")
Back to Journals » International Journal of Nanomedicine » Volume 14
Co-delivery doxorubicin and silybin for anti-hepatoma via enhanced oral hepatic-targeted efficiency
Authors Li Y, Yang D, Wang Y, Li Z, Zhu C
Received 17 September 2018
Accepted for publication 1 December 2018
Published 28 December 2018 Volume 2019:14 Pages 301—315
DOI https://doi.org/10.2147/IJN.S187888
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Mian Wang
Ying Li, Dandan Yang, Yian Wang, Zhan Li, Chunyan Zhu
Drug Delivery Research Center, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100193, People’s Republic of China
Background: To establish the combination of doxorubicin (DOX) and silybin (SLB) in oral hepatic-targeting liposomes with the goal of reducing cardiotoxic side effects and improve oral hepatoma treatment.
Methods: Distearoylphosphatidylethanolamine–polyethylene glycol–cholic acid-modified liposomes (CA-LP) were used to encapsulate DOX and SLB (CA-LP–DOX/SLB), and the hepatic targeting, efficacy against hepatoma and cardioprotective effects were evaluated by cell toxicity, scratch and apoptosis in vitro studies, and pharmacokinetics and pharmacodynamics in vivo studies.
Results: In vitro cell studies showed that CA-LP–DOX/SLB inhibited HepG2 cell proliferation and HCC97H cell migration, and protected H9c2 cells. In vivo pharmacokinetics demonstrated that the CA-LP–DOX/SLB-treated group showed higher liver accumulation and lower heart accumulation of DOX relative to those in the CA-LP–DOX and LP–DOX-treated groups. In vivo pharmacodynamic studies showed that the CA-LP–DOX/SLB-treated group not only efficiently inhibited growth but also induced significantly less tissue damage than that observed in the CA-LP–DOX-treated group.
Conclusion: Concurrent administration of DOX and SLB via CA-LP provided a viable strategy to mitigate acute DOX-induced cardiotoxicity.
Keywords: doxorubicin, silybin, hepatic targeting via oral administration, cholic acid transporter, anti-hepatoma, biodistribution in vivo
Introduction
Hepatic cancer (HCC) is one of the most widespread malignancies and is the third leading cause of tumor-related deaths worldwide. The prognosis of HCC patients remains poor because of metastasis and a high recurrence rate.1–3
Oral treatments are noninvasive and cost-effective and can be prescribed on an outpatient basis, which improves patient compliance and quality of life, particularly for elderly patients and patients with advanced or relapsed cancer. Doxorubicin (DOX) is a popular anti-HCC agent that causes DNA damage and activates apoptosis.4,5 The chemotherapeutic use of DOX has some limitations in oral antitumor treatment, mainly serious cytotoxic effects on normal tissues and poor membrane permeation, especially cardiotoxicity.6 Although the exact mechanism of cardiotoxicity remains unknown, it is believed that redox cycling of the quinolone ring in DOX generates reactive oxygen and nitrogen species resulting in extensive cardiac damage, which limits its full potential as a therapeutic option.7–10 Because DOX continues to be a mainstay in chemotherapy, the search for a safe and effective remedy to reverse DOX-induced cardiotoxicity remains a critical issue in both cardiology and oncology.
Combining DOX with other agents to mitigate highly cardiotoxic adverse effects as a chemotherapeutic strategy has strong merit. Various natural products have demonstrated cardioprotective effect in vitro and in vivo.11–14 Among such natural products, silybin (SLB), a kind of water-insoluble flavonoid molecule, has demonstrated cardioprotective effect.15–17 SLB is a strong antioxidant and free radical scavenger that affords protection against cardiovascular disease.18 SLB exhibits cardioprotective effects through various mechanisms, such as antioxidative, free radical scavenging, and lipid peroxidation inhibitory mechanisms. Despite the advantages of co-administration of DOX and SLB, the different physicochemical and pharmacokinetic profiles of each drug render optimal delivery challenging. To achieve maximal intracellular synergistic effects, it is expected that multiple drugs should be simultaneously delivered to the same cells at an optimized ratio. Co-delivery of DOX and SLB in an oral hepatic targeting drug delivery system is an effective strategy to increase efficacy and decrease adverse effects.
Cholic acid transporters, such as apical sodium-dependent bile salt transporter (ASBT) and Na+/taurocholate co-transporting polypeptide (NTCP), are promising targets for oral hepatocyte-specific delivery and are predominantly present in large numbers on intestinal epithelium cell and sinusoidal hepatocyte cell membranes, respectively. Hence, ASBT and NTCP targeting could provide a viable strategy for oral delivery of DOX to hepatocytes.19–23 Furthermore, cholic acid and its derivatives can specifically interact with ASBT and NTCP as drugs that can actively target the liver by using cholic acid as carriers.
In a previous study, we synthesized a novel biodegradable polymer, distearoylphosphatidylethanolamine (DSPE)–polyethylene glycol (PEG)–cholic acid, with oral hepatic targeting properties, and established DSPE–PEG–cholic acid-modified liposomes (CA-LP). Liposomes (LPs) can load water-soluble drugs and liposoluble drugs simultaneously. Oral hepatic targeting efficiency evaluation in vitro and in vivo results demonstrated that CA-LP efficiently delivered drugs to the liver.24,25
The present study aim was to develop an actively targeted co-delivery system of DOX and SLB with CA-LP (CA-LP–DOX/SLB) that could selectively target the liver, and be efficiently internalized into liver cells. CA-LP–DOX/SLB were prepared and characterized. The release in vitro, stability in vitro, cellular uptake, cytotoxicity, apoptosis, and the effect on cell migration were evaluated in vitro. Furthermore, the pharmacokinetics, biodistribution, antitumor effects, and systemic toxicity of different formulations were investigated in mice and tumor-bearing mice.
Methods
Animals
Male ICR mice (18–22 g) and male BALB/c nude mice (18–22 g) were purchased from Vital River Co. (Beijing, People’s Republic of China), and kept under specific pathogen-free condition for 1 week before the study, with free access to standard food and water. All studies in mice were performed in accordance with guidelines approved by the Ethics Committee of the Chinese Academy of Medical Sciences and Peking Union Medical College. The committee has approved the experiments.
Preparation of CA-LP–DOX/SLB
Briefly, DSPE–PEG–cholic acid, phospholipid (Lipoid, Ludwigshafen, Germany), and cholesterol (Sigma-Aldrich Co., St Louis, MO, USA) were combined into LPs. SLB (Sino-herb Bio-technology Co., Ltd, Xi’an, People’s Republic of China) was loaded into LPs by using the ethanol injection method. DOX (Beijing Huafeng United Technology, Co. Ltd., Beijing, People’s Republic of China) was actively loaded into preformed LPs by using the ammonium sulfate gradient method. Subsequently, DSPE–PEG–cholic acid (3 mg/mL), phospholipid (40 mg/mL), cholesterol (8 mg/mL), and SLB (5 mg/mL) were dissolved in ethanol, and the solutions were then mixed with aqueous ammonium sulfate and magnetically stirred at a speed of 1,000 rpm until the solvents were removed. The resulting multilamellar vesicles were stepwise extruded by using a Lipex extruder (Northern Lipids, Inc., Burnaby, BC, Canada). The transmembrane ammonium sulfate gradient was generated by removal of external ammonium sulfate and subsequent replacement in three dialysis steps. CA-LP-SLB and DOX solution (1 mg/mL) were then mixed and incubated. The free drugs were removed by ultrafiltration to give CA-LP–DOX/SLB.
Characterization of CA-LP–DOX/SLB
The particle sizes and polydispersity index (PDI) of CA-LP–DOX/SLB were measured by using a Nano Series ZS Zetasizer instrument (Malvern Instruments Ltd., Malvern, Worcestershire, UK). Subsequently, CA-LP–DOX/SLB were stained with 2% uranyl acetate, and the morphologies of CA-LP–DOX/SLB were then examined by using transmission electron microscopy (TEM; JEM-1400, JEOL, Tokyo, Japan). DOX and SLB contents were determined by using a HPLC system with an ultraviolet (UV) detector (Shimadzu, Kyoto, Japan) and a C18 column (Kromasil, 250×4.6 mm, 5 μm), with a column temperature of 30°C. The mobile phase comprised methanol and 1% acetic acid in water (52/48, v/v), and the flow rate was 1.0 mL/min. Analytes were detected at 233 nm, and all samples were analyzed in triplicate. To estimate the encapsulating efficiency (EE%) of DOX and SLB, the CA-LP–DOX/SLB suspensions were destroyed by adding methanol. The resulting solutions were then thoroughly diluted prior to HPLC analysis. The EE% of DOX or SLB was calculated as follows:
|
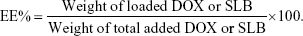
In vitro release
In vitro release of DOX and SLB from CA-LP–DOX/SLB was performed by using a dialysis method. Specifically, drug release study was conducted to determine the release profile of both DOX and SLB in PBS at pH 2 or 7.4. In brief, 1 mL of CA-LP–DOX/SLB suspension (SLB 1 mg/mL, DOX 0.33 mg/mL) was loaded into a dialysis bag (molecular weight cut off =10–14 kDa). The dialysis bag was immersed into 50 mL PBS (pH 7.4 or 2) containing SDS (3%, v/v) at 37°C under constant shaking in a water bath shaker. At predetermined time intervals, 1.0 mL of the dialysis medium outside of the dialysis bag was collected and replaced with 1.0 mL of the respective medium to keep the volume constant at 50 mL. Then, 1 mL of the release medium was filtered. The amounts of DOX and SLB released into the collected medium were quantified by using HPLC with UV detection. The in vitro release studies were conducted in triplicate. The data were expressed as the cumulative percentage of drug released as a function of time.
Stability study
A stability study was conducted according to the Chinese pharmacopeia, as follows: simulated gastric fluid (SGF): adding about 800 mL water and 10 g pepsin (800–2,500 U/mg, Sigma-Aldrich Co.) in 1 mol/L hydrochloric acid (16.4 mL), shaking well and diluting with water to 1,000 mL. Simulated intestinal fluid (SIF): taking potassium dihydrogen phosphate 6.8 g and adding 500 mL water. Adjusting pH to 6.8 with 0.4% sodium hydroxide solution. In addition, 10 g of trypsin (≥250 U/mg, Sigma-Aldrich Co.) was added with water to dissolve the solution. After mixing the two solutions, the resulting mixture was mixed with water to give a volume of 1,000 mL. An aliquot of 100 μL CA-LP–DOX/SLB was diluted with 1 mL SGF, SIF or serum, incubated at 37°C for 2, 6, and 12 hours, respectively, and then the particle size and EE% were measured at defined intervals.
In vitro cytotoxicity
The in vitro cytotoxicity of different DOX–SLB formulations in HepG2 (liver hepatocellular cells, National Infrastructure of Cell Line Resource, Beijing, People’s Republic of China) and H9c2 cells (a cardiac myoblast cell line, National Infrastructure of Cell Line Resource) was evaluated by MTT assay. Briefly, HepG2 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium containing a final concentration of 10% (v/v) FBS, 1% nonessential amino acid, and 1% antibiotics (penicillin, 100 U/mL plus streptomycin). H9c2 cells were cultured in minimum essential medium containing a final concentration of 10% (v/v) FBS, 1% nonessential amino acid, and 1% antibiotics (penicillin, 100 U/mL plus streptomycin). HepG2 and H9c2 cells were seeded at a density of 5×104 cells/well into 96-well plates. After 24 hours incubation, the medium was changed with SLB, DOX, CA-LP–DOX, the mixture of DOX and SLB, and CA-LP–DOX/SLB at different drug ratios for another 24 hours or 48 hours of co-incubation. The cells incubated with medium only were used as the control. Then, 20 μL of MTT solution (5 mg/mL) was added to each well and incubated for an additional 4 hours. After the medium was removed, 200 μL of dimethyl sulfoxide was added and then the fluorescence intensity was measured at 570 nm by using a microplate reader (Bio-Rad 500, Hercules, CA, USA). The cellular inhibition rate and half inhibitory concentration (IC50) values were calculated by using SPSS 17.0 software.
Tumor cell spheroids
HepG2 cells were used to prepare cancer cell spheroids with 200–300 mm diameters in 96-well plates by using a liquid overlay method.26 Briefly, RPMI 1640 medium with 1.5% of agarose was added to each well and the cells were seeded. After this, the plates were centrifuged for 15 minutes at 1,500 relative centrifugal force. Formation of spheroids was monitored by using a MF52-inverted microscope and an MS31 camera (Mingmei, Guangzhou, People’s Republic of China).
Scratch studies
To determine cell motility determination, HCC97H cells (hepatocellular carcinoma cells with high metastatic potential; Fu Heng Biology, Shanghai, People’s Republic of China, 1×105 cells/mL) were seeded in 6-well tissue culture plate, the cells were cultured in DMEM containing a final concentration of 10% (v/v) FBS, 1% nonessential amino acid, and 1% antibiotics (penicillin, 100 U/mL plus streptomycin). When the cells grew to 80%–90% confluence, the medium was aspirated, and the center of the cell monolayers was scraped by using a sterile micropipette tip to create a denuded zone (gap) of constant width. Subsequently, cellular debris was washed with PBS, and the HCC97H cells were exposed to DOX, CA-LP–DOX, and CA-LP–DOX/SLB. The wound closure was monitored and photographed at 24 hours by using an MF52 inverted microscope and an MS31 camera. To quantify the migrated cells, pictures of the initial wounded monolayers were compared with the corresponding pictures of cells at the end of incubation. The migrated distance rate across the white lines was calculated from each triplicate treatment, and the data are presented as the mean ± SD.
In vitro cell apoptosis studies
To further investigate the synergistic effects arising from co-delivery of DOX and SLB via CA-LP, apoptosis assays were performed for HepG2 cells. The HepG2 cells were seeded according to the cell viability assay in opaque 96-well plates. The wells were treated with free culture medium, DOX, LP–DOX/SLB, CA-LP–DOX, and CA-LP–DOX/SLB. After 24 hours treatment, flow cytometry was used to analyze the cells after staining with Annexin Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Cells in late apoptotic stages were labeled with both Annexin V-Alexa Fluor 488 and propidium iodide (PI). Early apoptotic cells with exposed phosphatidylserine but intact cell membranes bound to Annexin V Alexa Fluor 488 but excluded PI, whereas necrotic cells were exclusively stained by PI rather than with Annexin V Alexa Fluor 488.27,28
Uptake by HepG2 cells
HepG2 cells were seeded into a 6-well plate at a density of 1×105 cells. After attachment for 24 hours, the cells were washed and incubated with DOX, CA-LP–DOX, and CA-LP–DOX/SLB with different DOX concentrations for 1 hour at 4°C, 22°C, and 37°C. Then, the cells were washed three times with 4°C PBS (pH 7.4) and fixed with 4% paraformaldehyde for 10 minutes. Finally, the cells were observed by using a DeltaVision microscope (DV Elite, Pittsburgh, PA, USA).
Pharmacokinetic and in vivo distribution studies
Mice (five per group) were intragastrically administered DOX, LP–DOX, CA-LP–DOX, and CA-LP–DOX/SLB. The blood samples were withdrawn from the eye socket at several intervals post administration and immediately mixed with 10 μL of 1% heparin sodium to prevent clotting. Blood samples were centrifuged (10 minutes, 3,000 rpm) and plasma was collected and stored at 20°C until analysis. 100 μL of blood was mixed on a vortex mixer with 1.2 mL acetonitrile for 2 minutes and centrifuged at 13,000 rpm for 10 minutes. The supernatant was collected, transferred to a clean centrifuge tube, and dried with nitrogen. The resulting residue was dissolved in 100 μL methanol, vortex mixed for 3 minutes and centrifuged at 13,000 rpm for 10 minutes, and injected into an HPLC system for analysis.
The mice were killed, and the heart, liver, spleen, lung, and kidney were excised. For quantitative analysis, tissues, including those from heart, liver, spleen, lung, and kidney, were excised, weighed, and homogenized in normal saline solution, and then 200 μL homogenate was vortex mixed with 200 μL of 0.3 mol/L hydrochloric acid solution, and then vortex mixed with 1.2 mL acetonitrile for 2 minutes, and centrifuged at 13,000 rpm for 10 minutes. The supernatant was collected, transferred to a clean centrifuge tube, and dried with nitrogen. The resulting residue was dissolved in 200 μL methanol, vortex mixed for 3 minutes and centrifuged at 13,000 rpm for 10 minutes, and injected into an HPLC system for analysis.
In vivo antitumor effects
For subcutaneous mouse H22 xenograft models, male ICR mice (6 weeks, 18–22 g) were subcutaneously injected with H22 cells in the right armpit. When injected for 7 days, the tumors in the mice exceeded 0.3–0.5 cm3. The mice were then intragastrically administered every 2 days with CA-LP–DOX/SLB at a dose of 10 mg DOX/kg and 30 mg SLB/kg (five mice per group). The control group was administered 5% glucose, and the CA-LP–DOX group was administered CA-LP–DOX (free DOX at a dose 10 mg/kg). Body weight (BW) and tumor volume were monitored and recorded every 2 days after administration. At the end of the experiment, the mice were killed, and the tumors were excised, weighed, and photographed.
For orthotopic nude HepG2 xenograft models, BALB/c nude mice of 18–22 g midline incisions over the liver were performed. Subsequently, HepG2 cells were injected into the livers of nude mice, and the incisions were sutured. On the 12th day post-inoculation, the mice were administered every 2 days with CA-LP–DOX/SLB at the dose of 10 mg DOX/kg and 30 mg SLB/kg (five mice per group). The control group was administered 5% glucose, CA-LP–DOX group was administrated CA-LP–DOX (free DOX at a dose of 10 mg/kg). On the 24th day, the animals were immediately killed, and the livers were isolated. The livers were weighed and photographed and then fixed with 10% neutral formaldehyde, paraffin embedded, sectioned, stained with hematoxylin/eosin, and observed under a microscope according to standard procedures.
In addition, to investigate organ toxicity, at the end of the experiments, the heart, kidney, lung, and spleen tissues were excised and homogenized, and glutathione peroxidase (GSH-Px) and superoxide dismutase (SOD) activities and malonaldehyde (MDA) levels were determined in the supernatants. All procedures were performed according to the ethical requirements of the Nanjing Jiancheng Bioengineering Institute (Nanjing, People’s Republic of China).
Statistical analysis
At each experimental condition, the results were expressed as mean ± SD. Statistical analysis was conducted using one-way ANOVA. P<0.05 was considered significant.
Results and discussion
Preparation and characterization of CA-LP–DOX/SLB
DOX can be easily encapsulated into a hydrophilic core and SLB can be easily encapsulated into the hydrophobic lipid bilayer of nanoliposomes. The EE% values of DOX and SLB were 95.74%±9.09% and 93.07%±3.07%, respectively. The z-average diameter was 97.03±2.17 nm with a PDI of 0.239±0.026 (Figure 1A), which may be an optimal size for gastrointestinal permeation and intracellular uptake.29–31 TEM images showed the spherical shape of nanoliposomes (Figure 1B).
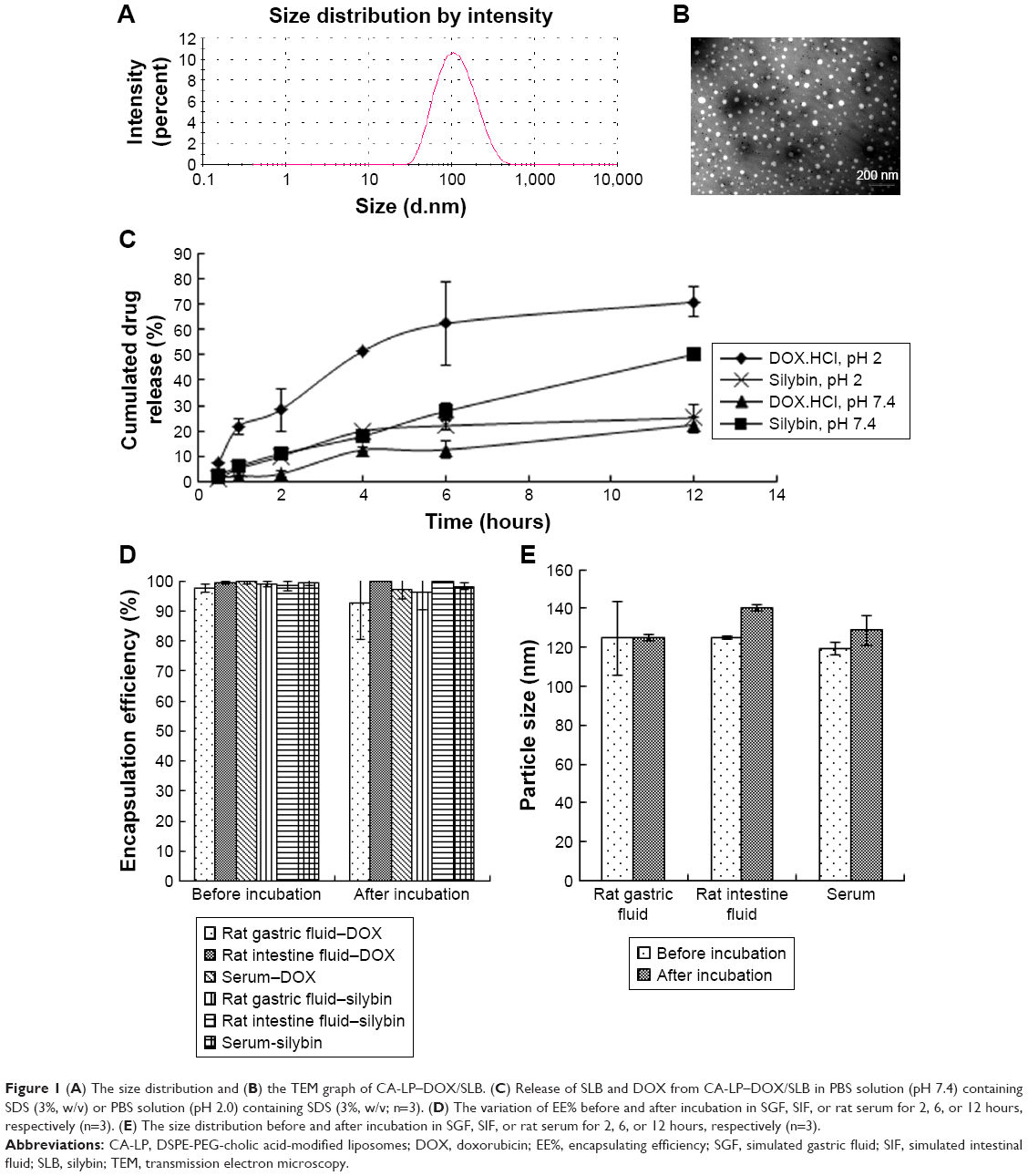 | Figure 1 (A) The size distribution and (B) the TEM graph of CA-LP–DOX/SLB. (C) Release of SLB and DOX from CA-LP–DOX/SLB in PBS solution (pH 7.4) containing SDS (3%, w/v) or PBS solution (pH 2.0) containing SDS (3%, w/v; n=3). (D) The variation of EE% before and after incubation in SGF, SIF, or rat serum for 2, 6, or 12 hours, respectively (n=3). (E) The size distribution before and after incubation in SGF, SIF, or rat serum for 2, 6, or 12 hours, respectively (n=3). |
To simulate the in vivo release conditions, the release behaviors of CA-LP–DOX/SLB were investigated at SGF and SIF, respectively. The release medium containing 3% SDS provided the appropriate DOX and SLB leakage conditions in the release study. The release profiles exhibiting the in vitro drug release patterns of DOX and SLB from CA-LP under different pH conditions are shown in Figure 1C. Both the drugs showed sustained release patterns. Most importantly, the release profiles displayed sequential release of DOX and SLB at both pH 7.4 and 2.0. The release rates of DOX soared at lower pH conditions mimicking the gastrointestinal environment. Such differential release pattern can be attributed to one drug being encapsulated in the lipid bilayer, whereas the other was enclosed within the lipid core.
To investigate the integrity of LP during the oral absorption process, the stability in SGF and SIF was studied. Figure 1D showed that the EE% still exceeded 80% after their incubations of CA-LP–DOX/SLB in rat gastric fluid, rat intestinal fluid, or rat serum for 2, 6, or 12 hours, respectively. Figure 1E showed that the particle size of CA-LP–DOX/SLB remained relatively stable before and after incubation of CA-LP–DOX/SLB in rat gastric fluid, rat intestinal fluid, or rat serum for 2, 6, or 12 hours, respectively. The size fluctuated from 120 to 140 nm. The results demonstrated that CA-LP–DOX/SLB can maintain its structural integrity in a gastrointestinal environment and prevent gastrointestinal enzyme destruction and protein adsorption in blood.
Intracellular uptake
Anticancer efficiency greatly depends on cellular internalization of drugs. In these experiments, Figure 2 demonstrated fluorescence signals of DOX (red). CA-LP–DOX/SLB and CA-LP–DOX entered the nuclei of the HepG2 cells, and DOX entered the cytomembrane at the same uptake time which further proved that CA-LP improved cellular uptake of DOX and that SLB did not interfere with the uptake of DOX into the nuclei of HepG2 cells. With increasing DOX concentration, CA-LP–DOX/SLB showed stronger fluorescence signals in the nuclei, indicating that the uptake of CA-LP–DOX/SLB was concentration dependent. Under the different incubating temperatures, CA-LP–DOX/SLB uptake by HepG2 cells at 37°C was greater than the uptakes at 4°C and 22°C. The uptake of CA-LP–DOX/SLB was influenced by temperatures, which suggested that the uptake of CA-LP–DOX/SLB is energy dependent.
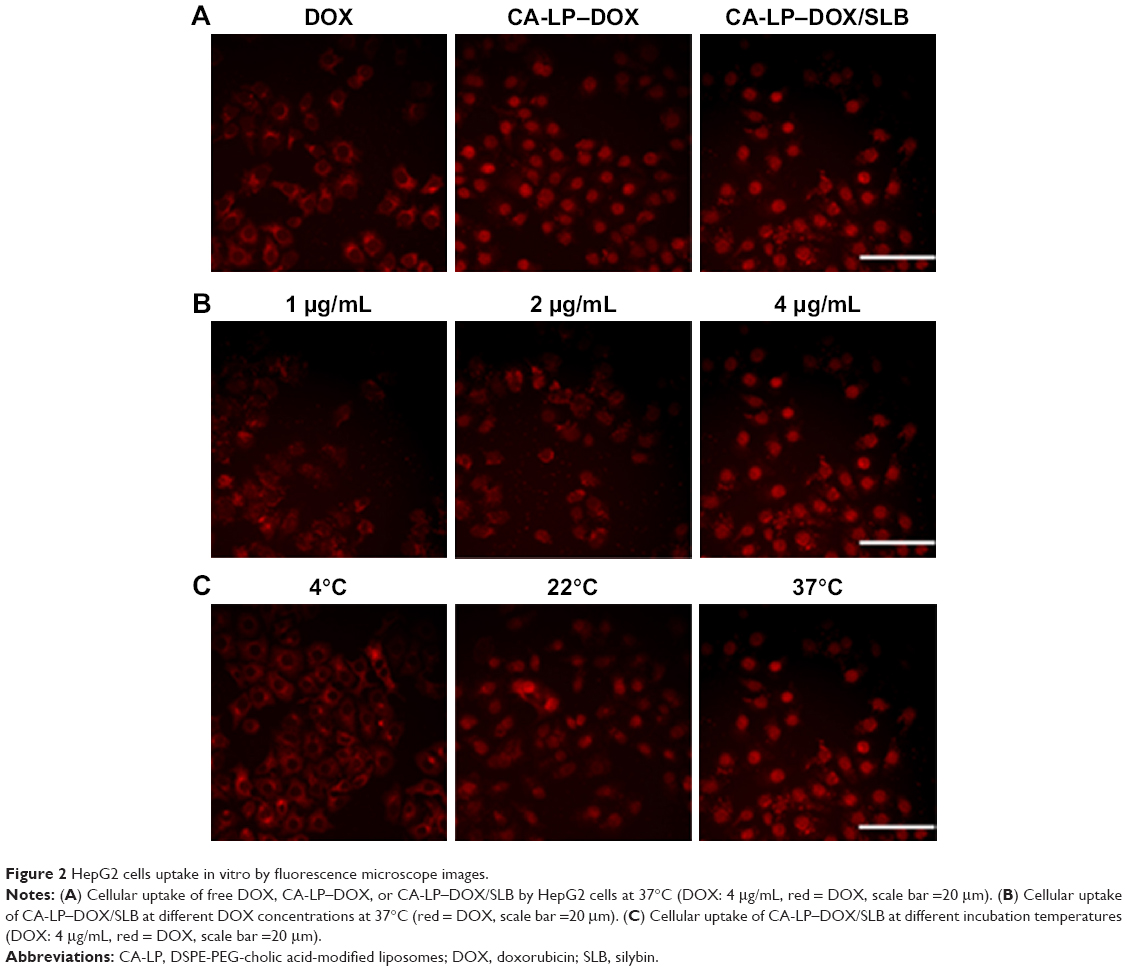 | Figure 2 HepG2 cells uptake in vitro by fluorescence microscope images. |
DOX exerts its effect when it is taken up into the nuclei of cells, where it binds with high affinity to DNA via intercalation between base pairs. There is good evidence to support DOX’s mechanism of action as a topoisomerase II inhibitor.32 Once DOX is intercalated into DNA, it perturbs the re-ligation step of topoisomerase II, which results in the formation of the “cleavable complex” and eventually results in double-strand DNA cleavage. Failure to repair DNA double-strand breaks results in an apoptotic response. Other cellular responses to DOX include the formation of DOX–DNA adducts33 and the inhibition of DNA methyltransferase.34 A range of several other diverse effects also have been mentioned, although the method of cell death remains unclear.
In vitro cytotoxicity
In vitro cytotoxicity for H9c2 and HepG2 cells
The IC50 values of SLB in HepG2 cells and H9c2 cells were 2.06×1010 and 6.57×106 μg/mL for 24 hours, and the IC50 values for 48 hours were 1.06×107 and 8.77×105 μg/mL, which demonstrated that SLB caused no significant cytotoxicity in HepG2 and H9c2 cells. The antiproliferative effects of DOX, CA-LP–DOX, DOX/SLB, CA-LP–DOX/SLB (1:3), CA-LP–DOX/SLB (1:6), CA-LP–DOX/SLB (1:10), and CA-LP–DOX/SLB (1:15) were compared in H9c2 cells (Figure 3A) and HepG2 cells after 24 or 48 hours cytotoxic treatment (Figure 3B). According to IC50 results, CA-LP–DOX/SLB (1:3, 5, 6, 10, 15) all showed lower cytotoxicity in H9c2 cells than did DOX, DOX/SLB, and CA-LP–DOX. CA-LP–DOX/SLB (1:3) showed lower cytotoxicity for HepG2 cells than did the other drug ratio groups. Therefore, we chose DOX/SLB (1:3) as the tested drug ratio. First, we optimized the drug ratio of DOX and SLB showing synergistic anti-HCC effects in HepG2 cells in vitro. The reduction in IC50 might be explained by the intracellular trafficking manner of CA-LP via endocytosis. The results confirmed that CA-LP–DOX/SLB showed a higher inhibition ratio against HepG2 cells than that of DOX by attaching CA to the surface of the carrier to increase the hepatoma-targeting effect. CA-LP–DOX/SLB showed less cytotoxicity against H9c2 cells by including the combination of SLB and DOX.
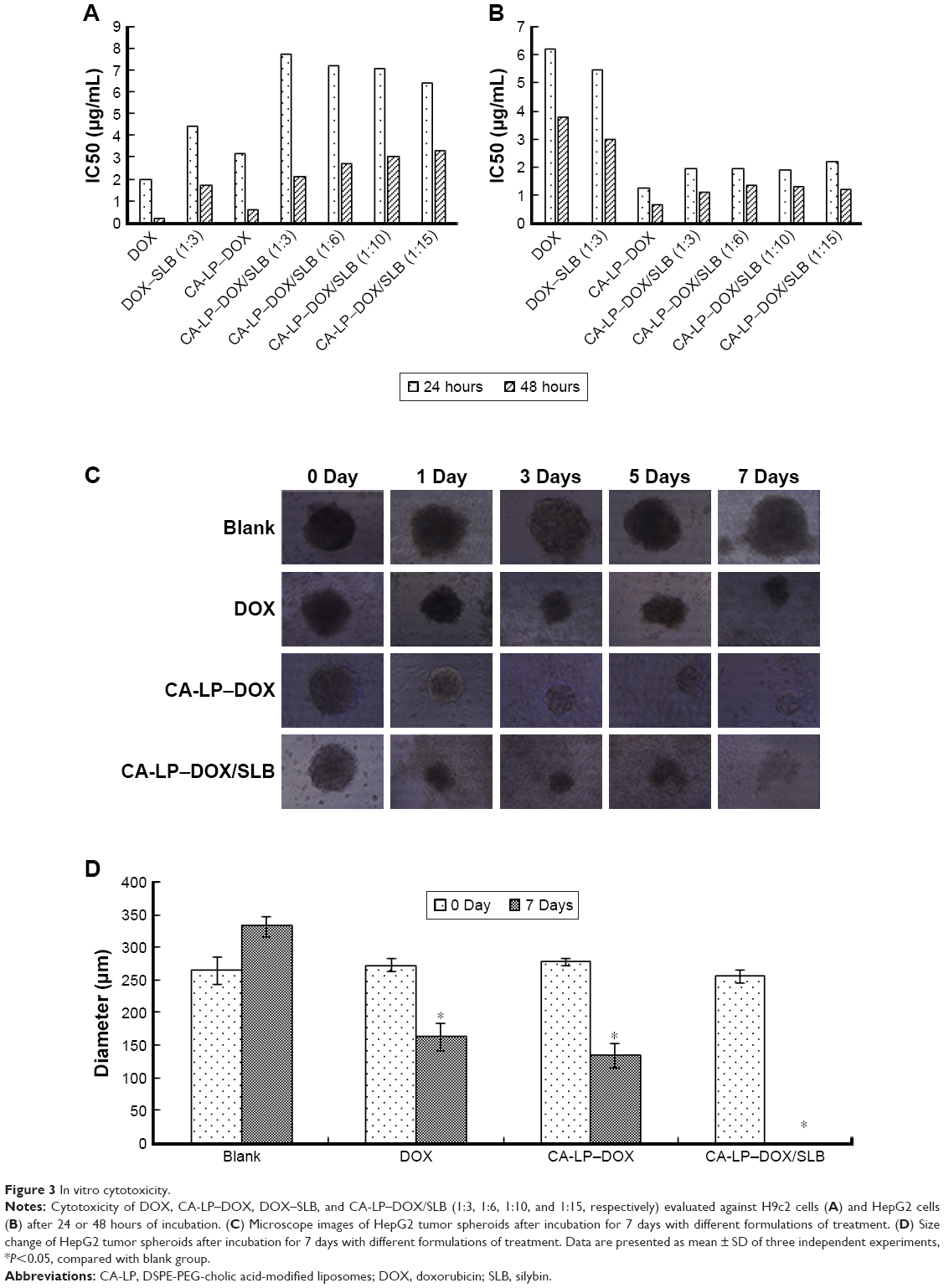 | Figure 3 In vitro cytotoxicity. |
Cytotoxicity in tumor cell spheroids
The diameters of the HepG2 cancer cell spheroids were 200–300 mm at day 7. Figure 3C showed images of the spheroids with respective diameters (μm) in different periods of time for HepG2 cells. The cytotoxicities of DOX, CA-LP–DOX, and CA-LP–DOX/SLB in tumor cell spheroids were assessed by evaluating microscope images. The data for HepG2 cells are shown in Figure 3D. For HepG2 tumor cell spheroids, after the treatment with different drug groups for 7 days, the cell spheroids for CA-LP–DOX/SLB nearly disappeared. CA-LP–DOX/SLB-induced cytotoxicity was much higher than that observed for the free drug and CA-LP–DOX (Figure 3C and D). It is important to note that there was a significant difference between CA-LP–DOX and CA-LP–DOX/SLB (P<0.05), which emphasized the importance of SLB.
Scratch studies
HCC is one of the most common malignancies worldwide, and has a high potential for vascular invasion, metastasis, and recurrence even after surgical resection, which leads to poor prognosis. HCC cells excrete proteinases, which breakdown extracellular matrix and enable invasion and metastasis.35 The HCC metastatic process involves many factors, so exploring the inhibitory effect of drugs on HCC metastasis is beneficial for HCC prognosis and reducing the recurrence. The scratch studies tested for metastasis in response to the inhibitory effect of CA-LP–DOX/SLB.
HCC97H cells represent one kind of high metastatic human hepatocellular carcinoma cell line,36 and were used for the cellular model in this study. The results are shown in Figure 4. The blank group can largely move and the gap becomes smaller and smaller over time. The cell migration rate of CA-LP–DOX/SLB was found to be less than those of DOX and CA-LP–DOX, which demonstrated that CA-LP–DOX/SLB has the better efficacy for suppressing cell motility in HCC97H cells.
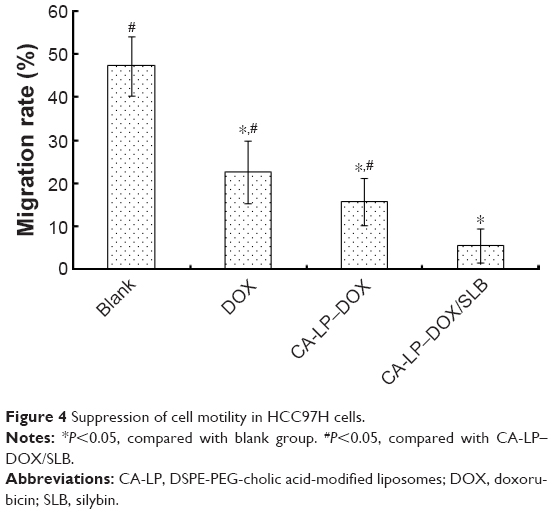 | Figure 4 Suppression of cell motility in HCC97H cells. |
In vitro cell apoptosis studies
Apoptosis is programmed cell death and an important self-regulatory mechanism for multicellular organisms to maintain homeostasis. Apoptosis is a key cell survival process when it inhibits only tumor cells.37 Therefore, promoting apoptosis has become the focus of anticancer research.
As shown in Figure 5, CA-LP–DOX/SLB showed remarkably higher rates of apoptosis in HepG2 cells than those of the free individual drugs and drug combination, with the majority of the cells in the late apoptotic phase (88.55%). In HepG2 cells, the late apoptotic rate after CA-LP–DOX/SLB treatment was higher than those after DOX treatment (33.88%), LP–DOX/SLB treatment (48.61%), and CA-LP–DOX treatment (60.63%). These results could be because of the higher uptake of CA-targeted nanoliposomes than that of the nontargeted ones by HepG2 cells. The cellular DOX uptake of CA-LP–DOX/SLB (Figure 2) indicated that loading DOX into CA-LP affected the uptake of DOX into the cells via the LPs.
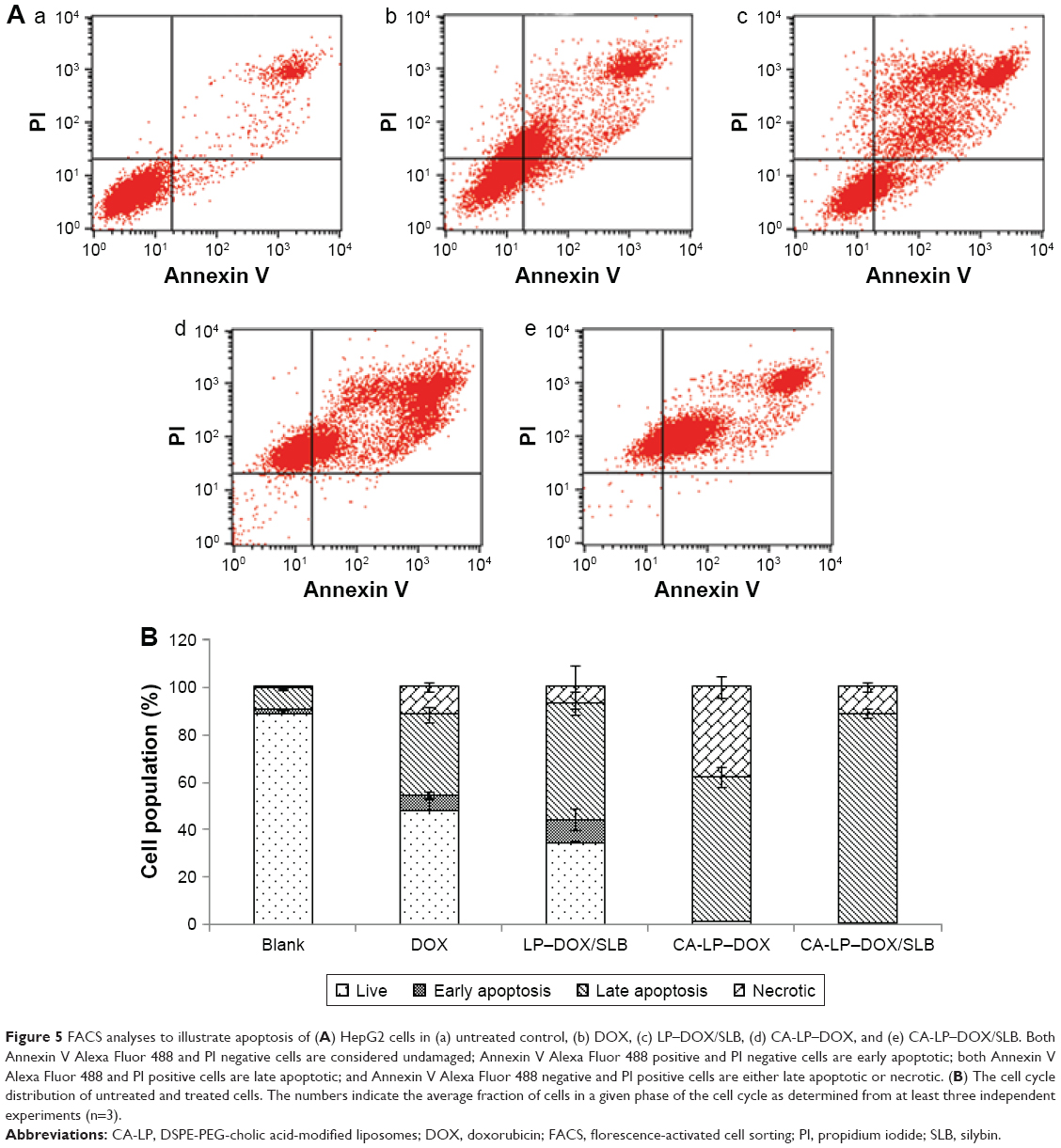 | Figure 5 FACS analyses to illustrate apoptosis of (A) HepG2 cells in (a) untreated control, (b) DOX, (c) LP–DOX/SLB, (d) CA-LP–DOX, and (e) CA-LP–DOX/SLB. Both Annexin V Alexa Fluor 488 and PI negative cells are considered undamaged; Annexin V Alexa Fluor 488 positive and PI negative cells are early apoptotic; both Annexin V Alexa Fluor 488 and PI positive cells are late apoptotic; and Annexin V Alexa Fluor 488 negative and PI positive cells are either late apoptotic or necrotic. (B) The cell cycle distribution of untreated and treated cells. The numbers indicate the average fraction of cells in a given phase of the cell cycle as determined from at least three independent experiments (n=3). |
In vivo pharmacokinetics and biodistribution
In vivo pharmacokinetics
The lower limit of detection for DOX by HPLC was 12.50 ng/mL, the lower limit of detection for SLB by HPLC was 39.00 ng/mL. The mean plasma concentration–time profiles of DOX after oral administration of different DOX formulations are illustrated in Figure 6A. As shown in Figure 6A, the plasma concentration of DOX in nanoliposomes, including LP–DOX, CA-LP–DOX, and CA-LP–DOX/SLB, was significantly increased compared with that in DOX. The area under the curve (AUC0-t) values in LP–DOX, CA-LP–DOX, and CA-LP–DOX/SLB were 1.44-, 1.79-, and 3.40-fold higher than that of DOX, and the half-life (t1/2) were 2.40-, 4.86-, and 3.06-fold longer than that in the DOX group, which indicated that DOX encapsulation into nanoliposomes dramatically enhanced drug absorption and extended circulation time in vivo.
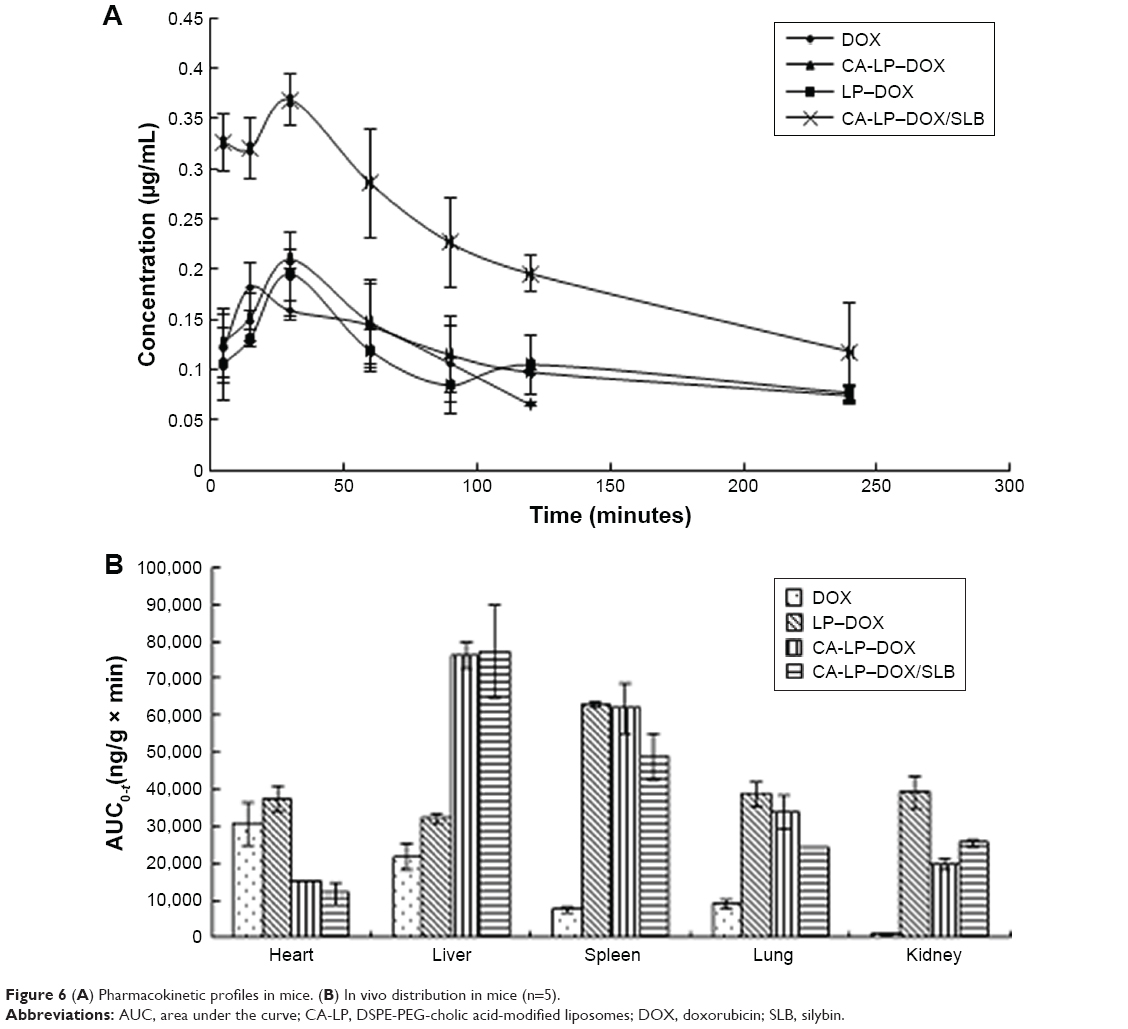 | Figure 6 (A) Pharmacokinetic profiles in mice. (B) In vivo distribution in mice (n=5). |
Furthermore, the CA-LP–DOX/SLB-treated group improved the plasma concentration relative to that for CA-LP–DOX with DOX alone. The pharmacokinetic parameter of AUC0-t in the CA-LP–DOX/SLB group was 1.90 times higher than that of the CA-LP–DOX group, which indicated that SLB increased the absorption of DOX in vivo. This finding demonstrated that co-delivery of LPs was more effective in enhancing DOX absorption in vivo because of the synergistic effects of released SLB from nanoliposomes.
The results further proved that DOX encapsulated in nanoliposomes obviously enhanced stability, extended blood circulation time, and improved plasma concentration, which gives drugs a greater chance to reach the targeted sites via the enhanced permeability and retention effect and transporter-mediated endocytosis in all likelihood. Overall, co-delivery nanoliposomes with DOX and SLB exhibited excellent synergistic effects on the absorption of DOX in vivo and would appear to be a promising vehicle for future studies and development.
In vivo biodistribution
The biodistribution of DOX in all DOX formulations was further studied in major organs, including the heart, liver, spleen, lung, and kidney. As shown in Figure 6B, CA-LP showed high liver accumulation, mainly because of the enhanced permeability, LP passive targeting, and CA targeting. However, the LPs also exhibited high accumulation in the spleen, the LPs unavoidable uptake by the reticuloendothelial system. Excitingly, the CA-LP–DOX/SLB-treated group showed decreased accumulation of DOX in the heart relative to those of the CA-LP–DOX and LP–DOX-treated groups, which indicated that co-delivery system could improve drug effects in vivo.
We showed that absorption of CA-LP–DOX/SLB across intestinal membranes and the consequent accumulation in liver was to a large part more than the absorption of CA-LP–DOX and DOX, otherwise, the consequent accumulation in the heart was to a large part less than those in CA-LP–DOX and DOX. Therefore, CA-LP and SLB generally led to higher absorption and further accumulation in the liver.
Furthermore, the t1/2 values of DOX and SLB in the liver was ~89 and 68 minutes, respectively. The approximate t1/2 values make it possible that the two drugs were simultaneously delivered to the same cells at an optimized ratio to obtain synergistic effects intracellularly.
Efficacy test
Antitumor effects in vivo
Since the serious cardiotoxicity of DOX has been a major obstacle to its clinical use, we measured BWs every other day to evaluate the systemic side effects of different DOX formulations. The subcutaneous H22-bearing mice model results are shown in Figure 7A–C, and the CA-LP–DOX group has been previously reported.25 Figure 7A demonstrated that almost no weight loss was observed in mice treated with CA-LP–DOX/SLB clearly, which indicated that co-delivery of chemotherapeutic drugs and tissue damage protectors in nanoliposomes caused no side effects. The tumor size and photos are shown in Figure 7B and C. The antitumor efficacies of different DOX formulations were tested in mice bearing H22 tumors. The tumor volumes in mice treated with saline grew rapidly, to ~700 mm3 by the 21st day. However, DOX- and LP–DOX-treated groups exhibited considerable tumor inhibition in vivo relative to that of the saline-treated group.25 Moreover, the tumor volumes of the mice exposed to CA-LP–DOX, and CA-LP–DOX/SLB was obviously reduced relative to those in the DOX and LP–DOX groups, which was attributed to the CA targeting effects of the LPs. However, there were no significant differences in the tumor inhibition effects of CA-LP–DOX and CA-LP–DOX/SLB, which demonstrated that SLB could not significantly influence the antitumor effects of DOX in vivo. CA-LP–DOX and CA-LP–DOX/SLB exhibited the most significant tumor inhibition effects of all DOX formulations (P<0.05 compared with DOX and LP–DOX).
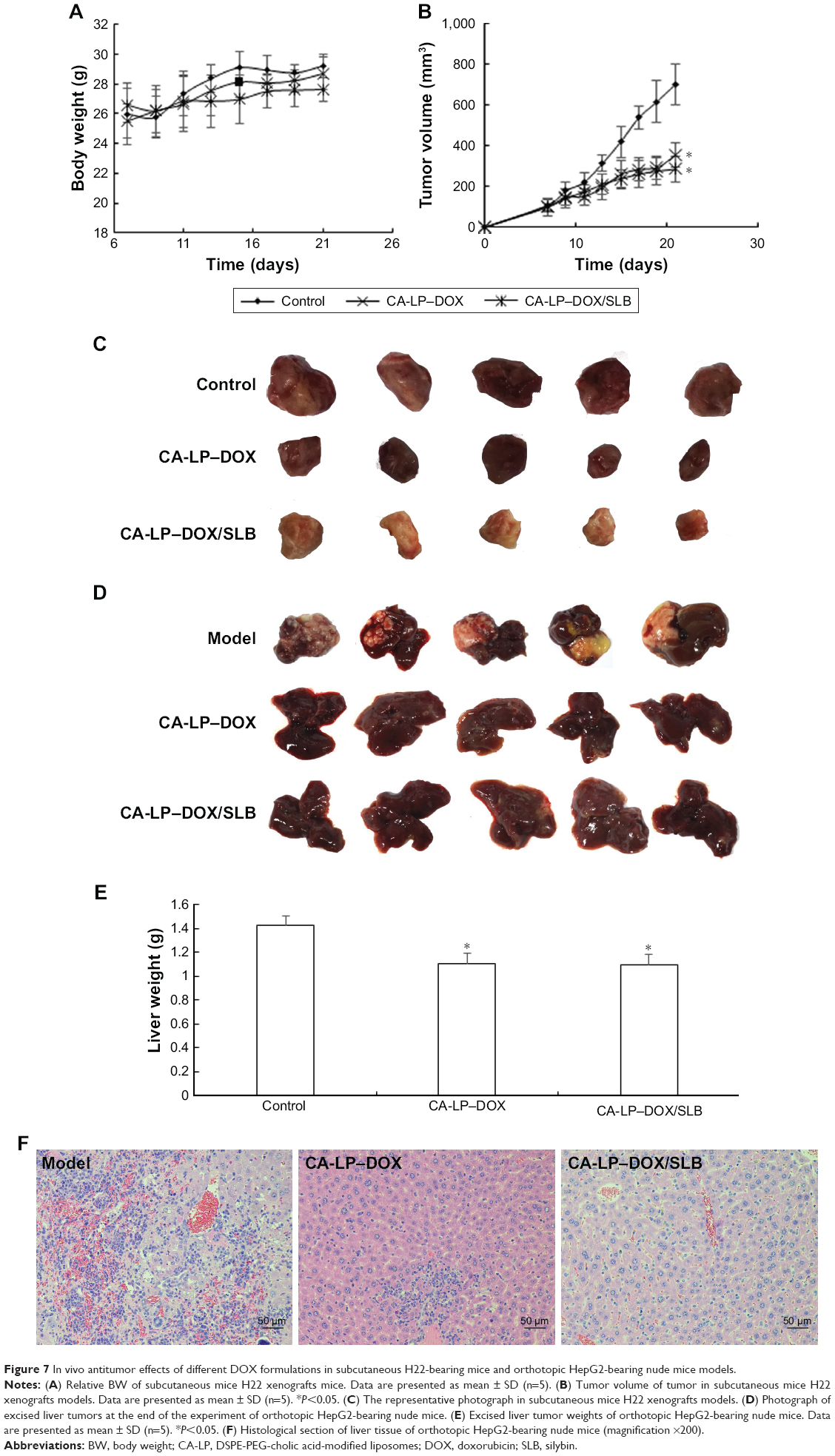 | Figure 7 In vivo antitumor effects of different DOX formulations in subcutaneous H22-bearing mice and orthotopic HepG2-bearing nude mice models. |
For the orthotopic HepG2-bearing nude mice models, the results are shown in Figure 7D–F. CA-LP–DOX and CA-LP–DOX/SLB led to significant decreases in tumor size relative to the model (Figure 7D). Moreover, the liver weights of CA-LP–DOX and CA-LP–DOX/SLB significantly differed from those of model (Figure 7E, P<0.05). Figure 7F demonstrated greater protection of liver tissue from tumor invasion in the CA-LP–DOX- and CA-LP–DOX/SLB-treated groups than in the model group. The CA-LP–DOX/SLB-treated group showed greater protection of liver tissue from tumor invasion than that in the CA-LP–DOX group.
Assessments of tissue toxicity
To further investigate the toxicity of different DOX formulations in vivo, the heart weight/body weight (HW/BW) ratios and the three clinical chemical parameters of GSH-Px, MDA, and SOD were used to evaluate the tissue toxicity. Reduced HW/BW ratios are a common sign of cardiotoxicity.38 SOD originates from active material in living organisms and can eliminate harmful substances produced during metabolism of organisms. Lipid peroxidation by free radicals generates thiobarbituric acid-reactive species that can be determined via MDA levels. GSH-Px is an important peroxide decomposition enzyme in the body. Selenium is part of the GSH-Px enzyme system and can catalyze GSH to oxidized glutathione and convert poisonous peroxide to a nonpoisonous hydroxy compound via a reduction reaction, which could protect cell membrane structures and functions from peroxide. Therefore, higher HW/BW, SOD, GSH-Px and lower MDA in tissues are less harmful. The results are shown in Figure 8, and the CA-LP–DOX group has been previously reported.25 The values of GSH and SOD in tumor-bearing mice exposed to CA-LP–DOX/SLB were significantly higher than those of the CA-LP–DOX-treated group, the values of MDA in tumor-bearing mice exposed to CA-LP–DOX/SLB were significantly lower than that of the CA-LP–DOX-treated group, which proved that the addition of SLB reduced tissue toxicity in vivo, an effect that may be related to the cardioprotective effect of SLB.
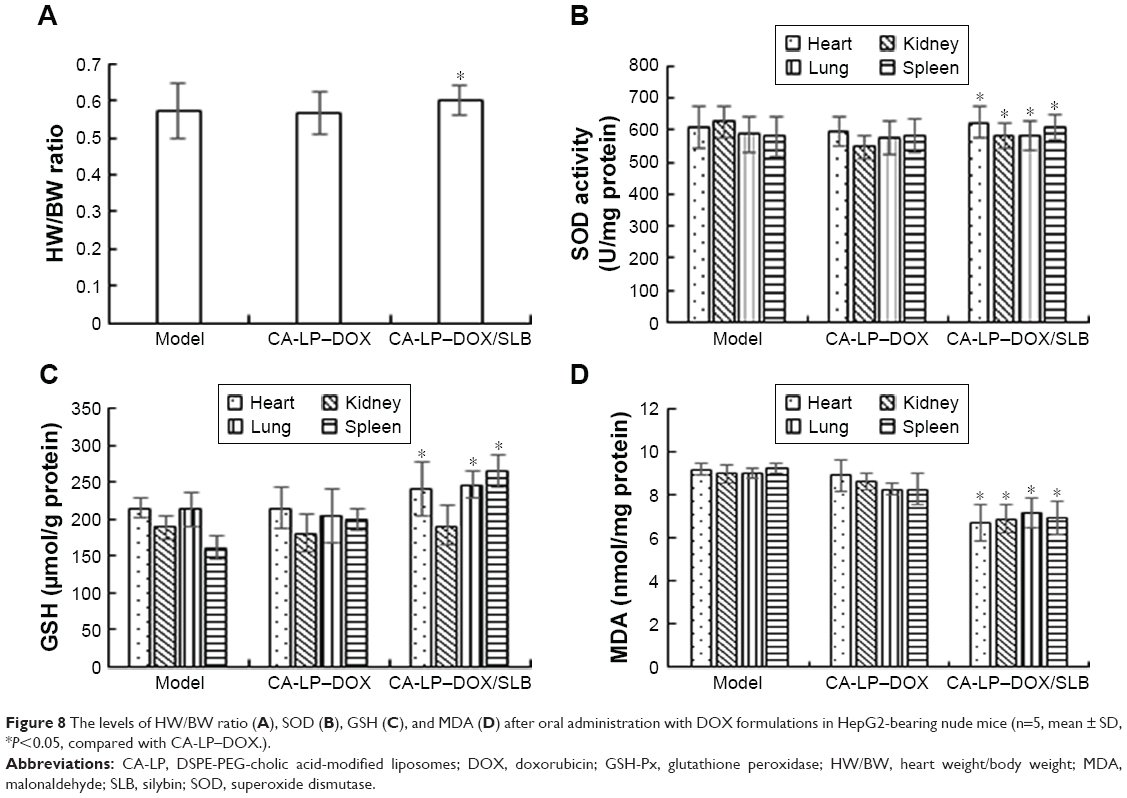 | Figure 8 The levels of HW/BW ratio (A), SOD (B), GSH (C), and MDA (D) after oral administration with DOX formulations in HepG2-bearing nude mice (n=5, mean ± SD, *P<0.05, compared with CA-LP–DOX.). |
It is worth noting that co-delivery CA-LP–DOX/SLB gave the best toxicity reduction efficiency among the nanoliposomes including CA-LP–DOX, which can be explained by the simultaneous release of SLB and DOX to the tumor cells and synergistic protection in tissues.
Conclusion
In summary, active hepatic-targeted co-delivery nanoliposomes with DOX as a therapeutic drug and SLB as an anti-cardiotoxic drug co-encapsulated into CA-LP (CA-LP–DOX/SLB) were prepared. The LPs exhibited spherical morphology with a uniform size distribution, had sustained release properties, and remained stable in SGF, SIF, and serum. Moreover, CA-LP–DOX/SLB exerted the strongest cytotoxic and highest cell apoptosis-inducing activities against HepG2 cells. CA-LP–DOX/SLB was more efficiently internalized into HepG2 cells, enhanced the cellular uptake of DOX, and significantly reduced cardiotoxicity effects via hepatic-targeted delivery and the synergistic effect of released SLB. More importantly, the in vivo results illustrated that CA-LP–DOX/SLB not only displayed better liver accumulation and liver targeting, and more efficient inhibition of liver tumor growth in H22 tumor-bearing mice and HepG2 tumor-bearing nude mice, but also induced significantly less pathological damage to the cardiac tissue relative to the damage caused by free DOX and CA-LP–DOX. Above all, these observations demonstrated that co-administration of DOX and SLB in CA-LP improved antitumor effects and minimized side effects in vivo. Therefore, co-delivery of anticancer drugs and tissue protection reagents in CA-mediated nanocarriers appear to be a promising approach to oral hepatoma therapy.
Acknowledgment
The authors would like to acknowledge the PUMC Youth Fund (2017350017).
Disclosure
The authors report no conflicts of interest in this work.
References
Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nat Rev Gastroenterol Hepatol. 2013;10(1):34–42. | ||
Zhang X, Ng HLH, Lu A, et al. Drug delivery system targeting advanced hepatocellular carcinoma: current and future. Nanomedicine. 2016;12(4):853–869. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Dong Y, Feng SS, Poly FSS. Poly(d,l-lactide-co-glycolide)/montmorillonite nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(30):6068–6076. | ||
Sun JB, Duan JH, Dai SL, et al. In vitro and in vivo antitumor effects of doxorubicin loaded with bacterial magnetosomes (DBMs) on H22 cells: the magnetic bio-nanoparticles as drug carriers. Cancer Lett. 2007;258(1):109–117. | ||
Lee JS, Hong EK. In vitro and in vivo antitumor effects of doxorubicin loaded with bacterial magnetosomes (DBMs) on H22 cells: the magnetic bionanoparticles as drug carriers. Cancer Lett. 2010;297:144–154. | ||
Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52(6):1213–1225. | ||
Agudelo D, Bérubé G, Tajmir-Riahi HA. An overview on the delivery of antitumor drug doxorubicin by carrier proteins. Int J Biol Macromol. 2016;88:354–360. | ||
Hayek ER, Speakman E, Rehmus E. Acute doxorubicin cardiotoxicity. N Engl J Med. 2005;352(23):2456–2457. | ||
Chen Y, Jungsuwadee P, Vore M, Butterfield DA, St Clair DK. Collateral damage in cancer chemotherapy: oxidative stress in nontargeted tissues. Mol Interv. 2007;7(3):147–156. | ||
Guo H, Zhang D, Li T, et al. In vitro and in vivo study of Gal-OS self-assembled nanoparticles for liver-targeting delivery of doxorubicin. J Pharm Sci. 2014;103(3):987–993. | ||
Wang SQ, Zhu XF, Wang XN, Shen T, Xiang F, Lou HX. Flavonoids from Malus hupehensis and their cardioprotective effects against doxorubicin-induced toxicity in H9c2 cells. Phytochemistry. 2013;87:119–125. | ||
Wang J, Chung MH, Xue B, Ma H, Ma C, Hattori M. Estrogenic and antiestrogenic activities of phloridzin. Biol Pharm Bull. 2010;33(4):592–597. | ||
Wang SQ, Han XZ, Li X, Ren DM, Wang XN, Lou HX. Flavonoids from Dracocephalum tanguticum and their cardioprotective effects against doxorubicin-induced toxicity in H9c2 cells. Bioorg Med Chem Lett. 2010;20(22):6411–6415. | ||
Zhang TH, Wang SY, Shen X. Effect of silybin on coxsackie B5 virus infected new born rat heart cells in culture. Acad J Second Military Med Univ. 1990;11(2):143. | ||
Chen H, Zhang TH, Wei T. Protective effects of silybin on cultured cardiomyocytes with deprivation of oxygen and glucose. Acad J Second Military Med Univ. 1990;11(2):147. | ||
Liang RL, Zy G, Qb G, et al. The effect of silibinin on Ca2+ channel activity of membrane of isolated myocardium cell from adult rat. Chin Pharm J. 1996;31(10):594. | ||
Salamone F, Galvano F, Marino Gammazza A, Marino A, et al. Silibinin improves hepatic and myocardial injury in mice with nonalcoholic steatohepatitis. Dig Liver Dis. 2012;44(4):334–342. | ||
Kolhatkar V, Polli JE. Structural requirements of bile acid transporters: C-3 and C-7 modifications of steroidal hydroxyl groups. Eur J Pharm Sci. 2012;46(1–2):86–99. | ||
Xiao L, Pan G. An important intestinal transporter that regulates the enterohepatic circulation of bile acids and cholesterol homeostasis: the apical sodium-dependent bile acid transporter (SLC10A2/ASBT). Clin Res Hepatol Gastroenterol. 2017;41(5):509–515. | ||
Schneider KM, Albers S, Trautwein C. Role of bile acids in the gut-liver axis. J Hepatol. 2018;68(5):1083–1085. | ||
Park J, Choi JU, Kim K, Byun Y. Bile acid transporter mediated endocytosis of oral bile acid conjugated nanocomplex. Biomaterials. 2017;147:145–154. | ||
Nurunnabi M, Khatun Z, Revuri V, et al. Design and strategies for bile acid mediated therapy and imaging. RSC Adv. 2016;6(78):73986–74002. | ||
Li Y, Zhu C. Enhanced hepatic-targeted delivery via oral administration using nanoliposomes functionalized with a novel DSPE–PEG–cholic acid conjugate. RSC Adv. 2016;6(33):28110–28120. | ||
Li Y, Yang D, Zhang Y, Zhu C. Novel DSPE-PEG-cholic acid-modified liposomes with hepatic targeting properties improve the anti-tumor efficacy of oral doxorubicin hydrochloride for liver tumor-bearing mice. J Biomed Nanotechnol. 2017;13(6):727–736. | ||
Perche F, Patel NR, Torchilin VP. Accumulation and toxicity of antibody-targeted doxorubicin-loaded PEG–PE micelles in ovarian cancer cell spheroid model. J Control Release. 2012;164(1):95–102. | ||
Ma ZJ, Lu L, Yang JJ. Lariciresinol induces apoptosis in HepG2 cells via mitochondrial-mediated apoptosis pathway. Eur J Pharmacol. 2018;821:1–10. | ||
Zhang Y, Zhao H, Di Y, et al. Antitumor activity of Pinoresinol in vitro: inducing apoptosis and inhibiting HepG2 invasion. J Funct Foods. 2018;45:206–214. | ||
Choi JS, Cao J, Naeem M, et al. Size-controlled biodegradable nanoparticles: preparation and size-dependent cellular uptake and tumor cell growth inhibition. Colloids Surf B Biointerfaces. 2014;122:545–551. | ||
Xing J, Liu D, Zhou G, et al. Liposomally formulated phospholipid-conjugated novel near-infrared fluorescence probe for particle size effect on cellular uptake and biodistribution in vivo. Colloid Surf B. 2018;161:588–596. | ||
Awaad A, Nakamura M, Ishimura K. Imaging of size-dependent uptake and identification of novel pathways in mouse Peyer’s patches using fluorescent organosilica particles. Nanomedicine. 2012;8(5):627–636. | ||
Cutts SM, Rephaeli A, Nudelman A, Hmelnitsky I, Phillips DR. Molecular basis for the synergistic interaction of Adriamycin with the formaldehyde-releasing prodrug pivaloyloxymethyl butyrate (AN-9). Cancer Res. 2001;61:8194–8202. | ||
Yokochi T, Robertson KD. Doxorubicin inhibits DNMT1, resulting in conditional apoptosis. Mol Pharmacol. 2004;66(6):1415–1420. | ||
Saito G, Swanson JA, Lee KD. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv Drug Deliv Rev. 2003;55(2):199–215. | ||
Arsura M, Cavin LG. Nuclear factor-kappaB and liver carcinogenesis. Cancer Lett. 2005;229(2):157–169. | ||
Tang J, Li L, Huang W, et al. miR-429 increases the metastatic capability of HCC via regulating classic Wnt pathway rather than epithelial-mesenchymal transition. Cancer Lett. 2015;364(1):33–43. | ||
Lin X, Wei J, Chen Y, et al. Isoorientin from Gypsophila elegans induces apoptosis in liver cancer cells via mitochondrial-mediated pathway. J Ethnopharmacol. 2016;187:187–194. | ||
Kumar NT, Liestøl K, Løberg EM, Reims HM, Mæhlen J. Postmortem heart weight: relation to body size and effects of cardiovascular disease and cancer. Cardiovasc Pathol. 2014;23(1):5–11. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.