NEDA status than teriflunomide and dimethyl fumarate.25 The main results from clinical trials of fingolimod are summarized in Table 2.
 | Table 2 Outcomes from Phase III studies: fingolimod, teriflunomide, and dimethyl fumarate at their respective recommended doses |
Teriflunomide
Two placebo-controlled Phase III trials, TEMSO and TOWER, have been conducted to investigate the efficacy and safety of teriflunomide. EMSO and TOWER studies demonstrated in the ARR and in disability progression in patients with RRMS the efficacy of teriflunomide at the approved dose. TEMSO was the first randomized, double-blind, Phase III trial by Sanofi-Aventis that compared two doses of teriflunomide (7 mg and 14 mg) with placebo. These doses were given once per day for 108 weeks.26 Study inclusion criteria were the following: RRMS patients, a relapsing course, with or without progression, 18–55 years of age, a 5.5 or lower EDSS score, with one relapse in the previous year or two relapses in the previous 2 years but not in the 60 days before randomization. Exclusion criteria were pregnancy and systemic diseases. The study found that teriflunomide 7 mg reduced ARR with a 31.2% relative reduction versus placebo. It had no significant changes on disability accumulation (23.7% reduction). The 14 mg dose decreased ARR with a 31.5% reduction. It also showed a positive result in 12-week confirmed disability progression with a 29.8% reduction.26 Compared with placebo, both doses of teriflunomide showed an improvement in MRI parameters. A bigger proportion of patients treated with both teriflunomide doses had less gadolium-enhanced lesions (P<0.001) and unique active lesions (P<0.001). No differences regarding the change in brain atrophy were found among the study groups. A post hoc analysis of this study showed that more patients treated with teriflunomide 7 and 14 mg compared to placebo achieved NEDA status (18%, 23% versus 14%).27 TOWER was the second randomized, multicentered, double-blind, Phase III study by Genzyme (a Sanofi company) that compared teriflunomide 7 and 14 mg with placebo for up to 48 weeks. The study included dosing regimens set forth in the previous trial. Inclusion criteria were as follows: RRMS, with or without underlying progression, 18–55 years of age, an EDSS score up to 5.5, and one relapse in the previous year or two relapses in the previous 2 years, but not in the 30 days before randomization. Main exclusion parameters were pregnancy, breastfeeding or intent to have a child during the study, other relevant diseases, previous use of cytokine therapy, interferons or GA (in the 3 months prior to randomization), and natalizumab or other immunosuppressants. In TOWER, teriflunomide 7 mg showed a 22.3% reduction in ARR, whereas in TEMSO, it did not show any improvement in disability accumulation. Teriflunomide 14 mg showed a 36.3% reduction in ARR and 31.5% reduction in the risk of confirmed disability progression (sustained for 12 weeks).28 Therefore, the results of these two studies were an improvement for both low and high teriflunomide doses in relapse parameters and a consistent improvement of the high dose in relapse rate and disability accumulation. Given the evidence provided by TEMSO, no MRI endpoint was included in this study. Post hoc analysis of TEMSO study observed in RRMS patients the positive effects of teriflunomide 14 mg, in particular the relapse-related neurological deficits. Compared with placebo, the 7 mg dose showed a 32% reduction in ARR associated with neurological sequelae (defined by EDSS/Functional System as ≥30 days post-relapse) and the 14 mg dose showed a 36% reduction. In the analysis of sequelae, only the 14 mg dose maintained these effects up to 180 days post relapse. Teriflunomide 14 mg also reduced emergency medical facility visits and annualized relapse of hospitalizations.29 A randomized, multicentered, double-blind placebo-controlled Phase III study, TOPIC, of Genzyme, was performed in patients (aged 18–55 years) with isolated syndrome defined by a first significant demyelinating episode occurring within 90 days prior to randomization and two or more T2 MRI lesions (at least 3 mm in diameter) for up 108 weeks. The studies that have been discussed so far included patients with active diseases established by McDonald criteria, an EDSS score ≥5.5, and at least two relapses in the previous two years or one in the previous year, but no relapse before randomization. Therefore, TOPIC study extended to other stages of RRMS the activity of teriflunomide. Exclusion criteria were pregnancy, breastfeeding or intent to have a child during the study, other relevant diseases, use of corticosteroids within 2 weeks prior to randomization, previous use of interferons, GA, teriflunomide or leflunomide, natalizumab, cladribine, mitoxantrone or other immunosuppressants, immunoglobulins, or cytokine therapy. In this study, teriflunomide delayed the onset of a second relapse, emphasizing the effect of this compound during the early stage of RRMS (compared with placebo, it reduced the risk of relapse or a new MRI lesion at 14 mg dose and 7 mg dose).30 The randomized, rater-blinded, Phase III study TENERE compared oral teriflunomide (7 and 14 mg daily) with IFN β 1a (8 μg for the first 2 weeks, 22 μg for the next 2 weeks, and 44 μg until the end of study). The study included patients ≥18 years old, with a relapsing clinical course, with or without progression, an EDSS score ≤5.5, and no relapse within 30 days prior to randomization. Exclusion criteria were pregnancy, breastfeeding or intent to have a child during the study, prior use of IFN β 1a, teriflunomide or leflunomide, natalizumab, cladribine, mitoxantrone or other immunosuppressants, use of other interferons, GA, and immunoglobulins or cytokine therapy within 3 months, and significant systemic diseases. In this study, teriflunomide 14 mg and IFN β 1a showed similar outcomes. The primary endpoint time to failure was 33% and 37%, respectively. ARR was 0.26 in teriflunomide 14 mg versus 0.22 in IFN β 1a (P=0.6). Teriflunomide 7 mg had higher ARR (0.410).31 This study confirmed the effect (regarding relapse rate and disability progression) seen in placebo-controlled clinical trials with the teriflunomide high dose compared with the low dose. Further, a post hoc analysis of TOWER study showed that high doses of teriflunomide compared with placebo reduced annualized rates of severe relapses (only teriflunomide 14 mg 52.5%), relapses with neurological sequelae (14 mg dose 36.6% and 7 mg dose 31.3%), and relapses leading to hospitalization (only the 14 mg dose 33.6%). These observations are similar to the previous findings on the efficacy of teriflunomide in treating disease.32 However, long-term studies are needed to consolidate these results. Major results from clinical trials of teriflunomide are shown in Table 2.
Dimethyl fumarate
The efficacy of dimethyl fumarate was investigated by two large, randomized, placebo-controlled Phase III studies DEFINE and CONFIRM carried out by Biogen Idec. Both studies confirmed the efficacy of dimethyl fumarate on relapse rates, and only DEFINE study confirmed the progression disability of the drug. Additional studies could evaluate the progression disability of dimethyl fumarate. DEFINE was a randomized, double-blind, Phase III study carried out in patients with RRMS who received dimethyl fumarate 240 mg BID or TID or placebo for a 2 year period.33 The study enrolled patients with the following criteria: a RRMS diagnosis, 18–55 years of age, an EDSS score up to 5, active disease defined by at least one relapse within 12 months before randomization or one MRI gadolinium-enhancing lesion within 6 weeks before randomization. Exclusion criteria were as follows: progressive forms of MS, other clinically relevant diseases, abnormalities in prespecified laboratory tests, and recent use of contraindicated drugs by the study protocol. In this study, the two dosage regimens 240 mg BID and TID of dimethyl fumarate reduced ARR compared to placebo, with a relative reduction of 53% and 48%, respectively. In the risk of confirmed disability progression, dimethyl fumarate 240 mg BID and TID showed 38% and 34% reductions, respectively.33 In addition, compared with placebo, it reduced the number of new or enlarged T2 lesions (85% BID and 74% TID), and more patients given dimethyl fumarate BID were free from gadolinium-enhanced lesions (93% BID and 62% placebo) over 2 years. In multicentered, double-blind CONFIRM trial, dimethyl fumarate 240 mg BID, TID, and placebo were administered in patients with RRMS for 96 weeks. Also the active agent GA 20 mg was given subcutaneously daily (rater blinded; in order to compare dimethyl fumarate and GA profile versus placebo, but not dimethyl fumarate versus GA).34 This second main trial showed that in reducing the number of relapses, teriflunomide was more effective than placebo. Inclusion and exclusion criteria of this study are the same as those of the DEFINE trial. Compared with placebo, both dosage regimens of dimethyl fumarate reduced ARR, with a relative reduction of 44% at 240 mg BID and 51% at 240 mg TID. Also GA compared with placebo showed a low relative reduction (29%). Disability progression did not significantly improve (unlike DEFINE, in this study rate of disability progression with placebo was lesser than in DEFINE) in dimethyl fumarate groups compared to placebo (relative reductions, 21%, 24%, and 7%).34 Both dosage regimens BID and TID compared with placebo reduced the mean number of new or enlarged T2 lesions (71% at 240 mg BID and 73% at 240 mg TID) and new T1 lesions (57% and 65%, respectively). In both the DEFINE and CONFIRM studies, MRI lesions were less in groups given dimethyl fumarate than placebo. Dimethyl fumarate had a more significant effect compared to GA in the following outcomes: at both dosage regimens BID and TID in the number of new or enlarged hyperintense T2 lesions, at the TID dosage regimen in ARR and in the number of new hypointense T1 lesions (but this comparison was not a study endpoint). In the ongoing long-term extension study of DEFINE, CONFIRM, and ENDORSE, low clinical and MRI outcomes were maintained in patients who continued the same dose of dimethyl fumarate 240 mg BID for the previous 2 years (plus 3 years of the extension). ARR was 0.202 at the first year, 0.163 at second, 0.139 at third, 0.143 at fourth, and 0.138 at fifth. Over 5 years, the MRI outcomes were as follows: 88% of patients were free from gadolinium-enhanced lesions, 73% from new T1 hypointense lesions, and 63% from new or enlarged T2 hyperintense lesions.35 Post hoc analysis of the two pivotal trials36,37 showed the positive effects of dimethyl fumarate 240 mg BID on NEDA. In 2 years, a high proportion of patients with BID dosage regimen compared to placebo achieved NEDA status (23% versus 12%).36,37 An integrated analysis of Phase III DEFINE and CONFIRM studies showed a high efficacy of dimethyl fumarate in young naïve patients with low EDSS score (also in all subgroups of patients),38 suggesting its clinical use even in these patients. The major outcomes of clinical trials of dimethyl fumarate are shown in Table 2.
Safety data
The incidence, frequency, and severity of the adverse effects of the three oral agents in Phase III trials are shown in Tables 3–5.
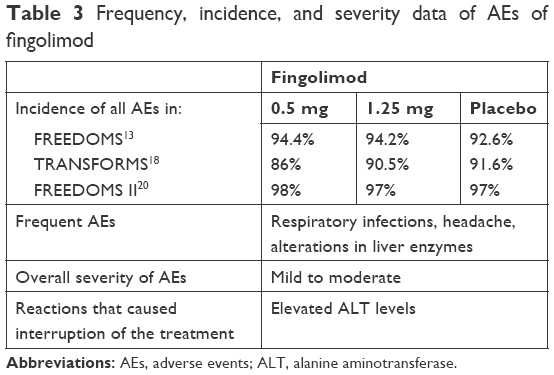 | Table 3 Frequency, incidence, and severity data of AEs of fingolimod |
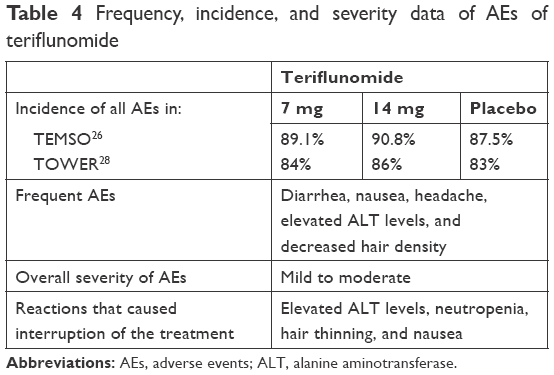 | Table 4 Frequency, incidence, and severity data of AEs of teriflunomide |
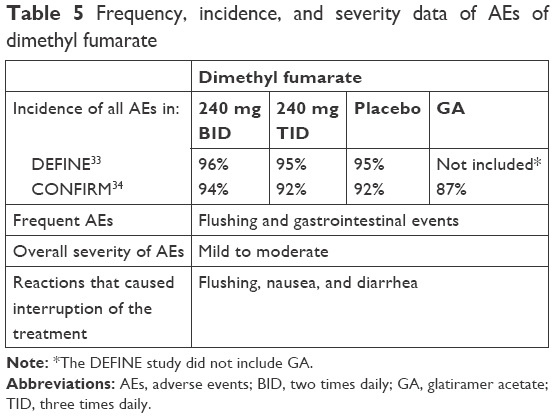 | Table 5 Frequency, incidence, and severity data of AEs of dimethyl fumarate |
Cardiovascular adverse effects
In Phase III clinical trials and their extension studies, fingolimod induced transient cardiac adverse effects on frequency and conduction: bradycardia and first- and second-degree atrioventricular block. The onset of these dose-dependent cardiac AEs occurred at the first administration of the drug, which returned to baseline value within 1 month of the start of the treatment. This might be due to the modulatory effects of fingolimod on S1P receptors in the heart.18 A randomized, open-label study, EPOC, demonstrated in a large population (over 900 RRMS patients treated with fingolimod and subgroups of patients treated for heart rate disorders or preexisting cardiac conditions) that the first-dose effects of these drugs on heart are transient and benign. Bradycardia had an incidence of 1%. All events were mild to moderate in severity and no case required suspension of fingolimod or treatment. Results on first-dose effects confirmed those that were obtained in the clinical trials.39 Other specific AEs associated with fingolimod, such as increased ALT levels, increased blood pressure, and macular edema, showed a dose-dependent effect.40 Seven patients had serious bradycardia in FREEDOMS study at first administration and seven patients in FREEDOMS II had second-degree atrioventricular block, among whom one withdrew from the study due to the severity of the disease.13,20 In TRANSFORMS study, no case was seriously affected by heart rhythm and conduction.18 Cardiac alterations, observed also in FREEDOMS extension study, were more frequent in groups switching from placebo to fingolimod 0.5 mg and 1.25 mg compared to those who continued the same dose of fingolimod in a pivotal study.21 Overall, these reactions were mild to moderate in severity and most did not require intervention. As a result of these effects, patients with cardiac or cardiovascular diseases or those who were taking medicines for lower heart rate may be at greater risk with fingolimod treatment. For this reason, regulatory guidelines recommend more attention or exclusion of patients with preexisting cardiac disease.20 In a long-term follow-up Phase II study, a 51-year-old woman with a history of dyspnea, depression, anxiety, Hashimoto’s thyroiditis, tachycardia, and pneumonia with respiratory failure episode (who had taken several medications) received teriflunomide 14 mg. After 4.8 years, she suddenly died of cardiac disease. She took benzodiazepines when AE occurred. Therefore, the death could have been caused by medications and comorbidities.41 In clinical Phase III trials, there was no evidence that teriflunomide and dimethyl fumarate caused clinically relevant cardiac effects (electrocardiogram evaluations with teriflunomide and dimethyl fumarate did not show clinically significant alterations). Another transient adverse effect was increased blood pressure. It was very common in groups taking fingolimod and teriflunomide but had caused no serious effects. In Phase III clinical trials and their extension studies of fingolimod, increased blood pressure frequently occurred in patients treated with both 0.5 and 1.25 mg doses.13,18,21 Also in clinical trials of teriflunomide, hypertension was more frequent in teriflunomide-treated group compared to placebo. In TEMSO study, teriflunomide 7 mg caused hypertension in 5.4% of patients and teriflunomide 14 mg in 5% compared to 3.1% of placebo. In TOWER study, two patients interrupted teriflunomide 7 mg due to increased blood pressure with additional AEs. After adjusting the hypertensive treatment, both patients recovered. An increase in average blood pressure occurred at the beginning of the treatment but got stabilized over time. Blood pressure must be monitored before the start of teriflunomide treatment and periodically. Hypertension was not reported with dimethyl fumarate treatment. Flushing was the vascular adverse effect frequently reported with dimethyl fumarate. Flushing symptoms, including redness, warmth, and burning, generally occur soon after starting dimethyl fumarate and improved or resolved over time. In placebo-controlled clinical trials,33,34 flushing was the most common reason for the discontinuation of treatment. However, most patients experienced only mild or moderate flushing. Administration of dimethyl fumarate with food may reduce the incidence of flushing. Cardiovascular events of fingolimod, teriflunomide, and dimethyl fumarate are shown in Table 6.
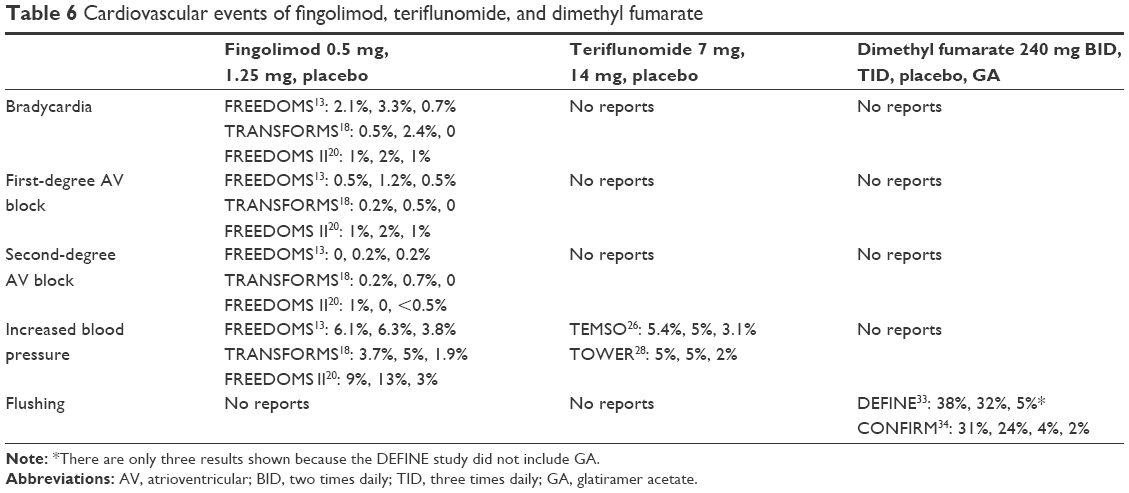 | Table 6 Cardiovascular events of fingolimod, teriflunomide, and dimethyl fumarate |
Hematochemical abnormalities
Increased ALT levels have been reported following fingolimod, teriflunomide, and dimethyl fumarate treatments in Phase III clinical trials. These abnormalities were very frequent with all the three oral treatments. Increase in ALT levels observed with fingolimod in Phase III studies had a dose-dependent effect. In FREEDOMS study, ALT levels were 12.5% with fingolimod 1.25 mg, 8.5% with 0.5 mg, and 1.7% with placebo; in TRANSFORMS study, 7% with fingolimod 1.25 mg, 8% with 0.5 mg, and 2% with IFN β 1a; and in FREEDOMS II study, 10% with fingolimod 1.25 mg, 8% with 0.5 mg, and 2% with placebo.13,18,20 Most of these changes occurred during the first month of the treatment, and the levels returned to normal ~2 months after fingolimod interruption. An increase in liver enzymes level was the most common reason for the discontinuation of treatment in Phase III and their extension studies. This did not affect significantly on the tolerability of the drug.40 Increased ALT recorded in the extension study of FREEDOMS was more frequently observed in the groups that switched from placebo to fingolimod 0.5 mg and 1.25 mg compared to groups that continued the same dose of fingolimod previously.21 Since this adverse effect occurred at the beginning of the treatment, it could be expected. No cases of severe hepatic toxicity have been reported in the clinical studies of fingolimod. Monitoring the levels of liver enzymes during the first month of treatment, every 3 months for 1 year, and then periodically is currently recommended.7 Although there is no evidence that patients with significant preexisting liver disease have a greater risk of developing these alterations, more attention should be paid to these patients. An increased incidence of ALT elevations was even reported in patients treated with teriflunomide. Both the doses of teriflunomide had similar incidence in clinical and extension studies (in TEMSO 6.3% and 6.7%; in TOWER 11% and 14%; in long-term follow-up of Phase II study 29.6% and 28.8%).26,28,41 According to the current recommendations, liver enzymes should be monitored prior to starting teriflunomide, every 2 weeks during the first 6 months of treatment, and then every 2 months.9 Elevations of liver enzymes also occurred with dimethyl fumarate during controlled clinical trials. The onset occurred in the first 6 months of treatment and then decreased. In CONFIRM study, these elevations had similar incidence in all study groups. Lymphopenia was another hematochemical abnormality caused by these oral drugs in clinical trials. Due to the mechanism of action, lymphopenia was an AE of treatment with fingolimod. It was very frequent but transitory. Also lymphopenia was one of the reasons that frequently resulted in the discontinuation of treatment with fingolimod in clinical and their extension studies.20,21 White blood cell count, mainly neutropenia and lymphopenia, was also reduced with teriflunomide. The onset ranged from the first 3–6 months of treatment and returned to normal level over time. In TEMSO study, this reaction was little more frequent in the group given a high dose of teriflunomide compared to those given a low dose.26 In TOWER study, a patient given teriflunomide 7 mg who was diagnosed with immune-mediated thrombocytopenia, discontinued the drug. After pharmacological treatment with corticosteroids and subsequently with rituximab, he recovered.28 Neutropenia, among the other reactions (along with increased ALT levels), more frequently caused the interruption of treatment (involving more patients in teriflunomide groups compared to placebo). In an open-label Phase II extension study (up to 8.5 years) of teriflunomide, leucopenia was more frequently observed with teriflunomide 14 mg than teriflunomide 7 mg (13.8% versus 3%).41 Overall, it was mild in severity in all clinical studies. Therefore, a complete blood count prior to starting teriflunomide has been recommended.9 Significant decreases in white blood cells, mainly of lymphocytes, were also associated with dimethyl fumarate. Mean lymphocyte counts decreased during the first year of treatment but remained stable with continued therapy. Since during clinical trials, lymphopenia was prolonged, lymphocytes counts should be monitored until their recovery. In clinical practice, monitoring of complete lymphocyte counts every 3 months before and during treatment with dimethyl fumarate is recommended.11 Hematochemical abnormalities are shown in Table 7.
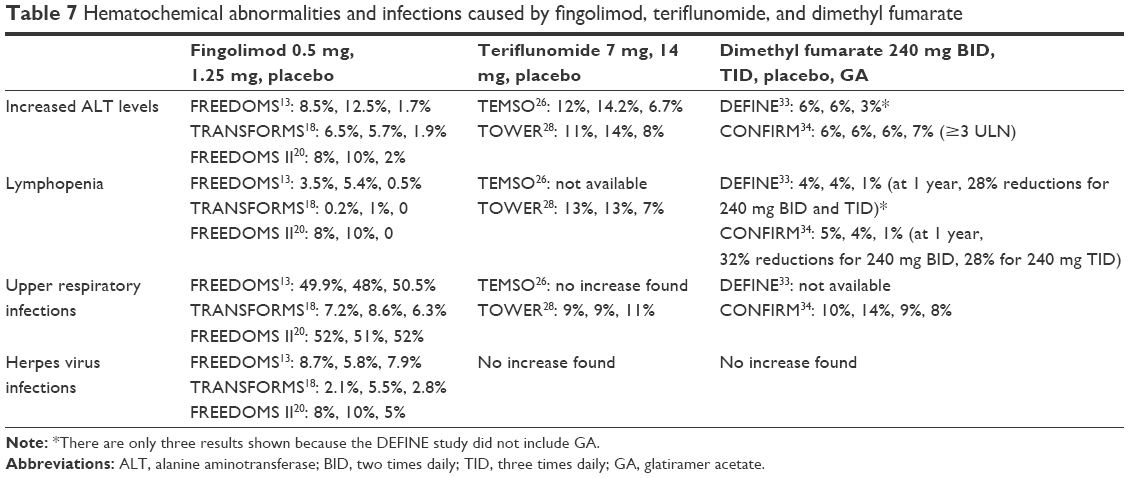 | Table 7 Hematochemical abnormalities and infections caused by fingolimod, teriflunomide, and dimethyl fumarate |
Infections
Immunomodulatory action of these drugs can increase the rate of infections. In all Phase III clinical trials, fingolimod in the treatment arms showed a similar rate of infections. In FREEDOMS study, infections occurred in a range of 69%–72%; in TRANSFORMS study, it was 51%–53% and in FREEDOMS II study 72%–74%. Infections of the upper and lower respiratory tract were most frequently found.13,18,20 Serious infections had a low incidence (1.6%–2.6% in FREEDOMS and 0.2%–1.7% in TRANSFORMS). These included herpes virus infections, such as genital herpes, herpes simplex labialis, and herpes zoster, urinary tract infection, and appendicitis. In TRANSFORMS study, two deaths were reported in fingolimod-treated group (1.25 mg) due to herpes simplex encephalitis and disseminated varicella zoster. The data from Phase II, Phase III, and their extension studies have shown an increase in varicella zoster infection in patients treated with fingolimod compared to placebo. This might be due to its action on the effector functions of CD8 T cell.42 In FREEDOMS II study, one cryptococcal infection was reported in a patient treated with fingolimod 0.5 mg 5 months after the interruption of the drug, and the patient recovered with itraconazole. At the end of the study, another patient developed symptoms of tuberculosis, and in subsequent extension study it was diagnosed.20 Two cases treated with fingolimod, who did not undergo any prior immunosuppression treatments, were diagnosed with PML. The first case, a 49-year-old patient who had MS for 5 years, was diagnosed with serious lymphopenia after 4 years of fingolimod treatment. The second case, a 54-year-old patient who had MS for 14 years, was diagnosed with ulcerative colitis after ~2.5 years of fingolimod and mesalazine treatment. He also developed serious lymphopenia.43,44 A third case treated with fingolimod also was confirmed to have PML. However, the incidence was low to define the risk of PML with this drug. Recently, 17 cases have been reported with fingolimod, after switch by natalizumab. These cases have been classified as suspected. In fact, the washout period before starting fingolimod was often shorter in patients with prior immunosuppressant therapy (eg, natalizumab).45,46 Current recommendations require an MRI scan before and during treatment with fingolimod. The frequency of infections and serious infections with teriflunomide was very low in Phase III clinical studies. The incidence of infections was similar in the treatment groups (serious infections in TEMSO 2.2% at 7 mg dose, 1.6% at 14 mg dose, 2.5% with placebo; in TOWER 3% in each group). In TEMSO study, serious pyelonephritis was observed in three patients with teriflunomide 14 mg, among whom one withdrew the drug. One patient in the placebo group had herpes zoster.26 In TOWER study, two deaths occurred due to infection: one was septicemia due to Gram-negative infection with intravascular coagulopathy in teriflunomide-treated group (14 mg) and the other was respiratory infection in placebo. No opportunistic infections occurred in clinical trials with teriflunomide. The two cases of opportunistic infections reported in TOWER study (one case of intestinal tuberculosis in teriflunomide 14 mg group and one case of hepatitis C with cytomegalovirus in placebo) have not been associated with teriflunomide.28 In long-term follow-up of Phase II study, a high rate (12.1%) of oral herpes has been recorded with teriflunomide 14 mg (with teriflunomide 7 mg 1.2%). In clinical trials, teriflunomide did not increase the risk of infection. The incidence of infections and serious infections was similar in patients treated with dimethyl fumarate or placebo in Phase III clinical studies (infections: in DEFINE study 64% at 240 mg BID, 68% at 240 mg TID, 65% with placebo; in CONFIRM 56% at the two dosage regimens of dimethyl fumarate, 50% with GA, and 50% with placebo; serious infections: in DEFINE and CONFIRM 1–2%). In general, infections were mild in severity. Gastroenteritis was the only infection classified as serious AEs in both the clinical trials.33,34 Upper respiratory tract infections (mainly nasopharyngitis, bronchitis, sinusitis), urinary tract infections, and gastroenteritis were the most frequently reported infections compared with placebo. Since PML has been reported in patients with psoriasis53,54 in the treatment with acid fumaric esters, lymphocyte counts were also monitored in RRMS with dimethyl fumarate. No increased incidence of serious infections was reported in clinical trials, although in ENDORSE extension study,35 one patients with prolonged lymphopenia developed PML. The patient had not been previously treated with natalizumab, which has been associated with PML. Overall, three PML cases, of which one was fatal in ENDORSE extension study,35 have been reported with dimethyl fumarate treatment. These occurred in patients <50 years, with persistent severe lymphopenia, and with no prior immunosuppressive treatments.47–49 Since a persisting severe lymphopenia may be a risk factor for PML, complete blood count must be regularly performed along with dimethyl fumarate treatment. At present PML cases are rare. Further studies should investigate the long-term effects of this drug. Infections are shown in Table 7.
Tumor incidence
Overall, fingolimod did not show an increase in cancer risk in Phase III clinical trials (0.9% increase in fingolimod-treated groups and 2.4% in placebo).40 FREEDOMS study reported four types of cancers in patients treated with fingolimod 1.25 mg, four in patients treated with 0.5 mg, and ten in patients treated with placebo. Majority of the cancers were skin cancers that included basal cell carcinoma, melanoma, and Bowen’s disease. Of these skin tumors, three occurred in patients treated with fingolimod 1.25 mg, four in patients with 0.5 mg, and four with placebo.13 Due to the monitoring introduced after the Phase II study, in TRANSFORM study 10 skin malignancies were diagnosed early. Six of these were basal cell carcinoma, of which two occurred in patients treated with fingolimod 1.25 mg, three with fingolimod 0.5 mg, and one with placebo. Three melanomas were found in fingolimod-treated group (0.5 mg) and one squamous cell carcinoma was diagnosed in IFN β 1a-treated group. Other malignancies reported were four breast carcinomas: three with onset after 4 months of treatment and one after 11 months. Among these, one patient died due to metastases.18 In FREEDOMS II study, 31 cancers were diagnosed. Most were skin cancers, of which six occurred with fingolimod 1.25 mg, ten with 0.5 mg, and two with placebo. In the other forms of cancer, fingolimod 0.5 mg had similar incidence to placebo. Long-term extension of TRANSFORMS study found six basal cell carcinomas and two breast neoplasms in the group that continued fingolimod 0.5 mg, and one basal cell carcinoma in the group that switched from IFN β 1a to fingolimod 0.5 mg.22 According to data from Phase III clinical trials, the rate of occurrence of basal cell carcinoma was high in these studies. The incidence was slightly high for fingolimod. In clinical practice with fingolimod (in the real world), 151 reports of basal cell carcinoma have been reported as at February 2015. For this reason, a medical examination of the skin at the beginning, after 1 year, and then annually is recommended, when a patient is being treated with fingolimod.7,50 Teriflunomide in Phase III clinical trials did not show an increased risk of cancer. One single cervical carcinoma occurred in TEMSO study in a patient treated with teriflunomide 14 mg, with onset after 18 months of treatment. Three different cancers – one in breast, one in thyroid, and one in cervix – occurred in the placebo group.26 In Phase III TOWER study, one occurrence of thyroid cancer was diagnosed with teriflunomide 14 mg.28 In long-term follow-up Phase II study, five different malignancies (one basal cell carcinoma, two renal cell carcinomas, and two breast neoplasms) have been detected with teriflunomide 7 mg. The incidence of these tumors is that predicted in the base population.41 In Phase III clinical studies of dimethyl fumarate, tumor incidence was low in all the treatment groups: specifically <1%. In DEFINE study, two basal cell carcinomas occurred, one with dimethyl fumarate 240 mg BID and one with placebo; two breast cancers, one with dimethyl fumarate 240 mg TID and one with placebo; one cervical carcinoma with dimethyl fumarate 240 mg TID; and one transitional cell carcinoma with dimethyl 240 mg BID. In CONFIRM study, no tumor was detected with dimethyl fumarate, whereas four different types of cancers with GA (basal cell carcinoma, cervical carcinoma, endometrial carcinoma, thyroid cancer) and one breast cancer were detected with placebo.33,34 Tumor incidence of each drug is shown in Table 8.
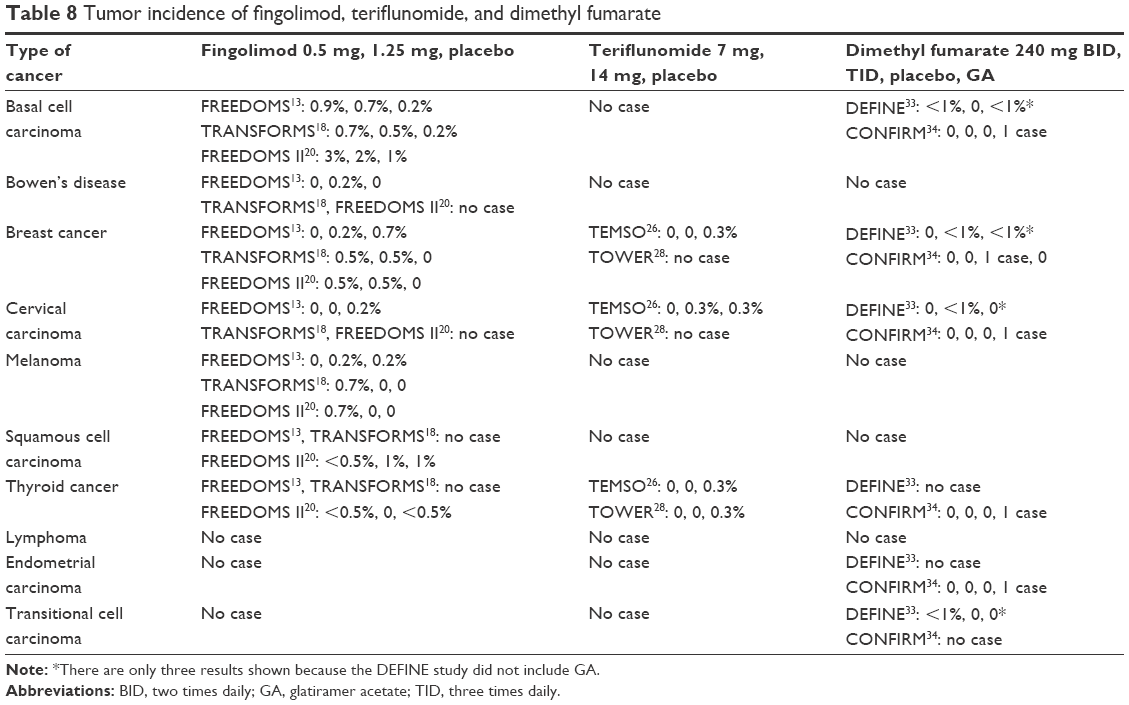 | Table 8 Tumor incidence of fingolimod, teriflunomide, and dimethyl fumarate |
Ocular adverse effects
Macular edema is a transient adverse effect that occurs during the early stage of treatment with fingolimod (in the first 3–4 months of treatment). This complication generally resolves with the interruption of the drug and it has been reported in all Phase III clinical trials. In FREEDOMS study, seven patients treated with fingolimod 1.25 mg developed macular edema, of which three were classified as serious AEs. In TRANSFORMS study, four patients treated with fingolimod 1.25 mg and two with fingolimod 0.5 mg developed macular edema. In FREEDOMS II study, four patients (two with fingolimod 1.25 mg, one with fingolimod 0.5 mg, and one with placebo) had serious ocular effects, two of which had sequelae.13,18,20 Its incidence in clinical trials was low (0.5%).51 However, an ophthalmologist examination in the first 3–4 months at the initial stage of the treatment is recommended.7 Patients with diabetes mellitus or inflammatory diseases of the eye (uveitis) have an increased risk of developing this disorder.7 An analysis of clinical trials of fingolimod (Phase II study, Phase III studies FREEDOMS, TRANSFORMS, and extension studies) regarding ophthalmological evaluations, estimated a low incidence of macular edema with fingolimod 0.5 mg. Four patients (0.3%) treated with fingolimod 0.5 mg developed macular edema, and of the total 2615 patients, 19 cases were found to have macular edema. This analysis showed that patients with a history of uveitis have a greater risk of developing macular edema. In fact, 5 (26%) patients in the 1.25 mg fingolimod-treated group had uveitis compared to 26 (1%) of all patients evaluated in the analysis.52 Patients with history of uveitis should undergo an ophthalmologist examination before starting therapy and follow up controls during treatment with fingolimod. Macular edema did not occur with teriflunomide and dimethyl fumarate treatments.
Respiratory adverse effects
Slight reductions in forced expiratory volume in 1 second (FEV1) have been reported with fingolimod 0.5 mg and 1.25 mg compared with placebo in Phase III clinical studies. These reductions were recorded at the first month of the treatment and did not worsen over time. These effects were not significant. In Phase III clinical trials, respiratory or pulmonary adverse effects with teriflunomide and dimethyl fumarate have not been reported. Interstitial lung disease (ILD) with leflunomide has been reported (indicated for rheumatoid arthritis). Since the plasma concentrations of teriflunomide and leflunomide are similar to that of teriflunomide, ILD could occur with teriflunomide.
Cutaneous reactions
Hair thinning was a more frequent adverse effect seen in clinical trials with teriflunomide. It generally occurred in the first 6 months of treatment. Most cases were prevalently mild to moderate in severity. In all clinical studies, incidence of hair thinning was similar in patients treated with teriflunomide. In TOWER, hair thinning occurred in 10% of patients treated with the low dose and 13% with high dose.28 In Phase II extension study, 21% of patients treated with low dose and 27.3% with high dose had hair thinning.41 Hair thinning did not occur in patients treated with fingolimod and dimethyl fumarate.
Immune reactions
Transient skin reactions and hypersensitivity have been reported with teriflunomide in Phase III studies. In TEMSO, these were more frequent with teriflunomide-treated group compared to placebo (10.3% and 11.2% versus 7.2%).26 Also long-term follow-up of Phase II study (up to 8.5 years) showed that these reactions were frequent with both teriflunomide doses. Rashes occurred in 22.2% of patients treated with teriflunomide 7 mg and in 16.7% with teriflunomide 14 mg.41 No severe reactions were noted in clinical studies. Instead, severe skin reactions including Stevens–Johnson syndrome and toxic epidermal necrolysis have been described in post-marketing reports.9 There have been no reports of these reactions with fingolimod and dimethyl fumarate in Phase III clinical trials.
Neurological adverse effects
Peripheral neuropathy, including poly- and mononeuropathy, was another frequent adverse effect of teriflunomide observed in Phase III clinical studies and in their extension studies. Similar incidence was observed with both teriflunomide doses (2%). In some cases, it led to drug discontinuation, but there were no serious events. In Phase II extension study, insomnia and depression were the more frequent adverse effects found with high doses of teriflunomide.41 These effects resolved with time. Depression and headache were also observed with fingolimod and dimethyl fumarate but were not relevant.
GI adverse effects
In Phase III clinical trials, GI adverse effects with teriflunomide treatment were among the most commonly reported reactions. They included nausea, diarrhea, abdominal pain, and vomiting. They were prevalently mild in severity and had similar incidence in both teriflunomide doses. In fact, in TOWER study, nausea occurred in 8% of patients treated with teriflunomide 7 mg and in 10% of patients with teriflunomide 14 mg; diarrhea occurred in 12% and 11% of patients, respectively.28 These reactions were also observed with dimethyl fumarate in Phase III clinical studies. Nausea and diarrhea were the most reported. Gastritis was the single GI effect classified as serious. The incidence of GI effects was higher in the first month of treatment and usually decreased with time. Nature of these reactions was transitory and generally mild to moderate in severity. Nausea was also a common adverse effect with fingolimod in clinical studies.
Renal AEs
Proteinuria was among the most frequently reported adverse effect of dimethyl fumarate in Phase III trials. Incidence was higher in patients treated with dimethyl fumarate compared to placebo. In DEFINE study, proteinuria occurred in 9% of patients with BID regimen dosage and in 12% with TID regimen dosage versus 8% of placebo.33 In CONFIRM study, it was 8% with BID regimen dosage and 10% with TID regimen dosage versus 7% of placebo.34 Overall, the incidence of renal adverse effects was similar in patients treated with dimethyl fumarate and placebo.33,34 Significant renal AEs have not been reported in clinical studies with fingolimod and teriflunomide.
Conclusion
Phase III clinical studies showed that all the three oral agents significantly reduced the relapse rates compared with placebo. In all clinical trials, fingolimod showed consistent reductions in relapse rate and lesions on MRI, and a sustained effect in RRMS activity. Teriflunomide showed efficient ARR and disability progression outcomes comparable to first-line drugs, whereas dimethyl fumarate showed only effective ARR outcomes, the progression of disability of which should be evaluated by additional studies. Therefore, it has been concluded that fingolimod had a higher efficacy in the treatment of RRMS compared to the other two oral agents and it is comparable to infusion medications. Since teriflunomide and dimethyl fumarate are only recently recommended for the treatment of RRMS, further studies are needed to fully evaluate their validity. Fingolimod encountered more safety issues than the other two drugs. Therefore, safety measures should focus on transitory effects, infections, and cancer risks, and require strict monitoring. Teriflunomide also should be monitored for certain parameters, eg, liver enzymes and lymphocytes count. However, given its characterized and manageable adverse effects, it can be considered secure. In conclusion, dimethyl fumarate seems to have a manageable profile. Post-marketing data should validate these findings.
Abbreviations
AE, adverse event; ALT, alanine aminotransferase; ARR, annualized relapse rate; BID, two times daily; Cmax, maximum plasma concentration; CNS, central nervous system; DHODH, dihydro-orotate dehydrogenase; EDSS, expanded disability status scale; EMA, European Medicines Agency; GA, glatiramer acetate; GI, gastrointestinal; IFN β 1a, interferon beta 1a; ILD, Interstitial lung disease; MMF, monomethyl fumarate; MRI, magnetic resonance imaging; MS, multiple sclerosis; MSFC, multiple sclerosis functional composite; NEDA, no evidence of disease activity; Nrf2, nuclear factor erythroid 2 derived factor 2; PML, progressive multifocal encephalopathy; RRMS, relapsing remitting multiple sclerosis; SIP, sphingosine 1 phosphate; TID, three times daily; Tmax, time to reach Cmax; US FDA, United States Food and Drug Administration.
Disclosure
The authors report no conflicts of interest in this work.
References
Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33(2):91–101. | ||
Cherwinski HM, Cohn RG, Cheung P, et al. The immunosuppressant leflunomide inhibits lymphocyte proliferation by inhibiting pyrimidine biosynthesis. J Pharmacol Exp Ther. 1995;275(2):1043–1049. | ||
Ruckemann K, Fairbanks LD, Carrey EA, et al. Leflunomide inhibits pyrimidine de novo synthesis in mitogen-stimulated T-lymphocytes from healthy humans. J Biol Chem. 1998;273(34):21682–21691. | ||
Bar-Or A, Pachner A, Menguy-Vacheron F, Kaplan J, Wiendl H. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs. 2014;74(6):659–674. | ||
Singer B, Comi G, Miller A, et al. Frequency of infections during treatment with teriflunomide: pooled data from three placebo-controlled teriflunomide studies. Neurology. 2013;80(7):Supplement P01.171. | ||
Scannevin RH, Chollate S, Jung MY, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341(1):274–284. | ||
European Medicines Agency (EMA). Allegato I. Riassunto delle carattersitiche del prodotto [Fingolimod summary of product characteristics]. Available from: http://www.ema.europa.eu/docs/it_IT/document_library/EPAR_-_Product_Information/human/002202/WC500104528.pdf. Accessed July 14, 2017. Italian. | ||
US FDA. GILENYA (fingolimod); highlights of prescribing information. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022527s008lbl.pdf. Accessed July 14, 2017. | ||
European Medicines Agency (EMA). Teriflunomide Summary of product characteristics. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002514/WC500148682.pdf. Accessed July 14, 2017. | ||
US FDA. AUBAGIO (teriflunomide); highlights of prescribing information. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202992s000lbl.pdf. Accessed July 14, 2017. | ||
European Medicines Agency (EMA). Dimethyl fumarate; summary of product characteristics. Available from: http://www.ema.europa.eu/docs/it_IT/document_library/EPAR_-_Product_Information/human/002601/WC500162069.pdf. Accessed July 14, 2017. | ||
US FDA. TECFIDERA™ (dimethyl fumarate); highlights of prescribing information. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204063lbl.pdf. Accessed July 14, 2017. | ||
Kappos L, Radue EW, O’Connor P, et al; FREEDOMS Study Group. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. New Engl J Med. 2010;362(5):387–401. | ||
Kappos L, Antel J, Comi G, et al; FTY720 D2201 Study Group. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355(11):1124–1140. | ||
Comi G, O’Connor P, Montalban X, et al; FTY720D2201 Study Group. Phase II study of oral fingolimod (FTY720) in multiple sclerosis: 3-year results. Mult Scler. 2010;16(2):197–207. | ||
Izquierdo G, O’Connor P, Montalban X, et al. Five-year results from a phase 2 study of oral fingolimod in relapsing multiple sclerosis. Mult Scler J. 2014;20(7):877–881. | ||
Kappos L, Radue EW, O’Connor P, et al. Fingolimod treatment increases the proportion of patients who are free from disease activity in multiple sclerosis: results from a phase 3, placebo-controlled study (FREEDOMS). Neurology. 2011;76(Suppl 4):A563. | ||
Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. | ||
Khatri B, Barkhof F, Comi G, Jin J, Francis G, Cohen J. Fingolimod treatment increases the proportion of patients who are free from disease activity in multiple sclerosis compared to IFN-b1a: results from a Phase 3, active-controlled study (TRANSFORMS). Neurology. 2012;78(1):Supplement PD5.006. | ||
Calabresi PA, Radue E-W, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(6):545–556. | ||
Kappos L, O’Connor P, Radue EW, et al. Long-term effects of fingolimod in multiple sclerosis: the randomized FREEDOMS extension trial. Neurology. 2015;84(15):1582–1591. | ||
Cohen JA, Khatri B, Barkhof F, et al; TRANSFORMS (TRial Assessing injectable interferoN vS. FTY720 Oral in RRMS) Study Group. Long-term (up to 4.5 years) treatment with fingolimod in multiple sclerosis: results from the extension of the randomised TRANSFORMS study. J Neurol Neurosurg Psychiatry. 2016;87(5):468–475. | ||
Kappos L, De Stefano N, Freedman MS, et al. Inclusion of brain volume loss in a revised measure of ‘no evidence of disease activity’ (NEDA-4) in relapsing-remitting multiple sclerosis. Mult Scler. 2016;22(10):1297–1305. | ||
Novartis. Novartis real-world data at AAN confirms benefit of Gilenya® on four key measures of disease activity in relapsing MS. Available from: https://novartis.gcs-web.com/static-files/435c32df-e6ed-434d-9a78-3178e9912b8d. Accessed July 14, 2017. | ||
Nixon R, Bergvall N, Tomic D, Sfikas N, Cutter G, Giovannoni G. No evidence of disease activity: indirect comparisons of oral therapies for the treatment of relapsing-remitting multiple sclerosis. Adv Ther. 2014;31(11):1134–1154. | ||
O’Connor P, Wolinsky JS, Confavreux C, et al; TEMSO Trial Group. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–1303. | ||
Freedman M, O’Connor P, Wolinsky J, et al. Teriflunomide increases the proportion of patients free from disease activity in the TEMSO Phase III study. Neurology. 2012;78(1):Supplement PD5.007. | ||
Confavreux C, O’Connor P, Comi G, et al; TOWER Trial Group. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(3):247–256. | ||
O’Connor PW, Lublin FD, Wolinsky JS, et al. Teriflunomide reduces relapse-related neurological sequelae, hospitalizations and steroid use. J Neurol. 2013;260(10):2472–2480. | ||
Miller AE, Wolinsky JS, Kappos L, et al; TOPIC Study Group. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(10):977–986. | ||
Vermersch P, Czlonkowska A, Grimaldi LME, et al; TENERE Trial Group. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler. 2014;20(6):705–716. | ||
Miller AE, Macdonell R, Comi G, et al. Teriflunomide reduces relapses with sequelae and relapses leading to hospitalizations: results from the TOWER study. J Neurol. 2014;261(9):1781–1788. | ||
Gold R, Kappos L, Arnold DL, et al; DEFINE Study Investigators. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. | ||
Fox RJ, Miller DH, Phillips JT, et al; CONFIRM Study Investigators. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. New Engl J Med. 2012;367(12):1087–1097. | ||
Gold R, Arnold DL, Bar-Or A, et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: interim analysis of ENDORSE, a randomized extension study. Mult Scler. 2017;23(2):253–265. | ||
Havrdova E, Gold R, Fox R, et al. BG-12 (dimethyl fumarate) treatment for relapsing-remitting multiple sclerosis (RRMS) increases the proportion of patients free of measured clinical and neuroradiologic disease activity in the phase 3 studies. Neurology. 2013;80(7):Supplement P07.106. | ||
Havrdova E, Giovannoni G, Gold R, et al. Effect of delayed-release dimethyl fumarate on no evidence of disease activity in relapsing-remitting multiple sclerosis: integrated analysis of the phase III DEFINE and CONFIRM studies. Eur J Neurol. 2017;24(5):726–733. | ||
Viglietta V, Miller D, Bar-Or A, et al. Efficacy of delayed-release dimethyl fumarate in relapsing-remitting multiple sclerosis: integrated analysis of the phase 3 trials. Ann Clin Transl Neurol. 2015;2(2):103–118. | ||
Hughes B, Cascione M, Freedman MS, et al; EPOC study investigators. First-dose effects of fingolimod after switching from injectable therapies in the randomized, open-label, multicenter, Evaluate Patient OutComes (EPOC) study in relapsing multiple sclerosis. Mult Scler Relat Disord. 2014;3(5):620–628. | ||
Kappos L, Cohen J, Collins W, et al. Fingolimod in relapsing multiple sclerosis: an integrated analysis of safety findings. Mult Scler Relat Dis. 2014;3(4):494–504. | ||
Confavreux C, Li DK, Freedman MS, et al; Teriflunomide Multiple Sclerosis Trial Group. Long-term follow-up of a phase 2 study of oral teriflunomide in relapsing multiple sclerosis: safety and efficacy results up to 8.5 years. Mult Scler. 2012;18(9):1278–1289. | ||
Arvin AM, Wolinsky JS, Kappos L, et al. Varicella-zoster virus infections in patients treated with fingolimod: risk assessment and consensus recommendations for management. JAMA Neurol. 2015;72(1):31–39. | ||
US FDA. FDA Drug Safety Communication: FDA warns about cases of rare brain infection with MS drug Gilenya (fingolimod) in two patients with no prior exposure to immunosuppressant drugs. Available from: http://www.fda.gov/downloads/Drugs/DrugSafety/UCM457120.pdf. Accessed July 14, 2017. | ||
EMA. New recommendations to minimise risks of the rare brain infection PML and a type of skin cancer with Gilenya. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/12/news_detail_002447.jsp&mid=WC0b01ac058004d5c1. Accessed July 14, 2017. | ||
Killestein J, Vennegoor A, van Golde AEL, Bourez RLJH, Wijlens MLB, Wattjes MP. PML-IRIS during Fingolimod Diagnosed after Natalizumab Discontinuation. Case Rep Neurol Med. 2014;2014:307872. | ||
Calic Z, Cappelen-Smith C, Hodgkinson SJ, McDougall A, Cuganesan R, Brew BJ. Treatment of progressive multifocal leukoencephalopathy-immune reconstitution inflammatory syndrome with intravenous immunoglobulin in a patient with multiple sclerosis treated with fingolimod after discontinuation of natalizumab. J Clin Neurosci. 2015;22(3):598–600. | ||
European Medicines Agency. Updated recommendations to minimise the risk of the rare brain infection PML with Tecfidera. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/10/news_detail_002423.jsp&mid=WC0b01ac058004d5c1. Accessed 14 July, 2017. | ||
Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med. 2015;372(15):1476–1478. | ||
Nieuwkamp DJ, Murk J-L, van Oosten BW, et al; PML in Dutch MS Patients Consortium. PML in a patient without severe lymphocytopenia receiving dimethyl fumarate. N Engl J Med. 2015;372(15):1474–1476. | ||
EMA, New recommendations to minimise risks of the rare brain infection PML and a type of skin cancer with Gilenya. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/12/news_detail_002447.jsp&mid=WC0b01ac058004d5c1. Accessed July 14, 2017. | ||
Jain N, Bhatti MT. Fingolimod-associated macular edema: incidence, detection, and management. Neurology. 2012;78(9):672–680. | ||
Zarbin MA, Jampol LM, Jager RD, et al. Ophthalmic evaluations in clinical studies of fingolimod (FTY720) in multiple sclerosis. Ophthalmology. 2013;120(7):1432–1439. | ||
Sweetser MT, Dawson KT, Bozic C. Manufacturer’s response to case reports of PML. N Engl J Med. 2013;368(17):1659–1661. | ||
Van Oosten BW, Killestein J, Barkhof F, Polman CH, Wattjes MP. PML in a patient treated with dimethyl fumarate from a compounding pharmacy. N Engl J Med. 2013;368(17):1658–1659. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.