")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Comparative fasting bioavailability of two clopidogrel formulations in healthy Mediterranean volunteers: an in vitro–in vivo correlation
Authors Zaid AN , Al Ramahi R, Bustami R, Mousa A, Khasawneh S
Received 3 December 2014
Accepted for publication 17 February 2015
Published 24 April 2015 Volume 2015:9 Pages 2359—2365
DOI https://doi.org/10.2147/DDDT.S78658
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Abdel Naser Zaid,1 Rowa’ Al Ramahi,1 Rana Bustami,2 Ayman Mousa,3 Sewar Khasawneh2
1Department of Pharmacy, Faculty of Medicine and Health Sciences, An-Najah National University, Nablus, Palestine; 2Pharmaceutical Research Unit, Amman, Jordan; 3R&D Department, Middle East Pharmaceutical Industries Co Ltd, Riyadh, Saudi Arabia
Objective: The aim of this study was to evaluate the bioequivalence of two drug products, generic clopidogrel bisulfate 75 mg film-coated tablets versus the reference Plavix® clopidogrel bisulfate 75 mg film-coated tablets.
Methods: Bioequivalence of tablets was tested by comparisons against the reference brand product in accordance with the requirements of the Declaration of Helsinki, the current Good Clinical Practice Guidelines, and the International Conference on Harmonization.
Results: The relationship between concentration and peak area ratio was found to be linear within the range 24.500–1,836.600 pg/mL for clopidogrel. The correlation coefficient (r) was always greater than 0.99 during the course of the validation. Statistical comparison of the main pharmacokinetic parameters showed no significant difference between test and reference. The point estimates (ratios of geometric mean) were 104.122%, 104.184%, and 109.091% for areas under the plasma concentration–time curve (AUC) AUC0-last, AUC0-∞, and peak plasma concentration Cmax, respectively. These pharmacokinetic parameter values of clopidogrel and its main metabolite lie within the bioequivalence limit (80%–125%) specified by the US Food and Drug Administration and the European Medicines Agency.
Conclusion: The tested drug product was bioequivalent to the reference drug under fasting conditions and had the same safety profile, which is important to achieve equivalent therapeutic effect with the reference.
Keywords: film coating, stability, clopidogrel, bioequivalence
Introduction
Clopidogrel is a novel thienopyridine inhibitor of adenosine diphosphate (ADP)–induced platelet activation (Figure 1). It blocks the activation of platelets by ADP by selectively and irreversibly inhibiting the binding of the agonist to its receptor on the platelet, thereby affecting ADP-dependent activation of the fibrinogen receptor present on the platelet surface.1
 | Figure 1 Chemical structure of clopidogrel. |
Clopidogrel is a prodrug and requires complex metabolic activation in the liver. Up to 85% of the absorbed drug can be transformed by carboxyl esterases into a carboxylic acid derivative of clopidogrel, the major metabolite circulating in the blood; the carboxylic acid derivative of clopidogrel is inactive. Only 15% of the absorbed clopidogrel dose is transformed by isoenzymes of cytochrome P450 (CYP), namely, CYP1A2, CYP2B6, CYP2C9, CYP2C19, and CYP3A4, to a thiol metabolite, which is responsible for the antithrombotic effect of clopidogrel.2,3
Clopidogrel is widely used these days, especially as a part of dual antiplatelet therapy with aspirin after coronary interventions and stent placement. It is standard of care in patients experiencing acute coronary syndromes.4 Clinical response to clopidogrel varies widely. This might be due to underdosing, drug interactions, or intrinsic interindividual differences caused by genetic polymorphisms.3 Clopidogrel has become generic after expiration of the pharmaceutical patent of clopidogrel in May 2012. Several commercial preparations are available now, and it is of critical importance to demonstrate that these preparations are bioequivalent and have similar inhibition of platelet reactivity.5 During the development of new generic formulations, the formulators try to adhere to the same excipients used in the brand product and the manufacturing process as much as they can to minimize problems with stability. However, it is important to conduct an in vivo study to find any difference between the generic product and the original brand in terms of safety and clinical outcome.
This is a bioequivalence study aimed at comparing two drug products (the reference brand clopidogrel bisulfate [Plavix®] and the test generic clopidogrel bisulfate), and the key pharmacokinetic (PK) parameters for both the drugs and their metabolites were assessed. This generic is expected to be marketed in the Mediterranean, so it is important to test its bioequivalency in this group of people because clopidogrel is highly affected by genetic polymorphisms.
Materials and methods
Chemicals
Clopidogrel bisulfate working standard was supplied by Dr Reddy’s Laboratories Ltd (Hyderabad, India), while Clopidogrel-d4 hydrogen sulfate internal standard (IS) was supplied by TRC (North York, Canada). High-performance liquid chromatography (HPLC)-grade methanol and acetonitrile were purchased from Romil (Cambridge, United Kingdom), isopropanol was obtained from Carbon Group (Cork, Ireland), extrapure formic acid was obtained from Scharlau (Port Adelaide, Australia), diethylether was brought from JHD (Guangzhou, People’s Republic of China), and HPLC-grade water was supplied by Sartorius Purified Water (Goettingen, Germany).
The study was a comparative, randomized, two-period, two-treatment, two-sequence, single-dose, open-label, crossover bioequivalence study of clopidogrel 75 mg film-coated tablets (Pladogrel, Avalon Pharma, Middle East Pharmaceutical Industries Co Ltd, Saudi Arabia) versus Plavix® 75 mg film-coated tablets (Sanofi Winthrop Industrie, Cedex, France) in healthy subjects under fasting conditions. Randomization tablet was generated using software SAS version 9.4.
Volunteers and clinical protocol
The study was conducted by Arab Pharmaceutical Industry Consulting Co Ltd, Jordan, in accordance with the requirements of the Declaration of Helsinki,6 the current Good Clinical Practice guidelines,7 and the International Conference on Harmonization8 guidelines. The study protocol and the informed consent forms were approved by the institutional review board. The study included 38 healthy, mixed skin Arab and Mediterranean subjects aged between 19 years and 46 years, with a body mass index of 18.9–30.0 kg/m2 (minimum of 52 kg weight), as well as nonsmokers or light smokers (smokers of not more than 10 cigarettes per day). The volunteers were subjected to a full medical and physical examination to confirm their health status and to verify that they were not on any medication during the study period for full adherence with the inclusion criteria. A written informed consent, which explained the nature of the study, was given to the participants. The participants were instructed to abstain from taking alcohol, caffeine, xanthine, grapefruit, or CO2-containing beverages during the study to avoid any induction or inhibition of liver enzymes. Smoking was prohibited for 11 hours prior to dosing and 4 hours after the drug administration, after which smoking of five cigarettes was allowed during each study period. The study used an open-label, randomized two-period crossover design with an 8-day washout period between doses in 38 healthy subjects under fasting conditions. The volunteers were randomly divided into two groups each of 19 subjects. The first group was given the reference brand, and the second group was given the test formulation with a crossover after the washout period. On the morning of the study, each volunteer gave a blood sample to serve as a blank for the drug assay. Each participant received an oral dose of the assigned formulation, given with 240 mL of water in the sitting position. During each period, 22 blood samples were collected; 8 mL was collected 1 hour before dosing, and 8 mL sample was withdrawn at the following time points: 0.25, 0.50, 0.75, 1.00, 1.33, 1.67, 2.00, 2.33, 2.67, 3.00, 3.50, 4.00, 5.00, 6.00, 8.00, 10.00, 12.00, 16.00, 24.00, 36.00, and 48.00 hours respectively, postdose during each study period. Samples were immediately collected into sodium-heparinized tubes and centrifuged at 5,000 rpm for 5 minutes at 4°C within 30 minutes of collection. The resulting plasma was immediately stored at −70°C until analysis. Methanol-free conditions were used in the extraction procedure to avoid a back-conversion of the metabolite clopidogrel carboxylic acid to the parent drug. Four hours after drug administration, a standard lunch containing soup (no carrots), half a chicken, and rice with mixed vegetables (no carrots), yogurt, a loaf of bread, and salad (tomato and cucumber) was served and subjects had free access to water 1 hour after drug administration.
Instruments and chromatographic separations
The analysis was performed using an HPLC system (Agilent 1200) coupled with a mass spectrometry (MS) system API4000. The stationary phase was ACE 5 C8 (50×2.10) mm, 5m. The mobile phase was acetonitrile: H2O: formic acid (60:40:0.1, v/v/v); measured pH for mobile phase =3.00±0.2.
The dissolution study was carried out using dissolution tester (Pharmatest, Model PTWS 610, Germany).
Preparation of standard solutions
A drug solution was prepared by dissolving 0.2008 g clopidogrel bisulfate (assay: 99.62%; limit of detectionfactor: 0.9983, and conversion factor: 0.7664) with methanol up to 50.00 mL final volume to make up a stock solution containing 306.095 μg/mL.
Sample preparations for HPLC injection
A 0.500 mL volume of each plasma sample was spiked with 100 μL of IS (concentration 3: 3.945 ng/mL), to get IS with final concentration of 0.658 ng/mL Clopidogrel-d4 (based on 0.600 mL final volume). Each sample was vortexed for 30 seconds.
Validation procedures
A simple, fast, selective, accurate, and precise HPLC method coupled with tandem MS–MS, for the determination of clopidogrel in human plasma has been developed and validated. In brief, the linearity study was carried out in the range of concentrations from 24.500 pg/mL to 1,836.600 pg/mL. The lower limit of quantitation (LLOQ) was estimated by analyzing known samples of clopidogrel at progressively lower concentrations. The coefficient of variation was used to determine assay precision.
Stock solution stability in mobile phase was assessed using two standard mixtures that are equivalent to the LLOQ and the upper limit of quantitation concentrations with IS.
Short- and long-term matrix-based stability levels were assessed using two quality control (QC) low and QC high clopidogrel concentrations.
Stability after freeze-and-thaw cycles was assessed using two sets of QC samples that were subjected to three freeze-thaw cycles (stability samples). Stability samples were processed with a freshly prepared calibration curve and analyzed in a single run with comparison QC sample (Comparison sample).
Whole blood stability was assessed by spiking whole-blood samples with two different drug concentrations. Recovery for the drug and IS was assessed using six extracts at three concentration levels (low, medium, and high).
Matrix effect was investigated for clopidogrel and the IS. The matrix factor (MF) was calculated in each lot of matrix, by calculating the ratio of the peak area in the presence of matrix, to the peak area in the absence of matrix (pure solution of clopidogrel). The IS-normalized MF was also calculated by dividing the MF of the analyte by the MF of the IS.
The QC samples were used to evaluate the performance of the assay. They were prepared by spiking blank plasma with clopidogrel. The QC samples were prepared to have low, medium, and high concentrations (clopidogrel: 73.500 pg/mL, 196.000 pg/mL, 918.300 pg/mL, and 1,377.450 pg/mL). Four QC samples were incorporated with each analysis run as unknown samples. The concentration in each QC sample was determined from the calibration curve, and it was compared with the nominal concentration. The analysis run was accepted if at least three out of four QC samples were within 15% of nominal concentration.
In vitro dissolution profile of clopidogrel tablet
In vitro dissolution study was carried out on 12 tablets from each of the tested drug and Plavix®. USP Type II apparatus (paddle assembly) was used for this test. Acid buffer (1,000 mL) at pH 2 was used as the dissolution medium. The dissolution medium in the paddles was mixed at a speed of 50 rpm, and the bath was maintained at a temperature of 37°C±0.5°C.9
Samples of 5 mL were withdrawn at regular intervals of times. Same volume of fresh dissolution medium was added to the paddles after each sample was taken.
The withdrawn samples were analyzed after suitable dilutions at a wavelength of 240 nm using an ultraviolet spectrophotometer (Thermo Scientific, Helios, Germany).
The in vitro drug release profile of the test was compared versus that of the brand using similarity factor f2 and nonsimilarity factor f1 according to the US Food and Drug Administration (FDA) guidelines.10
PK and statistical analysis
The PK parameters were estimated using standard noncompartmental methods. The peak plasma concentration (Cmax) and the corresponding time of peak plasma concentration (Tmax) were taken directly from the data. The elimination rate constant (ke) was calculated from the slope of the semilogarithmic plot of the terminal elimination phase of the plasma concentration–time curve calculated by linear regression. The elimination half-life (t1/2) was calculated using the formula t1/2= ln2/ke. The areas under the drug’s plasma concentration–time curves from AUC0-last and the area to infinity (AUC0-∞) were calculated by using the linear trapezoidal method. Extrapolation to infinity was done by dividing the last measurable plasma concentration Clast by the terminal rate constant ke (AUClast→inf = Clast/ke). The AUC0→inf value is the sum of the estimated and extrapolated parts (AUC0→inf = AUC0→last + AUClast→inf). For the purpose of bioequivalence analysis, one-way analysis of variance (ANOVA) was used to assess the effect of formulations, periods, sequences, and subjects on AUC0-last, AUC0-∞, and Cmax using Thermo Scientific Kinetica (version 5.1), a commercially available software package.
In vitro–in vivo correlation
Level A in vitro–in vivo correlation (IVIVC) was elaborated at different time intervals to assess whether a correlation existed between the in vitro drug release and the in vivo absorption of drug using the Wagner–Nelson equation.11
Results
Results of validation procedures
The relationship between concentration and peak area ratio was found to be linear within the range 24.500–1,836.600 pg/mL for clopidogrel. The correlation coefficient (r) was always greater than 0.99 during the course of the validation. The method was found to be sensitive, with LLOQ of 24.00 pg/mL; recovery of the drug ranged from 24.44% to 26.24%, and mean recovery of IS was 49.92%. Short-term stability analysis of the drug in plasma proved that the drug is stable for up to 24 hours at room temperature. Stock solution short-term stability proved that the drug is stable for up to 17 hours at room temperature. Postpreparative stability study showed that the drug is stable for up to 48 hours. Whole-blood stability study proved that the drug is stable for 1 hour. Samples with concentrations of up to 5,509.800 pg/mL can be diluted with dilution factor of three with adequate accuracy and precision. No carry-over or matrix effect was found.
In vitro drug-release studies were carried out because these are considered a useful tool for assessing in vivo drug behavior. The in vitro release of clopidogrel from the tested drug and Plavix® tablet formulations was within the acceptable limits. The calculated f2 and f1 values between these two products were 60 and 9, respectively.
Results of PK study
Both generic clopidogrel 75 mg film-coated tablets and Plavix® 75 mg film-coated tablets were well tolerated by all the subjects, and they were all discharged in good health. Figure 2 shows the plasma concentrations of both brands, indicating that the two brands are superimposable.
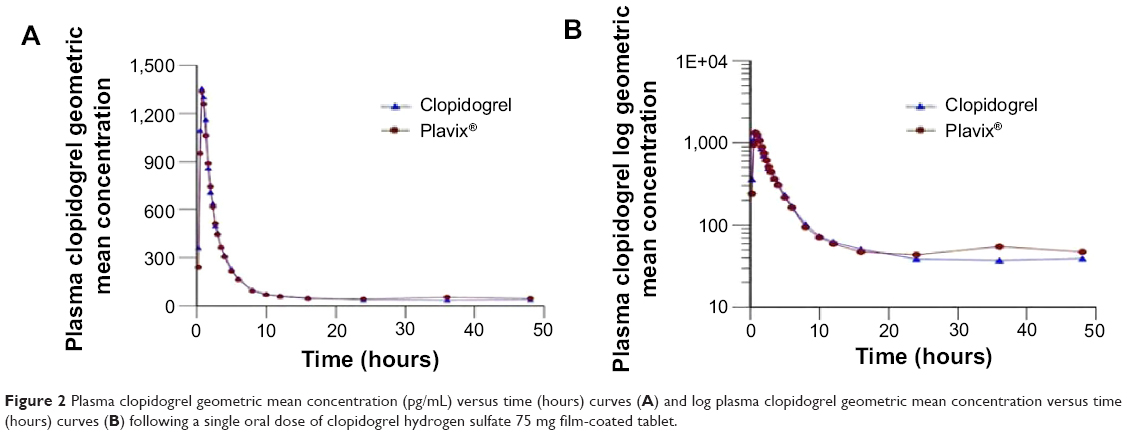 | Figure 2 Plasma clopidogrel geometric mean concentration (pg/mL) versus time (hours) curves (A) and log plasma clopidogrel geometric mean concentration versus time (hours) curves (B) following a single oral dose of clopidogrel hydrogen sulfate 75 mg film-coated tablet. |
All estimated PK parameters were in agreement with reported values. Tables 1 and 2 show a summary of the PK parameters for the two formulations of clopidogrel 75 mg and the metabolite clopidogrel carboxylic acid. The point estimates (ratios of geometric mean) were 104.122%, 104.184%, and 109.091% for AUC0-last, AUC0-∞, and Cmax, respectively. No statistically significant difference between the two formulations was found. These PK parameter values lie within the bioequivalence limits (80%–125%) specified by the FDA and the European Medicines Agency (EMEA).12,13 The results in this part of the study suggest equivalent clinical efficacy of the two brands. Moreover, no serious adverse effects were observed during this study. Overall, clopidogrel showed a good safety profile. In fact, bleeding is the most important adverse effect of clopidogrel, which might be even increased when clopidogrel is used in combination with salicyclic acid. No significant changes in the subjects’ state of health were reported.
 | Table 1 Summary of calculated pharmacokinetic parameters of clopidogrel in the bioequivalence study (n=38) |
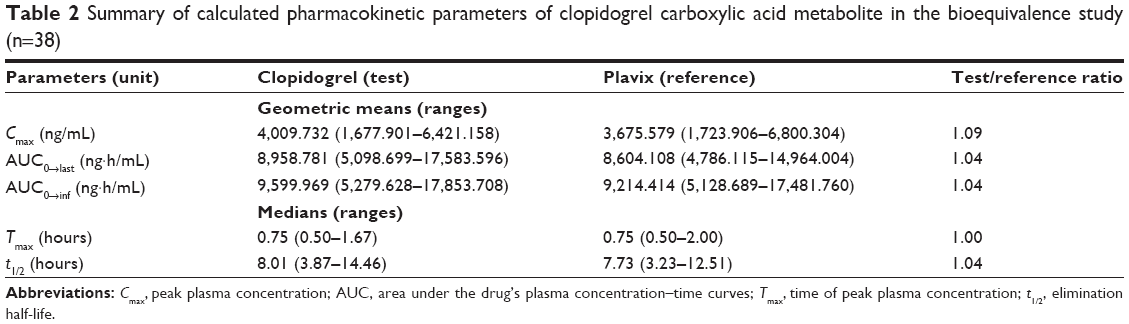 | Table 2 Summary of calculated pharmacokinetic parameters of clopidogrel carboxylic acid metabolite in the bioequivalence study (n=38) |
High level A IVIVC was found between the amount of absorbed clopidogrel and the amount released at the used time intervals. The correlation coefficient between the dissolved and absorbed clopidogrel was 0.99 and the P-value was 0.0018.
Discussion
According to The FDA, brand name drug is defined as a drug marketed under a proprietary, trademark-protected name and a generic drug is the same as a brand name drug in terms of active ingredient, dosage form, safety, strength, route of administration, quality, performance, and intended use and, additionally, it contains the same salt, ester, or chemical form. Generic versions of a drug can vary in shape, scoring configuration, packaging, and recipients. If all the previous criteria are met, then the two drugs are considered to be therapeutically equivalent.14
During the development phase of an oral solid dosage form, several preformulation and formulation trials, as well as physiochemical tests, are carried out in order to achieve a high-quality generic product that is interchangeable with the original brand in terms of efficacy and safety. Accordingly, in vitro dissolution in media of different pH values is conducted on the generic product and it must show similar dissolution profile or a profile overlapping that of the reference brand. Only if the generic shows a similar factor (f2) higher than 50% and a nonsimilar factor (f1) less than 15% in all these media can we predict a successful bioequivalence profile of the product, especially if the drug is a Class I product according to the Biopharmaceutics Classification System. In this case, bioequivalence studies can be waived and the above-mentioned in vitro dissolution can represent a surrogate to bioequivalence.15 Clopidogrel is a Class II product, which means that in vitro dissolution is not sufficient to prove that it is interchangeable with the original brand.16 Accordingly, clopidogrel generic tablets should be subjected to bioequivalence study in order to prove efficacy and safety comparable with those of the original brand.
Several previous studies have tested the bioequivalence of new generic clopidogrel against Plavix® tablets3,17,18 or the antiplatelet effect of other salts.5,19 The aim of this study was to test the bioavailability of new generic clopidogrel 75 mg tablet versus the reference Plavix. The two dosage forms were administered to 38 fasting volunteers in order to eliminate the influence of food on drug absorption. Clopidogrel is a prodrug that requires biotransformation to the active metabolite by the polymorphic CYP2C19. Carriage of genetic variants encoding for nonfunctional CYP2C19 enzyme was associated with an increased risk of stent thrombosis and the combined end point of cardiovascular death, myocardial infarction, or stroke. The FDA20 required a black box warning about the reduced effectiveness of clopidogrel in homozygous carriers of a CYP2C19 nonfunctional allele.
A previous study concluded that measurement of clopidogrel bisulfate concentrations alone should not be a reliable method for bioequivalence assessment due to the very low concentrations, very small and variable values of AUC, and high intrasubject variability. Thus, bioequivalence evaluation should be based on clopidogrel carboxylic acid metabolite determination.21 So, in this study, both were tested. The validated analytical methods described above were used for quantification of clopidogrel and its main metabolite. The validated analytical methods provided appropriate accuracy, sensitivity, and selectivity with high sample throughput and economically convenient procedure required for PK studies. For this study, several PK parameters were tested using a validated HPLC–MS method. In fact, all validation parameters were conducted according to the international guidelines and they were within the accepted limits, as reported in Table 1.
Regarding the efficacy of our generic product, statistical comparison of the main PK parameters – AUC0-last, AUC0-∞, Cmax, and Tmax – clearly indicated no significant difference between test and reference tablets in any of the calculated PK parameters for clopidogrel bisulfate and clopidogrel carboxylic acid metabolite. The obtained values were compliant with the FDA and EMEA requirements for bioequivalence of generic drugs since the AUC0-∞, AUC0-last, and Cmax mean ratios are within the 80%–125% interval.12,13 It was concluded that the test tablets (clopidogrel 75 mg film-coated tablets) are bioequivalent to the commercial Plavix® tablet in terms of both extent and rate of absorption after a single oral dose of each to healthy adult participants under fasting conditions. Evaluation based on clopidogrel carboxylic acid metabolite, unequivocally and with the proper power, confirmed the bioequivalence between the investigated clopidogrel products.
Safety is important also; in our study, all participants completed the study without showing any serious adverse effect and were discharged in good health.
The high level A of IVIVC indicates that the dissolution method presented in this study can be used for routine quality control and as an important tool in order to save money and time in any future production or development of varying-strength clopidogrel tablets.
Limitations of this study include the relatively small number of patients enrolled and the nonblinded method of clopidogrel administration.
Conclusion
The statistical analysis of the results performed on AUC0-last, AUC0-∞, and Cmax using the ANOVA method showed that both test tablets and reference tablets (Plavix® 75 mg) are bioequivalent since they deliver equivalent quantities of active ingredient to the systemic circulation at equivalent rates for both AUC0-last and Cmax ratios within the 80%–125% interval proposed by the FDA and the EMEA. These results show the good formulation of this new generic tablet, which is important to achieve equivalent therapeutic benefits and avoid any potential problems that may arise due to poor formulation.
Disclosure
The authors report no conflicts of interest in this work.
References
Rao TR, Usha PR, Naidu MU, Gogtay JA, Meena M. Bioequivalence and tolerability study of two brands of clopidogrel tablets, using inhibition of platelet aggregation and pharmacodynamic measures. Curr Ther Res Clin Exp. 2003;64(9):685–696. | ||
Karaźniewicz-Łada M, Danielak D, Burchardt P, et al. Clinical pharmacokinetics of clopidogrel and its metabolites in patients with cardiovascular diseases. Clin Pharmacokinet. 2014;53(2):155–164. | ||
Garcés-Eisele J, Ruiz-Argüelles A, Estrada-Marín L, et al. Pharmacogenetic selection of volunteers increases stringency of bioequivalence studies; the case of clopidogrel. Indian J Pharm Sci. 2014;76(4):281–286. | ||
O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;127(4):e362–e425. | ||
Oberhänsli M, Lehner C, Puricel S, et al. A randomized comparison of platelet reactivity in patients after treatment with various commercial clopidogrel preparations: the CLO-CLO trial. Arch Cardiovasc Dis. 2012;105(11):587–592. | ||
World Medical Association. “Declaration of Helsinike” as Amended by the 59th World Medical Assembly. Seoul, South Korea: World Medical Association, Inc; 2008. | ||
European Medicines Agency. Note for Guidance on Good Clinical Practice. London, UK: European Medicines Agency; 1997. (CPMP/ICH/135/95). | ||
ICH. ICH harmonization tripartite guideline. Guidelines for good clinical practice. In: International Conference of Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human uses; 1996; Geneva, Switzerland. | ||
USP 37–NF 32. Rockville, MD: The United States Pharmacopeial Convention, Inc; 2015:2424–2425. | ||
Food and Drug Administration, CDER, USA. Guidance for Industry Dissolution Testing for Immediate Release Solid Oral Dosage Forms. Silver Spring, MD: Food and Drug Administration, CDER; 1997. | ||
Shargel L, Wu-Pong S, Yu AB. Applied Biopharmaceutics and Pharmacokinetics. 5th ed. New York, NY: McGraw-Hill; 2005:123–131. | ||
Food and Drug Administration. Guidance for Industry: Statistical Approaches to Establishing Bioequivalence. 2001. Available from: http://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_SearchResults_Dissolutions.cfm?PrintAll=1 | ||
European Medicines Agency. Note for Guidance on the Investigation of Bioavailability and Bioequivalence. London, UK: European Medicines Agency; 2001. (CPMP/EWP/QWP/1401/98). | ||
Food and Drug Administration. Glossary of Terms. [webpage on the Internet]. U.S. Food and Drug Administration [cited February 02, 2012]. Available from: http://www.fda.gov/drugs/informationondrugs/ucm079436.htm | ||
WHO. General Notes on Biopharmaceutics Classification System (BCS)-Based Biowaiver Applications. Geneva: WHO Prequalification of Medicines Programme; 2011. | ||
Lassoued MA, Khemiss F, Sfar S. Comparative study of two in vitro methods for assessing drug absorption: Sartorius SM 16750 apparatus versus everted gut sac. J Pharm Pharm Sci. 2011;14(1):117–127. | ||
Brvar N, Lachance S, Lévesque A, et al. Comparative bioavailability of two oral formulations of clopidogrel: determination of clopidogrel and its carboxylic acid metabolite (SR26334) under fasting and fed conditions in healthy subjects. Acta Pharm. 2014;64(1):45–62. | ||
Setiawati E, Yunaidi DA, Handayani LR, Santoso ID, Setiawati A, Tjandrawinata RR. Bioequivalence study of two clopidogrel film-coated tablet formulations in healthy volunteers. Arzneimittelforschung. 2011;61(12):681–684. | ||
Sambu N, Radhakrishnan A, Curzen N. A randomized crossover study comparing the antiplatelet effect of plavix versus generic clopidogrel. J Cardiovasc Pharmacol. 2012;60(6):495–501. | ||
Wilffert B, Swen J, Mulder H, et al; KNMP Working Group Pharmacogenetics. Pharmacogenetics. From evidence based medicine to mechanism based medicine. Reviewing the role of pharmacogenetics. Int J Clin Pharm. 2013;35(3):369–375. | ||
Pawłowska M, Duda J, Tejchman-Małecka B, Bogiel M, Marzec A, Sieradzki E. Usefulness of the parent compound determination in bioequivalence evaluation of clopidogrel generic products. Arzneimittelforschung. 2009;59(6):289–296. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.