")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway
Authors Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, Cleverly AL, Desaiah D, Guba S, Benhadji KA, Slapak CA, Lahn M
Received 14 April 2015
Accepted for publication 19 May 2015
Published 10 August 2015 Volume 2015:9 Pages 4479—4499
DOI https://doi.org/10.2147/DDDT.S86621
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Video abstract presented by Susan Guba
Views: 912
Stephan Herbertz,1 J Scott Sawyer,2 Anja J Stauber,2 Ivelina Gueorguieva,3 Kyla E Driscoll,4 Shawn T Estrem,2 Ann L Cleverly,3 Durisala Desaiah,2 Susan C Guba,2 Karim A Benhadji,2 Christopher A Slapak,2 Michael M Lahn2
1Lilly Deutschland GmbH, Bad Homburg, Germany; 2Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, IN, USA; 3Lilly Research Laboratories, Eli Lilly and Company, Windlesham, Surrey, UK; 4Lilly Research Laboratories, Eli Lilly and Company, New York, NY, USA
Abstract: Transforming growth factor-beta (TGF-β) signaling regulates a wide range of biological processes. TGF-β plays an important role in tumorigenesis and contributes to the hallmarks of cancer, including tumor proliferation, invasion and metastasis, inflammation, angiogenesis, and escape of immune surveillance. There are several pharmacological approaches to block TGF-β signaling, such as monoclonal antibodies, vaccines, antisense oligonucleotides, and small molecule inhibitors. Galunisertib (LY2157299 monohydrate) is an oral small molecule inhibitor of the TGF-β receptor I kinase that specifically downregulates the phosphorylation of SMAD2, abrogating activation of the canonical pathway. Furthermore, galunisertib has antitumor activity in tumor-bearing animal models such as breast, colon, lung cancers, and hepatocellular carcinoma. Continuous long-term exposure to galunisertib caused cardiac toxicities in animals requiring adoption of a pharmacokinetic/pharmacodynamic-based dosing strategy to allow further development. The use of such a pharmacokinetic/pharmacodynamic model defined a therapeutic window with an appropriate safety profile that enabled the clinical investigation of galunisertib. These efforts resulted in an intermittent dosing regimen (14 days on/14 days off, on a 28-day cycle) of galunisertib for all ongoing trials. Galunisertib is being investigated either as monotherapy or in combination with standard antitumor regimens (including nivolumab) in patients with cancer with high unmet medical needs such as glioblastoma, pancreatic cancer, and hepatocellular carcinoma. The present review summarizes the past and current experiences with different pharmacological treatments that enabled galunisertib to be investigated in patients.
Keywords: TGF-β, TGF-βRI kinase inhibitor, ALK5, galunisertib, LY2157299, cancer, clinical trials
Introduction
Since the discovery of the cytokine transforming growth factor-beta 1 (TGF-β1),1 a diverse family of ligands, corresponding receptors, and signal transduction proteins known as the TGF-β superfamily were identified (Table 1).2 Although the members of the TGF-β superfamily share genetic and protein structures, some of these members have different physiological functions and play distinct roles in diseases, including cancer (Figure 1). For example, the activin receptor-like kinase 1 (ALK1) receptor and the ALK5 receptor, also known as TGF-β receptor type I (TGF-βRI), can both promote angiogenesis but have different intracellular activation pathways with different effects on angiogenesis (for details, see the section TGF-β/ALK5 signaling pathway and its role in cancer). Here, we will focus on the role of the TGF-β/ALK5-mediated pathway and describe the experiences of developing galunisertib.
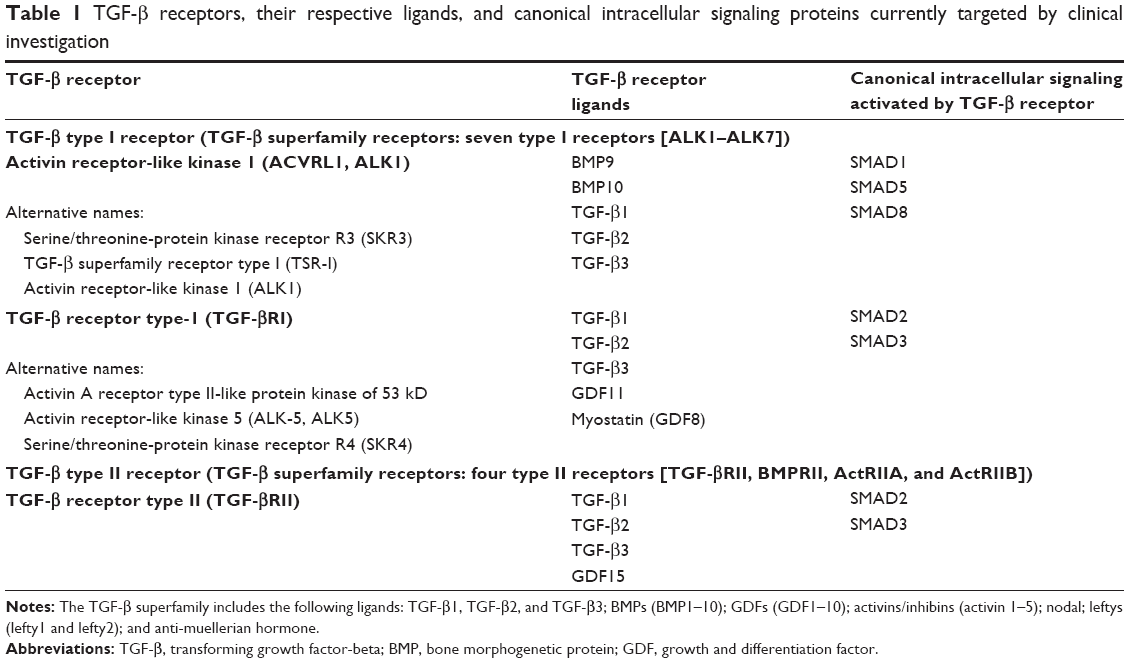 | Table 1 TGF-β receptors, their respective ligands, and canonical intracellular signaling proteins currently targeted by clinical investigation |
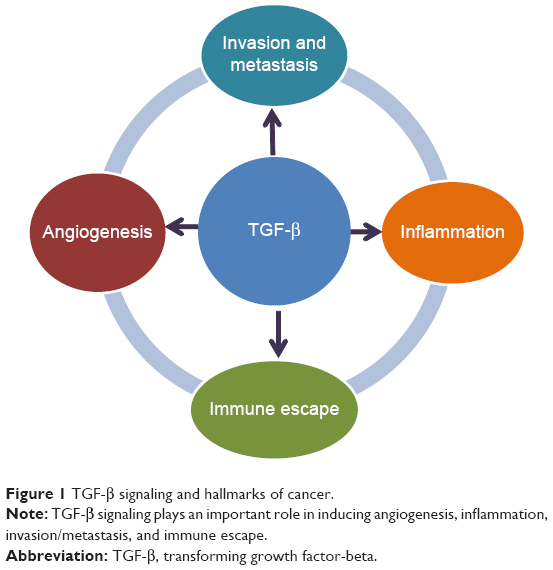 | Figure 1 TGF-β signaling and hallmarks of cancer. |
TGF-β/ALK5 signaling pathway and its role in cancer
In the early 1980s, TGF-β was biochemically isolated from tumor cells and was named after its ability to transform normal rat kidney fibroblasts.1 This approximately 13 kDa polypeptide was later designated as TGF-β1.3 TGF-β ligands include TGF-β1, TGF-β2, and TGF-β3 (Table 1), all of which regulate diverse biological functions.4,5 All three ligands can independently engage the specific receptor TGF-βRI/ALK5, which then undergoes dimerization with TGF-β receptor type II (TGF-βRII).6 Recent studies have shown that two TGF-β ligand molecules bind as homodimers to two TGF-βRI and two TGF-βRII molecules (ie, a homodimer bound to a heterotetramer), creating a heterodimer complex consisting of six distinct molecules.7 For simplicity reasons, figures often do not show the duplicity of the heterodimer (Figure 2). This heterodimer complex phosphorylates the intracellular proteins SMAD2 and SMAD3, activating a signaling cascade to induce several nuclear transduction proteins. The induction of these proteins leads to cellular proliferation, differentiation, motility, survival, and apoptosis in tumor cells.8 This TGF-β/ALK5 signaling pathway is commonly referred to as the canonical signaling pathway (Figure 1). In addition to this canonical pathway, a number of accessory receptors have been identified, which can separately interact with the TGF-RI and TGF-RII. One example is the TGF-βRIII, to which the glycoprotein endoglin (CD105) can bind with high affinity.9
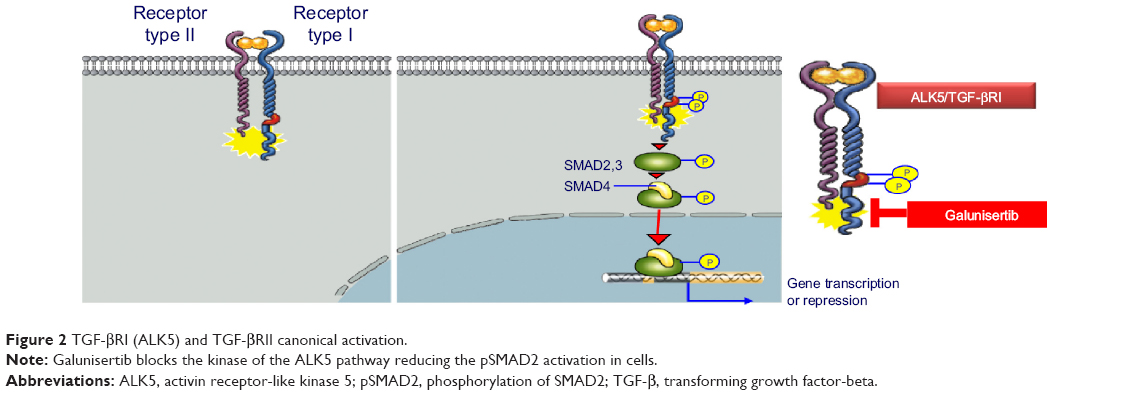 | Figure 2 TGF-βRI (ALK5) and TGF-βRII canonical activation. |
Given the complexity of the TGF-β signaling pathway, its physiological role is diverse and appears to be dependent on the disease setting and cellular context. In cancer, TGF-β can affect tumor growth directly (referred to as intrinsic effect of TGF-β signaling) or indirectly (referred to as extrinsic effect) by promoting tumor growth, inducing epithelial-mesenchymal transition (EMT), blocking antitumor immune responses, increasing tumor-associated fibrosis and enhancing angiogenesis (Figure 1).10 Although TGF-β1 was originally found to transform normal cells,1 subsequent studies revealed that it also behaved as a tumor suppressor in epithelial cells. The factors determining whether TGF-β signaling has a tumor promoter or suppressor function are a matter of intense research.8 Currently, it is postulated that the tumor suppressor function of TGF-β signaling is lost in early stages of cancer similar to recessive loss-of-function mutations in other tumor suppressors.5 Such a loss of function has been associated with drug resistance in tumors and reported in colon and lung cancer cell lines.11–14 The switch to becoming a tumor promoter with resulting drug resistance often occurs in the presence of EMT.15 TGF-β signaling plays a prominent role in EMT by influencing key transcription factors, including Snail, zinc-finger E-box-binding, and basic helix–loop–helix transcription factors. Besides these genetic and physiological changes, TGF-β signaling plays a critical role in the regulation of immune cell function in normal and tumor-associated lymphocytes, in particular, the activation of T-regulatory cells.16,17
In addition to its role in regulating immune cells, TGF-β signaling is a strong inducer of fibrosis.18 It is an activator of cancer-associated fibroblasts (CAFs)19 that can be a major source of collagen type I and other fibrogenic factors. CAFs and their associated production of fibrogenic factors may further foster a microenvironment that decreases immune responses and increases drug resistance and tumor angiogenesis.20
The role of TGF-β signaling in angiogenesis is well recognized during ontogeny and in tumor growth.21 For example, murine embryos deficient in TGF-βRI exhibit severe defects in vascular development; TGF-β signaling acts as a key regulator of development of both vascular endothelial and smooth muscle cells.21
In summary, TGF-β signaling plays a key role in pathways that are considered hallmarks of cancer22 by aiding cancer cells in their evasion of immunosurveillance, increasing inflammation (in part by modulating cytokine responses, tumor migration, and angiogenesis) (Figure 1).5,23,24 Thus, targeting this TGF-β-mediated signal transduction in tumors may provide a novel approach to controlling tumor growth by blocking a central activation node with its connected downstream signaling pathways.25
Pharmacological approaches: blocking the TGF-β signaling pathway
Specific inhibitors are key for the pharmacological development of ALK5 inhibitors:
Given the structural and genetic similarities of the different TGF-β signaling pathways, inhibitors must be highly specific to block the intended activation pathway. For example, ALK5 and ALK1 pathways both increase tumor angiogenesis (Table 1).26 ALK1 inhibitors block the interaction of bone morphogenetic protein (BMP)-9 and -10 with ALK1, which interrupts the subsequent phosphorylation of SMAD1 (pSMAD1)/pSMAD5/pSMAD8 (Table 1). PF-03446962 is an ALK1 neutralizing antibody that does not bind other ALKs. Currently, PF-03446962 is being evaluated in Phase II trials in patients with solid tumors to determine its ability to block angiogenesis (Table 2). Another molecule directed against ALK1 is dalantercept/ACE-041, a chimeric protein consisting of the ALK1 ligand binding extracellular domain and an Fc portion (ALK1-Fc). This ligand trap is a soluble receptor that can bind circulating ligands and prevents their engagement with cell surface receptors. Dalantercept efficiently blocks BMP-9 and BMP-10-induced SMAD1 phosphorylation and SMAD1-dependent transcription. Dalantercept is currently in Phase II trials as monotherapy and in combination with vascular endothelial growth factor inhibitors.27 In contrast to the ALK1 inhibitors, the inhibition of the ALK5 pathway blocks activation of different intracellular proteins (eg, SMAD2/3) and alters the vascular and smooth muscle cell compartment.21 This may improve delivery of chemotherapy28 although it may have a reduced inhibitory effect on pro-angiogenic factors, such as vascular endothelial growth factor and basic fibroblast growth factor. In contrast to ALK1 inhibitors, ALK5 inhibitors increase angiogenesis in cell cultures of normal endothelial cells.29
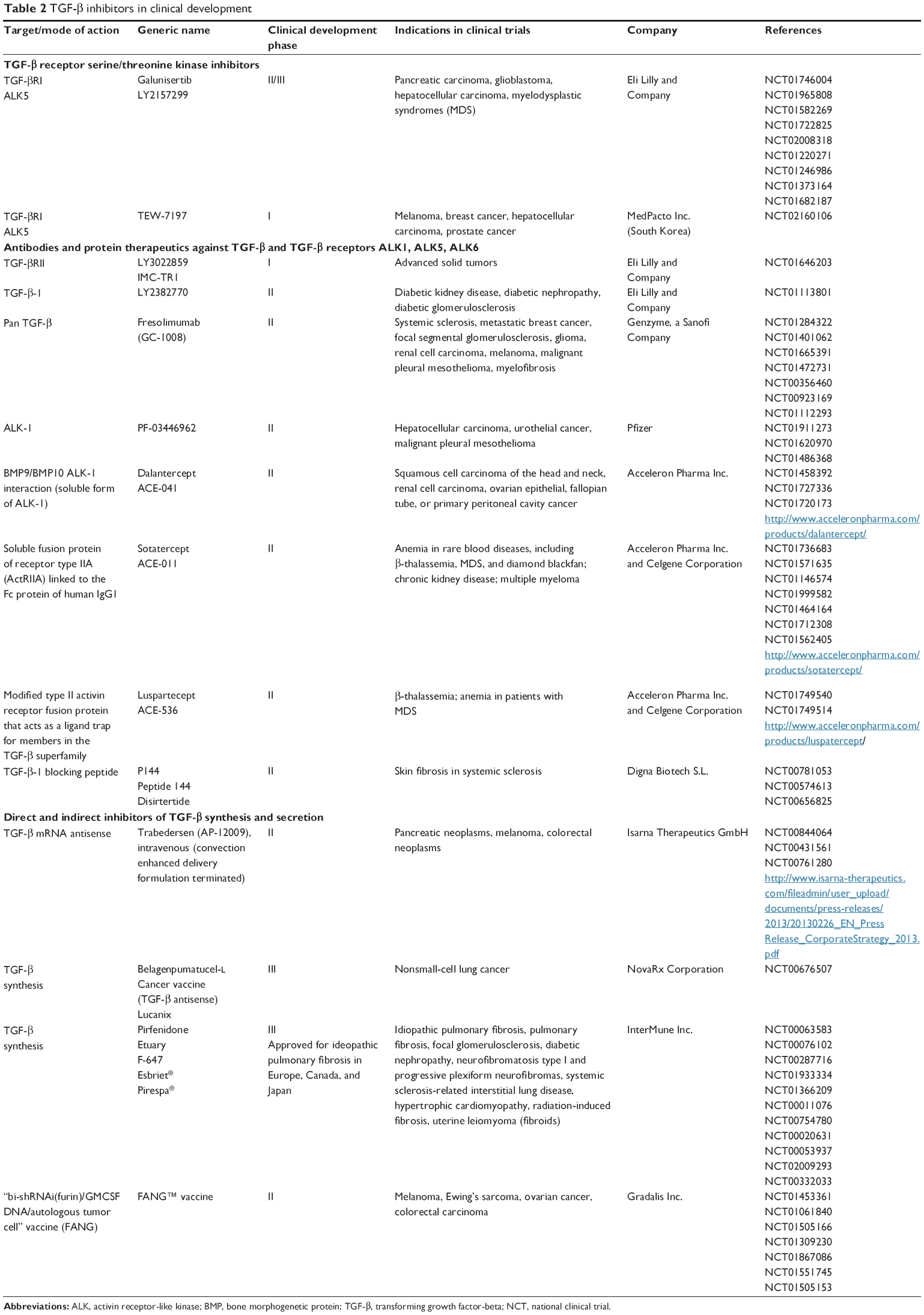 | Table 2 TGF-β inhibitors in clinical development |
Monoclonal antibodies: Arguably, monoclonal antibodies are considered highly specific and can provide the best approach to develop selective inhibitors to the TGF-β signaling pathway. Fresolimumab (formerly GC1008) is one of the first pan-TGF-β ligand monoclonal antibodies to be investigated in cancer patients.30 Fresolimumab is being developed for patients with renal fibrosis and until recently, for patients with metastatic cancer.30–33 In a Phase I trial, fresolimumab was given to 29 patients with malignant melanoma and renal cell carcinoma administered in doses up to 15 mg/kg given first every 28 days then subsequently by biweekly dosing to patients with stable disease. Reversible cutaneous keratoacanthomas/squamous cell carcinoma (four patients) and hyperkeratosis were reported as major drug-related adverse events. One patient with malignant melanoma achieved a partial response lasting 44.4 weeks, and six melanoma patients had stable disease. Of the seven patients with partial response or stable disease, six patients had received ≤3 mg/kg of fresolimumab. Given that patients with higher doses (up to 15 mg/kg) had no responses or stable disease, the authors postulated that clinical activity was associated with lower doses.30 In a second Phase II study, fresolimumab was given to 13 patients with malignant pleural mesothelioma at 3 mg/kg given every 21 days, the anticipated most active dose. None of the patients had radiographic responses, and three patients had stable disease at 3 months. However, five patients developed new antibodies against tumor lysates isolated from malignant pleural mesothelioma, suggesting a possible immune response. After the 13 patients were treated, the manufacturer terminated further development of fresolimumab for oncology indications.33 Eli Lilly and Company also developed a pan-TGF-β ligand inhibitor but did not pursue its development due to severe toxicity in animals (data on file, Eli Lilly and Company, Indianapolis, IN, USA). In addition, a TGF-β1-specific monoclonal antibody (TβM1 or LY2382770) was evaluated in a Phase I study in patients with cancer. LY2382770 showed no radiographic responses once escalated to the predefined dose of 240 mg (flat dose) given every 28 days, which was thought to be efficacious based on a pharmacokinetic/pharmacodynamic (PK/PD) model for LY2382770.34 There are also monoclonal antibodies that were designed to block the TGF-β receptors, such as the monoclonal antibody TR1.35 TR1 blocks the TGF-βRII; this monoclonal antibody is currently being evaluated in a first-in-human dose (FHD) study. Given the toxicity observed with small molecule inhibitors (SMIs) blocking ALK5 (see section SMIs of the TGF-bRI/ALK5 and the early development of galunisertib), specific inhibitors of TGF-βRII using monoclonal antibodies were thought to have more manageable toxicity or perhaps even a reduced toxicity profile.
Antisense oligonucleotides (ASOs): Another approach to selectively blocking the TGF-β signaling is the use of ASOs. For example, the first-generation ASO trabedersen (formerly AP12009) was developed to block the TGF-β2 production in glioma cells and was advanced to clinical investigation.36 Although the safety profile was manageable, the antitumor activity of trabedersen seemed to be limited to a subset of patients, mainly those with World Health Organization grade III glioma. Similar to fresolimumab, lower doses appeared to be associated with better radiographic responses. However, the clinical development of trabedersen with its convection-enhanced delivery of compound in patients with glioblastoma was terminated. The convection-enhanced delivery was necessary for trabedersen because first-generation ASOs generally achieve their optimal pharmacological activity if they are continuously applied and administered directly to the tumor. Because of this added complexity for the administration of an ASO, newer ASOs with improved chemistry are being generated for future investigation in a range of indications, including cancer.37
Vaccines strengthening the patient’s microenvironment:
Another approach to blocking the TGF-β signaling pathway is based on vaccines. For example, belagenpumatucel-L is a cancer vaccine (NovaRX Corporation, San Diego, CA, USA) that inhibits the synthesis of TGF-β2. Genetically modified whole tumor cells stimulate the patient’s own immune system to attack the tumor. The vaccine contains four allogenic and irradiated nonsmall-cell lung cancer (NSCLC) cell lines expressing a TGF-β2 synthetic antisense gene. The recently reported Phase III “STOP” trial in patients with stage III/IV NSCLC did not meet its predefined primary endpoint of overall survival (OS). However, an increase in OS was observed in a subgroup of patients who had started treatment with belagenpumatucel-L within 12 weeks of completing frontline chemotherapy. Also, patients with nonadenocarcinoma NSCLC (= squamous cell carcinoma and large cell lung cancer) had an OS of 19.9 months when treated with belagenpumatucel-L compared with an OS of 12.3 months for patients treated with placebo (hazard ratio 0.55, P=0.036). The authors of the study concluded that belagenpumatucel-L should be further evaluated in lung cancer.38
SMIs of the TGF-βRI/ALK5 and the early development of galunisertib (LY2157299 monohydrate)
Among the TGF-β inhibitors, SMIs represent a large and diverse group of chemical entities that are designed to block the activation of the signaling cascade downstream of the TGF-β receptor type I kinase (TGF-βRI or ALK5) or type II (TGF-βRII) by inhibiting the serine/threonine kinase. There is a growing list of SMIs blocking the TGF-βRI,39–42 including recent SMIs such as Ki2689443 and TEW-7197.44,45
At Eli Lilly and Company, several SMIs were identified in the past years (Table 3). A large library of SMIs was screened in vitro using a TGF-β-dependent cell-based assay. Selected compounds were further evaluated for their ability to inhibit autophosphorylation of the isolated human TGF-βR-I kinase domain. The TGF-βR-I kinase domain was expressed as a constitutively active construct (T204D mutation) by Sf9 insect cells.46,47 For example, the diheteroaryl-substituted pyrazole 1 (LY364947) was identified as a potent inhibitor (IC50=51 nM) (Table 3). Compounds were further evaluated by measuring their inhibitory effect in a TGF-β-dependent luciferase assay produced in mink lung cells (p3TP Lux) and their growth inhibition in mouse fibroblasts (NIH3T3).48 Compounds such as LY580276,49 LY364947,50 and LY210976151 share with LY2157299 monohydrate the selectivity profile and inhibition of the ALK5.52–54 Overall, the kinase selectivity profile of galunisertib met the desired characteristics (Table 4).
 | Table 3 Clinical and surrogate compounds developed by Eli Lilly and Company (data on file) |
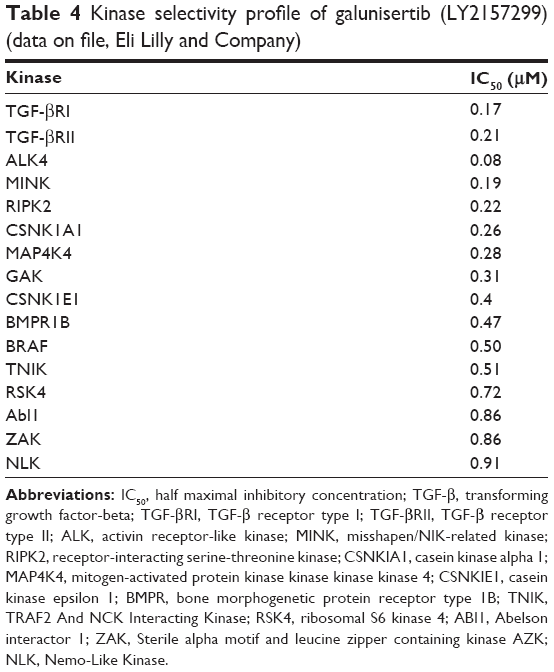 | Table 4 Kinase selectivity profile of galunisertib (LY2157299) (data on file, Eli Lilly and Company) |
Compared with other SMIs, galunisertib (LY2157299) monohydrate had reduced cardiovascular toxicity in animals and appeared to be less potent in inhibiting pSMAD2 levels in vitro (Table 3).49,55–57 Galunisertib (pronounced: gal-ue’ ni-ser-tib) is now the United States Adopted Name and International Nonproprietary Name designation for LY2157299 monohydrate. The first part of the name refers to the Greek-Roman physician Aelius Galenus (born: 129 AD; died: c.200/c.219 AD), also known as Galen of Pergamon.58 Apart from galunisertib that started its clinical development in 2006, only TEW-7197 is known to be in clinical investigation. In mid-2014, a Phase I study of TEW-7197 was initiated in patients with breast cancer, melanoma, hepatocellular carcinoma (HCC), and glioblastoma.59
Despite the long list of ALK5 SMI inhibitors, few ALK5 inhibitors have been moved to clinical investigation, perhaps because of the observed severe cardiac toxicities in animals.41,60 Galunisertib overcame this barrier by addressing the following: (a) running preclinical toxicology studies with administration schedules that identified a sufficient margin of safety, (b) developing a predictive PK/PD model using animal pharmacology information, and (c) developing assays of PD markers in patients with cancer to confirm the therapeutic window as predicted by the PK/PD model (Figure 3).
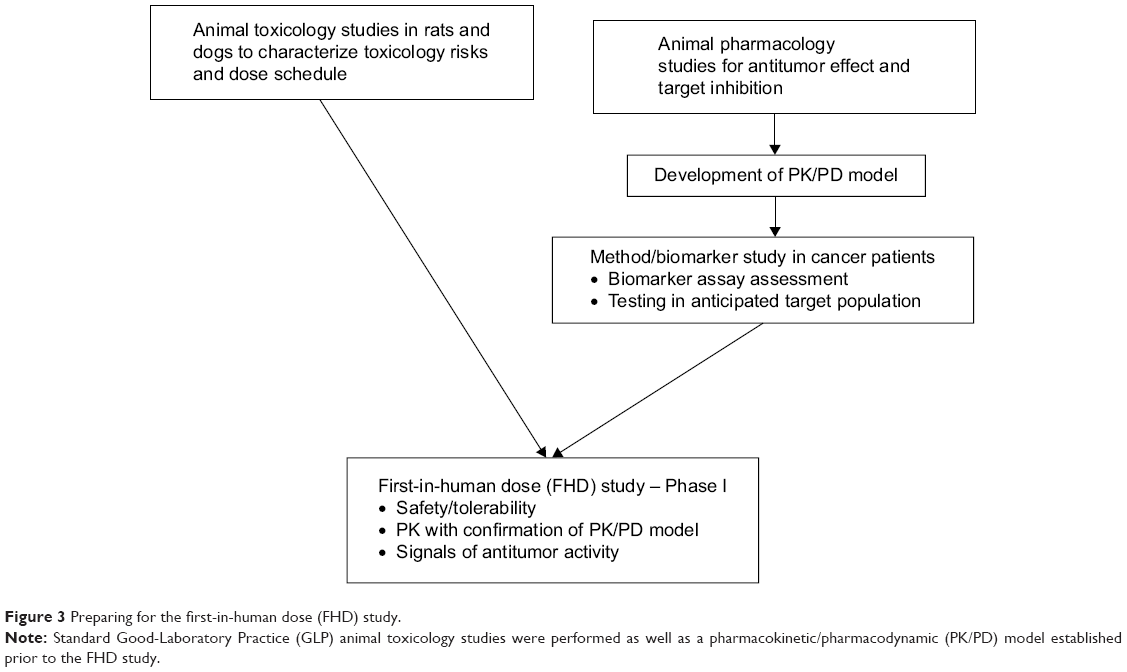 | Figure 3 Preparing for the first-in-human dose (FHD) study. |
Toxicology
In animal studies, several ALK5 inhibitors showed an increased incidence of hemorrhagic, degenerative, and inflammatory lesions in heart valves. Galunisertib was selected for clinical investigation based on its profile in animal toxicology studies in rats and dogs.49,50,57,61,62 The lesions appeared either at very high doses (1 month of continuous dosing) or during continuous dosing for 6 months. Intermittent dosing provided a sufficient margin of safety to advance galunisertib into clinical development.57 In general, rats appeared to be more sensitive to galunisertib toxicity than dogs. Because of the difference in heart rate and also intravascular blood pressure, there is a possibility that the cardiac lesions were related to shear stress-associated intravascular remodeling. Under such conditions, the TGF-β signaling pathway is activated in endothelial and smooth muscle cells.63,64 Based upon these findings, it is possible that the lesions observed in the valves resulted from an inhibition of a physiologic TGF-β-dependent mechanism, which is normally needed to repair the injury caused by shear/stress at the outflow of the heart. When valves and ascending aorta were examined, they showed activation of the TGF-β pathway after administration of a TGF-β inhibitor as measured by pSMAD2 staining. Patients with Loeys–Dietz syndrome have a similar paradox where a loss of functional mutation in the TGF-β signaling gene is present; however, in the tissue, an activation of the TGF-β pathway is observed.65,66 One hypothesis for this paradox is that alternative pathways are activated.67 A second hypothesis proposes a changed receptor/ligand processing as observed in studies with cells.68,69 If the second hypothesis based on a possible pathway adaptation is correct, then the intermittent dosing of TGF-β inhibitors provides a safe dosing regimen.
Galunisertib affects bone development and alters inflammatory responses in the skin or gut of rats and dogs. It is well known that blocking TGF-β signaling can cause chronic inflammation in skin and gut, which in turn can lead to precancerous conditions.70,71 Intermittent or continuous regimens with low doses may reduce such a risk of developing chronic inflammation and thus allow long-term administration of a TGF-β inhibitor.
Pharmacology in cellular assays and animals
In addition to the animal toxicology studies, evidence of target inhibition and preclinical antitumor efficacy was needed for developing a predictive PK/PD model.72,73 The initial antitumor activity of galunisertib was characterized in three different cancer models: two breast cancer models, MX1 and 4T1, and a NSCLC model using Calu6. Dosing (75 mg/kg twice daily by oral gavage) was initiated 4–6 days postinoculation and continued for 20 days in the xenograft models (MX1 and Calu6) for the entire length of the survival study with the 4T1 model.74 A statistically significant tumor growth delay of 10.1 days was observed in the MX1 model, and the 4T1 tumor model exhibited a 4.5-day survival advantage. In the Calu6 xenograft, galunisertib significantly delayed tumor growth.
Despite these encouraging observations with some cell lines in standard xenografts (eg, Calu6 and U87MG), it appears that only select tumor cell lines respond to galunisertib treatment in standard, immune-deficient xenografts. In patient-derived xenografts (PDXs), the antitumor response to galunisertib treatment is also limited.75 Furthermore, in some immune-deficient PDX models, tumor growth appeared to be enhanced, which was previously raised as a potential risk for administering TGF-β inhibitors to patients.76,77 However, it is possible that such growth is only observed in immune-compromised animals; in immune-competent murine models, no such tumor growth augmentation has been observed. Tumor-bearing, immune-competent mice treated with the monoclonal antibody against TGF-βRII, TR1, had antitumor effects that were at least partly dependent on the presence of T-cell subsets.35 Finally, mice receiving long-term exposure (over 1 year of administration) of a soluble TGF-β antagonist had no adverse events.78 All these observations imply that immune-competent animal models may be more predictive to evaluate the activity of TGF-β inhibitors. It appears that an active immune response is essential to assess the effect of TGF-β signaling inhibition in animal models; thus, models using immune-compromised animals may have limited use in screening for TGF-β inhibitors.
Because of the difficulties of using animal models as a screening method for identifying novel agents, alternative in vitro models were considered. Tumor clonogenic assays with PDX were evaluated, but it became evident that many PDX had a disrupted canonical pathway.75 Hence, this assay was found not to be adequate to screen large libraries of novel SMI TGF-β inhibitors. Recently, experiments with a 3D in vitro model were used to assess the effect of galunisertib.79 In such 3D culture systems, galunisertib had activity whereas it had none in standard monolayer assays. An advantage of the 3D culture model was the possibility to evaluate some aspects of the immune response. Whether such 3D in vitro assays may also offer an alternative to in vivo models is not yet determined.
Drug resistance mechanism and implications on drug combination
Based on the biology of TGF-β signaling, the associated mechanisms that affect drug resistance are likely to be dependent on the tumor microenvironment. One such example are the CAFs: these produce large amounts of the TGF-β ligands and are associated with fibrosis and with resistance to 5-fluorouracil (5-FU).20 Recently, drug resistance related to TGF-β signaling was reported to be associated with loss of the MED12 gene in tumors.80 MED12 loss induces not only an EMT-like phenotype that results in chemotherapy resistance to 5-FU but also resistance to the epidermal growth factor receptor tyrosine kinase inhibitor (EGFR TKI) gefitinib. Treatment with galunisertib in MED12-deficient cells restored the sensitivity to both chemotherapy and EGFR TKI. In addition to drug resistance to 5-FU and EGFR TKIs, there were reports connecting TGF-β signaling to paclitaxel resistance in triple-negative breast cancer.81 In all these observations, it appears that EMT or EMT-like phenotype of the tumor cells plays a critical role to drug resistance associated with TGF-β signaling.
PK/PD model – predicting a therapeutic window in patients with an acceptable safety profile
The development of preclinical PK/PD models have been invaluable in guiding early clinical trial design.82,83 A similar model was built using preclinical data on pSMAD2 inhibition, antitumor activity of galunisertib in Calu6 xenografts, and the observed PK in mice, rats, and dogs.72,73 The half-life of galunisertib in animals was less than 3 hours (Table 3). An observed moderate variation in PK was, in part, attributable to the formulation of galunisertib.84 Allometric PK scaling of galunisertib allowed a reliable prediction of both the exposure in humans within the expected range to produce antitumor activity. The drug effect continued even after the systemic disappearance of the drug: the PD effect of reducing pSMAD2 was still detectable in tumor tissue and peripheral blood mononuclear cells (PBMCs) up to 7 days after stopping galunisertib and when galunisertib was no longer detected in the plasma. This delayed PD effect was also seen when treated with the monoclonal antibody against TGF-βRII, TR1, suggesting that this phenomenon is not limited to SMIs (data on file, Eli Lilly and Company).
The simultaneous inhibition of pSMAD2 inhibition in tumor and surrogate tissue (ie, PBMCs) led to the development of a PD detection assay using peripheral blood. This assay was developed to monitor and confirm the PK/PD relationship during the FHD study. To avoid toxicity and maintain antitumor activity, the galunisertib exposure had to be limited to a pSMAD2 inhibition of approximately 30% over 24 hours, combined with a maximum inhibition of 50%. This was achieved by a twice-daily (BID) dose schedule that produced a modulatory exposure.85
Dosing considerations for galunisertib
Based on the PK/PD modeling and the toxicity observation, we decided to use a BID dosing schedule and a 14-day on/14-day off schedule. In preclinical models and later in the Phase I study, we had observed that pSMAD2 inhibition was extended up to 7 days after galunisertib was stopped. Given that continuous dosing may increase the risk for chronic toxicity, the 14-day treatment with an anticipated prolonged pSMAD2 inhibition of 7 days was the most acceptable regimen for long-term treatment. To avoid high single-day exposures, a morning and evening dosing schedule was instituted. All these interventions were designed to avoid a steady-state or continuous on-target inhibition.
Early biomarker development
The biomarker work early in development focused on two main objectives: a) biomarkers for patient selection and b) pharmacodynamic response markers. For patient selection, three groups were considered: those whose tumors produced high amounts of TGF-β1, (eg, in renal cell carcinoma,86 prostate cancer,87 and breast cancer25); those in whom TGF-β inhibition had shown clinical responses with other TGF-β inhibitors (such as glioma36), and those with skeletal metastasis. In such conditions, TGF-β is being mobilized from the bone matrix, and increased TGF-β1 can serve as a marker of tumor progression.88
A pSMAD2 assay to measure the reduction of pSMAD2 in PBMCs during the FHD trial was established.89 This pSMAD2 enzyme-linked immunosorbent assay (ELISA) used a polyclonal antisera and was tested on serum from patients with skeletal metastasis (a nondrug interventional trial). The intrapatient variability was determined to be less than 30%.90 This variability was considered acceptable for the FHD trial.
A plasma TGF-β1 ELISA was developed and subsequently used for PD assessments. This assay provided reliable information on plasma TGF-β1 levels when citrate-theophylline-adenosine-dipyridamole tubes were used for collection.91 The assay detects the activated TGF-β1 form in a standard ELISA format (R&D Systems, DB100B). The original hypothesis assumed that TGF-β1 levels will be reduced if galunisertib interrupts a possible autocrine or paracrine growth signal.
Other plasma proteins such as parathyroid hormone-related protein, von Willebrand factor, and interleukin-10 correlated with TGF-β1 levels.90 Given this connection, the measurements of such indirect plasma markers were thought to help with understanding of the downstream effect of the TGF-β signaling inhibition with galunisertib. Thus, they were considered as alternative PD markers for future clinical trials.
Gene expression profiling of PBMCs was initially pursued as an additional option to assess PD changes,92,93 but previously observed changes in PBMCs in ex vivo experiments were not able to be reproduced in subsequent clinical trials. In earlier ex vivo studies, T-cells were activated with CD3/CD28 costimulation to obtain reliable gene expression profiles.92 By contrast, PBMCs obtained from patients with skeletal metastasis were only stimulated with exogenous TGF-β1.93 The stimulation with exogenous TGF-β1 led to a rapid and subtle change in gene expression, which may have been too transient to be detected in patients treated with galunisertib.
Several attempts were also made to develop patient selection strategies based on gene expression profiles from tumor-bearing animal or PDX models.75,94,95 These profiles were compared with publicly available gene expression data sets, but it was not possible to define a clear profile. In part, this was due to the minimal antitumor activity with galunisertib in immune-deficient animal models or monolayer cell line experiments.
FHD study and early clinical development of galunisertib in cancer patients, including patients with glioblastoma
Given the potential for toxicity and general safety concerns, throughout the course of the FHD study the predictive PK/PD model was continuously updated, and results were shared with the scientific community (a summary of all clinical studies with galunisertib is presented in Table 5). The decision to select galunisertib as a clinical candidate was presented in 2005,72 and the FHD study was disclosed at a conference in 2006.56 Over the subsequent years, the progress of the FHD study and the patient safety information were routinely presented.96–99 Additionally, preclinical studies of galunisertib were regularly updated.72,74,94
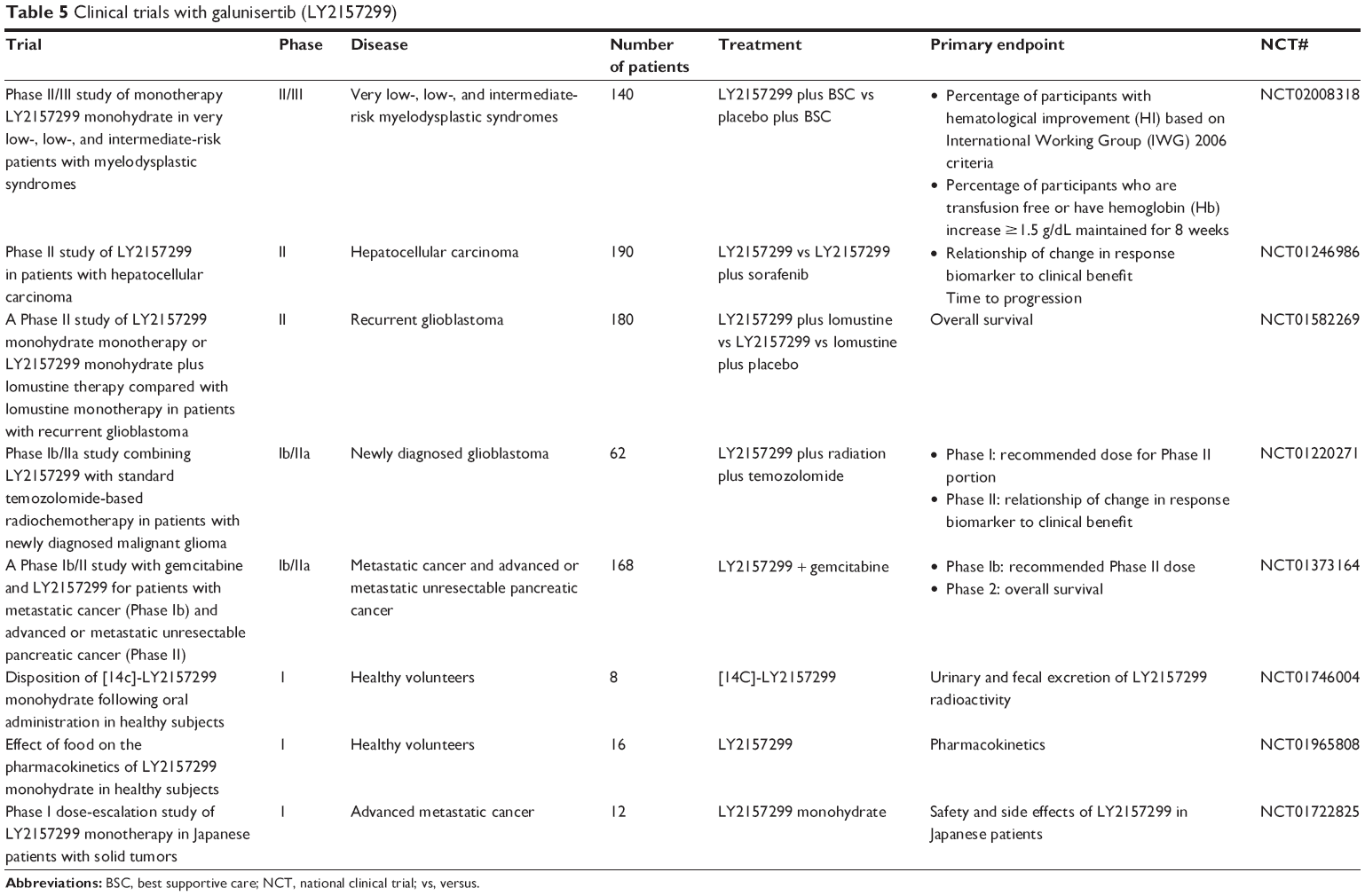 | Table 5 Clinical trials with galunisertib (LY2157299) |
One of the most important objectives of the FHD study was to assess the safety of galunisertib when administered to reach the predicted therapeutic window. Based on the PK/PD model, the therapeutic window was anticipated to be achieved when compound was dosed between 160 and 360 mg.73 Within the initial cohorts of the FHD study, galunisertib was given daily continuously based on the standard 1-month animal toxicology studies. Although not generally required for oncology Phase I trials, 6-month animal toxicology studies were begun to assess the risk of more prolonged exposures. This study started concurrently with enrollment of the patients into the FHD study. Unexpectedly, new toxicities emerged after the evaluation of 6-month animal toxicology studies. As a result of these new findings, the FHD study was placed on clinical hold and additional animal toxicology studies began using an intermittent dosing schedule. The intermittent dosing schedule in the most sensitive species (rats) provided an acceptable margin of safety. These data supported lifting the clinical hold; the FHD study resumed but was restricted to patients with glioblastoma, in part because of the previously reported activity of trabedersen.36
During the FHD study, all patients underwent comprehensive cardiac monitoring. These studies consisted of baseline and every-other-month echocardiography/Doppler imaging, baseline and monthly serial plasma assessment for Troponin I, brain natriuretic peptide, and additional cardiac measurements.100 No drug-related cardiovascular toxicity was observed during the entire course of the FHD study. The use of a central echocardiography/Doppler assessment along with cardiac serial serum marker evaluations proved valuable and was supplemented by the help of dedicated cardiologists on call at each site. In this study, one postmortem examination was performed on a patient who died from progressive malignant glioma: although the aorta was found to be abnormal, the ante-mortem echocardiography/Doppler was normal. There was an absence of pathological cardiac serum markers, and there were no histopathologic changes in the heart valves. The case was also peer reviewed by an external vascular pathologist who found the changes to be consistent with routine degenerative findings in age-matched patients.101
During the course of the FHD study, radiographic responses were observed; some subjects obtained complete responses that proved durable with patients on study drug more than 2 years. Some of these individuals with glioblastoma included patients with relapsed disease (World Health Organization grade II and III).102,103 Responses tended to occur after a minimum of two cycles of galunisertib. Also, in two patients with radiographic responses, a site-specific imaging study found that tumor blood flow was changed, similar to that seen with antiangiogenic agents.104 However, compared with other antiangiogenic drugs, the blood flow changes occurred at a later time.
Tissue was available from 8 of 20 patients with secondary/low-grade glioma. Of these eight patients, five had an IDH1/2 mutated tumor; within this cohort, four patients (80%) had either radiographic response or stable disease ≥ six cycles. This suggests that the TGF-β pathway plays an important role in this subgroup of glioma patients.103 This observation is intriguing because of the similarities described with trabedersen.36 Furthermore, in vitro, the IDH1 R131H variant has been associated with induction of mesenchymal gene expression phenotype.105 In addition to the patients with an IDH1 mutation and response, the other patients who appeared to benefit had no clear genetic pattern of a mutation. The recently defined mesenchymal subtype of glioma may be driven by TGF-β signaling given the well-recognized association between TGF-β and the EMT.106 As such, the mesenchymal phenotype or IDH1 variant may enrich for patients likely to respond to galunisertib.
In PBMCs, pSMAD2 was reduced in most patients treated with galunisertib.97 A post-treatment biopsy in one patient showed a reduction in Id1 and CD44 gene expression, both of which are upregulated by TGF-β signaling.107 These observations support that galunisertib achieved its pharmacologic effect by its presumed central mode of action (by blocking the TGF-β signaling). The data from PBMCs further indicated that peripheral blood cells could be used to determine the PD effect of TGF-β inhibitors; however, we did not identify whether this effect is mainly related to the effect on neutrophils, macrophages or lymphocyte subsets.
Using continuous assessment of PK and PD, the preclinical prediction of PK and PD was confirmed in humans, and the therapeutic window was deemed to be safe for further development.85 For the Phase II studies, a 150-mg BID dose (300 mg/day; given intermittently 14 days on/14 days off on a 28-day schedule) was selected, with few Grade 3 or 4 toxicities observed to date.103
Glioblastoma (glioma)
Nonclinical observations
The TGF-β signaling pathway and, especially, the pSMAD2 expression are associated with poor prognosis in glioblastoma.108–110 Thus, it was hypothesized that tumors overexpressing the TGF-β signaling pathway will respond to a TGF-β SMI. Also, data suggested that patients with low-grade glioma have higher plasma TGF-β1 levels than patients with high-grade gliomas. If TGF-β1 levels are decreased after treatment to the primary (such as surgical tumor removal), patients may benefit even if the surgery is not definitive.111 The source of the TGF-β signaling in glioma has not been identified and may not originate from the tumor cell itself. For example, TGF-β signaling is found in cancer-initiating stem cells, which are a small subset within the tumor tissue. Galunisertib may inhibit these cancer-initiating stem cells and thus arrests TGF-β-dependent tumor cell growth and migration.107,112,113
Although we observed no responses to galunisertib in most PDX and tumor cell lines, the glioma models such as U87MG did show direct treatment responses to galunisertib. Hence, the combination of galunisertib with concurrent temozolomide (TMZ) and radiation was evaluated in this model: an additive antitumor effect was observed.114,115 Also, the combination of galunisertib and lomustine showed additive antitumor activity in this same xenograft model. Interestingly, lomustine by itself reduced pSMAD2 in glioblastoma cell lines.116 This was an unexpected finding, further suggesting that a combination of lomustine and galunisertib may have additive pharmacologic effect.
Clinical experiences
Based on the totality of the data,102 galunisertib received orphan drug designation by the European Medical Agency (EMA) (May) (EMA/COMP/175329/2013) and the Food and Drug Administration (March) in 2013.117 Two Phase II studies in glioblastoma patients were concurrently initiated: 1) a blinded, randomized three-arm study comparing galunisertib to lomustine and the combination of galunisertib and lomustine in patients receiving second-line systemic treatment after first relapse118 and 2) a randomized Phase I/II study in patients with newly diagnosed glioblastoma receiving standard first-line therapy of chemoradiation with TMZ (Table 5).119 Although both studies are still ongoing, interim data have been presented. At American Society of Clinical Oncology 2013, safety data of the second-line study were presented, and no added toxicity across the entire study was reported.120 The first-line study investigates clinical benefit in relationship to biomarker changes such as total lymphocyte, CD3+, CD4+, CD4+CD25+, CD127+, Foxp3+, and CD8+ T-cells counts.121 At interim, galunisertib administration appeared to conserve or even increase CD8+ counts after standard chemoradiation had been completed, and patients were receiving adjuvant TMZ and galunisertib. This finding was not observed in patients receiving only adjuvant TMZ.
Hepatocellular carcinoma
Nonclinical observations
TGF-β expression is associated with both early and late progression of HCC.122–126 HCC cells can secrete TGF-β1 in an autocrine manner; blocking this feedback loop may have a beneficial impact on controlling tumor progression.123 In contrast to other HCC treatments, in vitro galunisertib had no cytotoxicity at the putative therapeutic concentrations; however, galunisertib was inhibitory in cell invasion and migration assays.125,127–133 This effect is seen at a 1–10 nanomolar concentration in several HCC cell lines.
Similar to the impact of sorafenib, galunisertib reduces alpha-fetoprotein (AFP) and Ki67 expression in patient-derived HCC liver slices.134 Recently, the combination of sorafenib and galunisertib has demonstrated an additive effect in immune-competent C57BL6/ASV-B mice,135 a murine model that spontaneously develops HCC as a result of SV40 antigen overexpression in the liver. Whether the additive effect of sorafenib and galunisertib is also related to the ability of sorafenib to inhibit the TGF-β pathway at high concentrations is unclear.136 Galunisertib is also active in HCC cells that are resistant to multikinase inhibitors such as sorafenib and sunitinib.137,138 This implies that galunisertib may have activity in conditions where other kinase inhibitors are inactive. In vitro studies further suggest that galunisertib has inhibitory activity on kinases downstream of SMAD2 when treatment is extended beyond 24 hours.132 Although the relevance of this finding on the antitumor activity of galunisertib remains to be further evaluated, these long-term cultures with galunisertib had no cytotoxic effect on cells, despite the changes of multiple kinases. This observation further supports that galunisertib exerts its antitumor effect not by cell killing but by altering the phenotype of the cells and by reducing their EMT phenotype. In summary, galunisertib has shown an antitumor effect in HCC that appears mainly based on the ability to inhibit migration and invasion rather than direct tumor cell killing.
Clinical studies
In 2013, galunisertib received orphan drug designation in HCC by the EMA (March) (EMA/COMP/95768/2013) and the Food and Drug Administration (April).117 Using the preclinical information, a two-arm dose comparison Phase II study139 was designed to compare the lower (160 mg/day) dose with the higher (300 mg/day) dose. The objective was to determine whether the lower dose was superior to the higher dose given that other TGF-β inhibitors may have been more effective at lower doses.30,36 All patients in this dose-comparison study had to have elevated AFP (≥1.5× upper limit of normal) and reduction in AFP was initially used to select the dose for future clinical development. All patients had to have failed prior sorafenib or be ineligible to receive sorafenib (Table 5). Preliminary results of this trial showed that there were no differences between the doses in an intent-to-treat analysis, although patients receiving 300 mg/day (including patients with poor prognosis) had slightly different demographic characteristics, such as Child Pugh B7 and prior liver transplantation. Given this lack of difference between the doses, all new patients were enrolled on 300 mg/day.140,141 Consistent with the in vitro observations, some patients had a reduction in plasma AFP, E-cadherin, and TGF-β1 levels. Patients with reduction in any or all of the three markers had a longer time-to-tumor progression and OS. Patients with ≥20% reduction from baseline in AFP had a median OS of approximately 21 months. This was observed in about 25% of all patients who had elevated AFP at baseline. Furthermore, 50% of the patients had elevated TGF-β1 levels (above 3,400 pg/mL). In 66% of these patients, a TGF-β1 reduction of ≥20% was observed, and median OS was about 12 months. Given the poor prognosis generally associated with elevated AFP, the survival information suggested that galunisertib was a potentially active agent in a difficult-to-treat population with poor prognosis.
Pancreatic cancer
Nonclinical observations
TGF-β signaling pathway is active in metastatic/advanced pancreatic cancer.142 In a xenograft model, LY2109761 was reported to have antitumor activity in combination with gemcitabine.143 This observation was confirmed using galunisertib (data on file, Eli Lilly and Company). One possible explanation for this additive antitumor effect is the improved delivery of chemotherapy via vessel normalization.28 It is also possible that TGF-β signaling blockade reduces the metastasis to the liver, which may independently improve survival.144 Galunisertib was able to block the renewal of cancer stem cells isolated from pancreatic cancers and was additive in the combination with a Hedgehog inhibitor.145 In addition, blocking the TGF-β signaling in the stroma alone in immune-competent or immune-deficient orthotopic pancreatic models was shown to reduce metastasis, reduce pancreas weight, and inhibit protumoral fibroblast activity.146 Thus, TGF-β inhibition appears to have antitumor activity by improving chemotherapy delivery, blocking metastasis, and inhibiting renewal of cancer-initiating stem cells.
Clinical studies
A blinded, randomized Phase I/II study was initiated to determine whether the activity observed in animals is also observed in patients with advanced or metastatic pancreatic cancer (Table 5). The combination (Phase I) had the expected manageable toxicity of gemcitabine,147 but the final efficacy data are not yet available.
Myelodysplastic syndromes
Nonclinical observation
Myelodysplastic syndrome (MDS) is characterized by bone marrow cytological dysplasia and ineffective hematopoiesis, in which TGF-β (through induction of inflammatory and myelosuppressive cytokines) may play a role.148 The natural inhibitor of the TGF-β signaling pathway, SMAD7, is suppressed in MDS progenitor cells due to increased miR21 expression, which in turn upregulates the TGF-β pathway. This leads to increased expression of myelosuppressive cytokines and decreased burst forming unit-erythroid (BFU-E) and colony forming unit- granulocyte, erythrocyte, monocyte, megakaryocyte (CFU-GEMM). These same cytokines are immunosuppressive and likely contribute to the fatigue seen in MDS. Blockade of the TGF-β pathway by galunisertib in vitro and in vivo murine models resulted in increased erythropoiesis and myeloid cell lineage CFUs, restoring normal hematopoiesis.149 Animals overexpressing TGF-β were treated with galunisertib, and hemoglobin levels improved in these mice. Human CD34+ cells from patients with MDS were also cultured with and without galunisertib; cells cultured with galunisertib demonstrated restoration of normal BFU-E and CFU-GEMM colony formation.
Clinical studies
The preclinical observation together with its favorable toxicity profile (<5% of drug-related Grade 3/4 toxicities) of galunisertib justified the start of a Phase II trial in patients with very low, low, and intermediate risk MDS to evaluate the activity of galunisertib (Table 5).150,103 The study is currently enroling patients.
Leukemia
LY2109761 was evaluated in models of leukemia with the hypothesis that it may block leukemic growth from the microenvironment.151 However, the antileukemic effect was limited, and no additional experiments were conducted. Currently, there are no studies evaluating the interdependency between the leukemic cells and the microenvironment in the bone marrow. However, the importance of the stroma as a source of TGF-β signaling has recently been highlighted. Blocking the TGF-β signaling after chemotherapy accelerated the hematopoietic reconstitution and delayed the return of cycling hematopoietic stem cells to quiescence. This was not observed during homeostasis, suggesting that TGF-β signaling is context dependent.152
Lung cancer
The TGF-β inhibition with LY2109761 reduced metastatic spread of NSCLC cell lines.153 The TGF-β inhibitor LY364947 was also able to overcome the EGFR resistance in cell lines that had no T790M mutation.154 Furthermore, LY2109761 showed antimigratory effects on a murine adenocarcinoma NSCLC cell line.155 Given these early observations, TGF-β signaling may play an important role in drug resistance in NSCLC and hence not be limited to certain lung cancer subtypes, as defined either by EGFR mutations or by histology (such as adenocarcinoma or squamous cell carcinoma).
Colorectal cancer
Using gene expression profiling, a mesenchymal subgroup among patients with colorectal cancer was recently identified.156 This subgroup had poor prognosis and did not benefit from adjuvant chemotherapy (5-FU). Additionally, MED12 loss can result in activation of TGF-β signaling and a mesenchymal phenotype that is also associated with resistance to fluoropyrimidine-based therapy.80 In cell-line experiments, the addition of galunisertib restored chemosensitivity.80 Galunisertib was also used to alter the gene expression of cells from the microenvironment, such as cancer-adjacent fibroblasts.157 From these experiments, a different gene expression profile for colorectal cancer was developed. Overall, it appears that the mesenchymal phenotype is the key characteristic for resistance to standard treatment in colorectal cancer. Whether the aggressiveness of the colorectal cancer is driven by the mesenchymal phenotype of the tumor cells or the microenvironment remains unclear at this time.
Breast cancer
In breast cancer, TGF-β signaling appears to act as an autocrine growth factor.158–160 Thus, TGF-β inhibition may have multiple effects, including the reduction of bone metastasis.88 For example, LY2109761 can minimize osteolytic bone metastasis.161 Because of its inhibition on metastatic spread, TGF-β inhibitors were active across different breast cancer subtypes.162,163 Basal-like breast cancer responds to TGF-β inhibition that prevents metastatic spread in animal models.164 In addition, models with human epidermal growth factor receptor 2 (HER2)/neu expression respond to TGF-β inhibition.165–167 Furthermore, paclitaxel combined with galunisertib has additive antitumor activity in ER/PR/HER2 nonexpressing “triple-negative” cell lines.168 Because triple-negative cell lines also have characteristics of cells undergoing EMT, TGF-β inhibition may be effective in limiting EMT in such cells. Hence, it is possible that the main target of TGF-β inhibitors is the alteration of the tumor cell phenotype rather than direct tumor cytotoxicity. Although these observations support the use of a TGF-β inhibitor in breast cancer, long-term administration of LY2109761 has also been associated with the outgrowth of chemoresistant breast cancer tumors.169 Whether this is an artifact of the model used in this particular study or reflects a possible long-term adverse effect related to continuous dosing remains controversial.
Prostate cancer
In prostate cancer, TGF-β expression in tissue or plasma has been associated with poor survival.87,170–172 LY2109761 has shown antitumor activity in animals by inhibiting bone metastasis in both MDAPCa2b and PC-3.173 At the same time, the bone density was increased in severe combined immunodeficiency mice harboring either tumor cell lines.
Melanoma
There are several mechanisms by which TGF-β may promote tumor growth in melanoma, including immune modulation/inhibition, remodeling, and drug resistance. For example, TGF-β can cause resistance to proto-oncogene B-Raf (BRAF) inhibition, and melanoma cell lines with a BRAF mutation also often have a mesenchymal phenotype.174,175 While pSMAD2 was inhibited in PDX of melanoma, no growth inhibitory effect was observed in the same PDX using a standard tumor colony assay.75 However, no antimigratory or anti-invasive effect of galunisertib was evaluated in these PDX. Recently, an antitumor effect of the combination galunisertib and ipilimumab was reported in a murine melanoma model, while each inhibitor had no antitumor effect in this immune-competent mouse model.176
Radiation and fibrosis
Radiation therapy is associated with both tumor cell killing and normal tissue fibrosis within the radiation field and with associated increases in TGF-β1 in the plasma and tissues.177,178 Also, increases in TGF-β signaling are not only associated with fibrosis or tumor cell killing but also with tumor cell activation inducing tumors to metastasize after radiation.179–181 LY2109761 administration before, during, and after radiation treatment inhibited lung fibrosis in irradiated animals.179 This provides a rationale to investigate the role of galunisertib in radiation-associated fibrosis or other fibrotic diseases.
Conclusion
The preclinical and clinical research on galunisertib, including the treatment of over 300 patients, has taught us some important lessons. First, SMIs of TGF-β can safely be developed for clinical testing, provided there is an adequate understanding of the PK/PD relationship. Most of the toxicities in animal models that were of concern prior to the start of clinical development of galunisertib have not been observed in humans. Second, the biology of the TGF-β inhibition is largely dependent on the microenvironment, perhaps more than originally anticipated. Thus, a focus on direct tumor cell cytotoxicity may be misleading and provide inconclusive observations that will not be helpful to advance clinical development of future TGF-β inhibitors. Consequently, more novel preclinical testing assays are required than those traditionally used in oncology research. Third, it appears that the activity of TGF-β inhibition is dependent on a subtle modulation of the EMT, stem cell function, and immune function. Hence, it will be important to investigate new biomarkers that are related to EMT, stem cell function, and immune responses. For example, it appears that mesenchymal phenotypes contain TGF-β-associated gene expression profiles regardless of the histologic tumor. Whether this applies to all tumors is not clear at this time but it may help with patient selection in future studies. For clinical development, radiographic tumor responses will likely occur only in those patients who have the opportunity to receive longer treatment periods. Thus, patient selection tools, defining who will most likely benefit from TGF-β inhibition, remain one of the most challenging questions to date. Alternatively, combining TGF-β inhibitors with other agents will pose other challenges.
Disclosure
All authors are employees or were employees of Eli Lilly and Company. The authors report no other conflicts of interest in this work.
References
Roberts AB, Lamb LC, Newton DL, Sporn MB, De Larco JE, Todaro GJ. Transforming growth factors: isolation of polypeptides from virally and chemically transformed cells by acid/ethanol extraction. Proc Natl Acad Sci U S A. 1980;77(6):3494–3498. | ||
Piek E, Heldin CH, Ten Dijke P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13(15):2105–2124. | ||
Anzano MA, Roberts AB, Meyers CA, et al. Synergistic interaction of two classes of transforming growth factors from murine sarcoma cells. Cancer Res. 1982;42(11):4776–4778. | ||
Roberts AB, Anzano MA, Lamb LC, Smith JM, Sporn MB. New class of transforming growth factors potentiated by epidermal growth factor: isolation from non-neoplastic tissues. Proc Natl Acad Sci U S A. 1981;78(9):5339–5343. | ||
Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103(2):295–309. | ||
Cheifetz S, Weatherbee JA, Tsang ML, et al. The transforming growth factor-beta system, a complex pattern of cross-reactive ligands and receptors. Cell. 1987;48(3):409–415. | ||
Huang T, David L, Mendoza V, et al. TGF-β signalling is mediated by two autonomously functioning TβRI:TβRII pairs. EMBO J. 2011;30(7):1263–1276. | ||
Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. | ||
Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008;1782(4):197–228. | ||
Yingling, JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3(12):1011–1022. | ||
Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271(5247):350–353. | ||
Grady WM, Myeroff LL, Swinler SE, et al. Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res. 1999;59(2):320–324. | ||
Kim WS, Park C, Jung YS, et al. Reduced transforming growth factor-beta type II receptor (TGF-beta RII) expression in adenocarcinoma of the lung. Anticancer Res. 1999;19(1A):301–306. | ||
Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17(1–2):41–58. | ||
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. | ||
Kehrl JH, Wakefield LM, Roberts AB, et al. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. 1986;163(5):1037–1050. | ||
Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10(8):554–567. | ||
Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18(7):816–827. | ||
Horie M, Saito A, Noguchi S, et al. Differential knockdown of TGF-beta ligands in a three-dimensional co-culture tumor–stromal interaction model of lung cancer. BMC Cancer. 2014;14:580. | ||
Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116(7):1955–1962. | ||
Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009;19(1):116–127. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3(11):807–821. | ||
Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013;13(11):788–799. | ||
Biswas S, Criswell TL, Wang SE, Arteaga CL. Inhibition of transforming growth factor-beta signaling in human cancer: targeting a tumor suppressor network as a therapeutic strategy. Clin Cancer Res. 2006;12(14 Pt 1):4142–4146. | ||
Bharathy S, Xie W, Yingling JM, Reiss M. Cancer-associated transforming growth factor beta type II receptor gene mutant causes activation of bone morphogenic protein-Smads and invasive phenotype. Cancer Res. 2008;68(6):1656–1666. | ||
Hawinkels LJ, Garcia de Vinuesa A, Ten Dijke P. Activin receptor-like kinase 1 as a target for anti-angiogenesis therapy. Expert Opin Investig Drugs. 2013;22(11):1371–1383. | ||
Kano MR, Bae Y, Iwata C, et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci U S A. 2007;104(9):3460–3465. | ||
Liu Z, Kobayashi K, van Dinther M, et al. VEGF and inhibitors of TGFbeta type-I receptor kinase synergistically promote blood-vessel formation by inducing alpha5-integrin expression. J Cell Sci. 2009;122(Pt 18):3294–3302. | ||
Morris JC, Tan AR, Olencki TE, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9(3):e90353. | ||
Lonning S, Mannick J, McPherson JM. Antibody targeting of TGF-beta in cancer patients. Curr Pharm Biotechnol. 2011;12(12):2176–2189. | ||
Trachtman H, Fervenza FC, Gipson DS, et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011;79(11):1236–1243. | ||
Stevenson JP, Kindler HL, Papasavvas E, et al. Immunological effects of the TGFβ-blocking antibody GC1008 in malignant pleural mesothelioma patients. Onco Immunology. 2013;2:e26218. | ||
Cohn A, Lahn M, Williams K, et al. A phase I dose-escalation study to a predefined dose of a transforming growth factor-β1 monoclonal antibody (TβM1) in patients with metastatic cancer. Int J Oncol. 2014;45(6):2221–2231. | ||
Zhong Z, Carroll KD, Policarpio D, et al. Anti-transforming growth factor beta receptor II antibody has therapeutic efficacy against primary tumor growth and metastasis through multieffects on cancer, stroma, and immune cells. Clin Cancer Res. 2010;16(4):1191–1205. | ||
Bogdahn U, Hau P, Stockhammer G, et al. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol. 2011;13(1):132–142. | ||
Isarna-therapeutics.com [homepage on the Internet]. Antisense Pharma Unveils its Corporate Strategy for 2013. Available from: http://www.isarna-therapeutics.com/fileadmin/user_upload/documents/press-releases/2013/20130226_EN_PressRelease_CorporateStrategy_2013.pdf. Accessed March 3, 2015. | ||
Giaccone G, Bazhenova L, Nemunaitis J, et al. A Phase III study of belagenpumatucel-L therapeutic tumor cell vaccine for non-small cell lung cancer (NSCLC). Presented at: Presidential Session I: Best and Late Breaking Abstracts, 17th ECCO – 38th ESMO – 32nd ESTRO European Cancer Congress; 2013; Amsterdam. Abstract E17-7081. | ||
Lahn M, Kloeker S, Berry BS. TGF-beta inhibitors for the treatment of cancer. Expert Opin Investig Drugs. 2005;14(6):629–643. | ||
Seoane J. The TGFBeta pathway as a therapeutic target in cancer. Clin Transl Oncol. 2008;10(1):14–19. | ||
Garber K. Companies waver in efforts to target transforming growth factor beta in cancer. J Natl Cancer Inst. 2009;101(24):1664–1667. | ||
Nagaraj NS, Datta PK. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin Investig Drugs. 2010;19(1):77–91. | ||
Ehata S, Hanyu A, Fujime M, et al. Ki26894, a novel transforming growth factor-beta type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. 2007;98(1):127–133. | ||
Jin CH, Krishnaiah M, Sreenu D, et al. Discovery of N-((4-([1,2,4]Triazolo[1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)-2-fluoroaniline (EW-7197): a highly potent, selective, and orally bioavailable inhibitor of TGF-beta type I receptor kinase as cancer immunotherapeutic/antifibrotic agent. J Med Chem. 2014;57(10):4213–4238. | ||
Son JY, Park SY, Kim SJ, et al. EW-7197, a novel ALK-5 Kinase inhibitor, potently inhibits breast to lung metastasis. Mol Cancer Ther. 2014;13(7):1704–1716. | ||
Wrana JL, Attisano L, Carcamo J, et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71(6):1003–1014. | ||
Wieser R, Wrana JL, J Massague J. GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO J. 1995;14(10):2199–2208. | ||
Leof EB, Proper JA, Goustin AS, Shipley GD, DiCorleto PE, Moses HL. Induction of c-sis mRNA and activity similar to platelet-derived growth factor by transforming growth factor beta: a proposed model for indirect mitogenesis involving autocrine activity. Proc Natl Acad Sci U S A. 1986;83(8):2453–2457. | ||
Sawyer JS, Beight DW, Britt KS, et al. Synthesis and activity of new aryl- and heteroaryl-substituted 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. Bioorg Med Chem Lett. 2004;14(13):3581–3584. | ||
Sawyer JS, Anderson BD, Beight DW, et al. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J Med Chem. 2003;46(19):3953–3956. | ||
Li HY, McMillen WT, Heap CR, et al. Optimization of a dihydropyrrolopyrazole series of transforming growth factor-beta type I receptor kinase domain inhibitors: discovery of an orally bioavailable transforming growth factor-beta receptor type I inhibitor as antitumor agent. J Med Chem. 2008;51(7):2302–2306. | ||
Yingling JM, Yan L, Lu K, et al. Characterization or Substituted Pyrazoles as ATP-Competitive TGF-beta Receptor Kinase Inhibitors: the TGF-beta superfamily. La Jolla (CA): American Association for Cancer Research; 2003. | ||
Peng SB, Yan L, Xia X, et al. Kinetic characterization of novel pyrazole TGF-beta receptor I kinase inhibitors and their blockade of the epithelial-mesenchymal transition. Biochemistry. 2005;44(7):2293–2304. | ||
Muraoka-Cook RS, Shin I, Yi JY, et al. Activated type I TGFbeta receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene. 2006;25(24):3408–3423. | ||
Li HY, Wang Y, Yan L, et al. Novel and potent transforming growth factor beta type I receptor kinase domain inhibitor: 7-amino 4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)-quinolines. Bioorg Med Chem Lett. 2004;14(13):3585–3588. | ||
Lahn M. Transforming growth factor beta (TGF-beta) inhibitors and their development in the treatment of cancer. In: 3rd Cancer Drug Research and Development GTCbio, 2006, San Francisco, CA. | ||
Stauber AJ, Credille KM, Truex LL, Ehlhardt WJ, Young JK. Nonclinical safety evaluation of a transforming growth factor β receptor I kinase inhibitor in fischer 344 rats and beagle dogs. J Clin Pract. 2014;4:196. | ||
Mattern SP, editor. The Prince of Medicine: Galen in the Roman Empire. Oxford: Oxford University Press; 2013. | ||
MedPacto, Inc. First-in-human dose-escalation study of TEW-7197 monotherapy in subjects with advanced stage solid tumors. Available from: https://clinicaltrials.gov/ct2/show/NCT02160106?term=nct02160106&rank=1. NLM identifier: NCT02160106. Accessed March 3, 2015. | ||
Anderton MJ, Mellor HR, Bell A, et al. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol. 2011;39(6):916–924. | ||
Singh J, Ling LE, Sawyer JS, Lee WC, Zhang F, Yingling JM. Transforming the TGFbeta pathway: convergence of distinct lead generation strategies on a novel kinase pharmacophore for TbetaRI (ALK5). Curr Opin Drug Discov Devel. 2004;7(4):437–445. | ||
Stauber AJ, Zimmermann JL, Berridge BR. Pathobiology of a valvulopathy in fischer 344 rats given a transforming growth factor-b RI kinase inhibitor. In: SOT 45th Annual Meeting and Tox Expo™; 2006. San Diego (CA): Oxford University Press. Abstract 290. | ||
Ueba H, Kawakami M, Yaginuma T. Shear stress as an inhibitor of vascular smooth muscle cell proliferation. Role of transforming growth factor-beta 1 and tissue-type plasminogen activator. Arterioscler Thromb Vasc Biol. 1997;17(8):1512–1516. | ||
Walshe TE, dela Paz NG, D’Amore PA. The role of shear-induced transforming growth factor-beta signaling in the endothelium. Arterioscler Thromb Vasc Biol. 2013;33(11):2608–2617. | ||
Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355(8):788–798. | ||
Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44(8):922–927. | ||
Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332(6027):358–361. | ||
Vilar JM, Jansen R, Sander C. Signal processing in the TGF-beta superfamily ligand-receptor network. PLoS Comput Biol. 2006;2(1):e3. | ||
Vizan P, Miller DS, Gori I, Das D, Schmierer B, Hill CS. Controlling long-term signaling: receptor dynamics determine attenuation and refractory behavior of the TGF-beta pathway. Sci Signal. 2013;6(305):ra106. | ||
Feagins LA. Role of transforming growth factor-beta in inflammatory bowel disease and colitis-associated colon cancer. Inflamm Bowel Dis. 2010;16(11):1963–1968. | ||
Hong S, Lee HJ, Kim SJ, Hahm KB. Connection between inflammation and carcinogenesis in gastrointestinal tract: focus on TGF-beta signaling. World J Gastroenterol. 2010;16(17):2080–2093. | ||
Yingling J. Targeting the TGF-β RI kinase with LY2157299: a PK/PD-driven drug discovery and clinical development program. Presented at: AACR Meeting; May 1; 2005. Abstract 1463a. | ||
Bueno L, de Alwis DP, Pitou C, et al. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type I receptor TGF-beta kinase antagonist, in mice. Eur J Cancer. 2008;44(1):142–150. | ||
Yingling JM, Shou J, Xia X, et al. A small molecule inhibitor of TGFβ RI kinase potentiates VEGF dependent angiogenesis in vitro. Proc Am Assoc Cancer Res. 2006. Abstract 250. | ||
Maier A, Peille AL, Vuaroqueaux V, Lahn M. Anti-tumor activity of the TGF-beta receptor kinase inhibitor galunisertib (LY2157299 monohydrate) in patient-derived tumor xenografts. Cell Oncol (Dordr). Epub 2015 Jan 9. | ||
Akhurst RJ, Derynck R. TGF-beta signaling in cancer–a double-edged sword. Trends Cell Biol. 2001;11(11):S44–S51. | ||
Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29(2):117–129. | ||
Yang YA, Dukhanina O, Tang B, et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002;109(12):1607–1615. | ||
Li CY, Wood DK, Huang JH, Bhatia SN. Flow-based pipeline for systematic modulation and analysis of 3D tumor microenvironments. Lab Chip. 2013;13(10):1969–1978. | ||
Huang S, Holzel M, Knijnenburg T, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell. 2012;151(5):937–950. | ||
Bhola NE, Balko JM, Dugger TC, et al. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123(3):1348–1358. | ||
Simeoni M, Magni P, Cammia C, et al. Predictive pharmacokinetic-pharmacodynamic modeling of tumor growth kinetics in xenograft models after administration of anticancer agents. Cancer Res. 2004;64(3):1094–1101. | ||
Rocchetti M, Poggesi I, Germani M, et al. A pharmacokinetic-pharmacodynamic model for predicting tumour growth inhibition in mice: a useful tool in oncology drug development. Basic Clin Pharmacol Toxicol. 2005;96(3):265–268. | ||
Bhattachar SN, Perkins EJ, Tan JS, Burns LJ. Effect of gastric pH on the pharmacokinetics of a BCS class II compound in dogs: utilization of an artificial stomach and duodenum dissolution model and GastroPlus, simulations to predict absorption. J Pharm Sci. 2011;100(11):4756–4765. | ||
Gueorguieva I, Cleverly AL, Stauber A, et al. Defining a therapeutic window for the novel TGF-β inhibitor LY2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br J Clin Pharmacol. 2014;77(5):796–807. | ||
Lahn M, Kunzmann R, Kohler G, et al. Comparison of cytogenetics, cytokine secretion, and oncogene expression in primary cultures of renal carcinoma cells. Oncology. 1997;54(5):429–437. | ||
Shariat SF, Shalev M, Menesses-Diaz A, et al. Preoperative plasma levels of transforming growth factor beta(1) (TGF-beta(1)) strongly predict progression in patients undergoing radical prostatectomy. J Clin Oncol. 2001;19(11):2856–2864. | ||
Bandyopadhyay A, Agyin JK, Wang L, et al. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006;66(13):6714–6721. | ||
Farrington DL, Yingling JM, Fill JA, et al. Development and validation of a phosphorylated SMAD ex vivo stimulation assay. Biomarkers. 2007;12(3):313–330. | ||
Baselga J, Rothenberg ML, Tabernero J, et al. TGF-beta signalling-related markers in cancer patients with bone metastasis. Biomarkers. 2008;13(2):217–236. | ||
O’Brien PJ, Ramanathan R, Yingling JM, et al. Analysis and variability of TGFbeta measurements in cancer patients with skeletal metastases. Biologics. 2008;2(3):563–569. | ||
Classen S, Muth C, Debey-Pascher S, et al. Application of T cell-based transcriptomics to identify three candidate biomarkers for monitoring anti-TGFbetaR therapy. Pharmacogenet Genomics. 2010;20(3):147–156. | ||
Kadam S, Cleverly AL, Farmen M, Grondin J, Cox YI, Lahn M. A canonical transforming growth factor beta-dependent signaling pathway is present in peripheral blood cells of cancer patients with skeletal metastasis. J Mol Biomark Diagn. 2013;4(3):153–161. | ||
Kanjilal V, Yingling JM, Yan L, et al. Integrative analysis of genomic RNA expression data from in vitro and in vivo models of TGF-beta receptor kinase inhibitors to identify novel informative biomarkers of TGF-beta signaling. Cancer Res. 2006;(1):204. | ||
Duan K, Kanjilal V, Yan L, Yingling J, Patel K, Shou J. Genomic analysis of TGFβ treated cell lines with phenotypically distinct outcomes to identify transcriptional signatures for assessment in stratification of cancers. Presented at: First AACR Centennial Conference on Translational Cancer Medicine; Nov 4–8; 2007; Singapore. Abstract B57. | ||
Calvo-Aller E, Baselga J, Glatt S, et al. First human dose escalation study in patients with metastatic malignancies to determine safety and pharmacokinetics of LY2157299, a small molecule inhibitor of the transforming growth factor-beta receptor I kinase. Poster presented at: ASCO Annual Meeting; 2008; Chicago, IL. J Clin Oncol. Abstract 14554. | ||
Rodon Ahnert J, Baselga J, Calvo E, et al. First human dose (FHD) study of the oral transforming growth factor-beta receptor I kinase inhibitor LY2157299 in patients with treatment-refractory malignant glioma. Presented at: ASCO Annual Meeting; 2011; Chicago, IL. J Clin Oncol. Abstract 3011. | ||
Azaro A, Baselga J, Sepúlveda JM, et al. The oral transforming growth factor-beta (TGF-β) receptor I kinase inhibitor LY2157299 plus lomustine in patients with treatment-refractory malignant glioma: the first human dose study. Poster presented at: ASCO Annual Meeting; 2012; Chicago, IL. J Clin Oncol. Abstract Number 2042. | ||
Rodon J, Carducci MA, Sepúlveda JM, et al. Integrated data review of the first-in-human dose (FHD) study evaluating safety, pharmacokinetics (PK), and pharmacodynamics (PD) of the oral transforming growth factor-beta (TGF-β) receptor I kinase inhibitor, LY2157299 monohydrate (LY). Presented at: ASCO Annual Meeting; 2013; Chicago, IL. Abstract 2016. | ||
Kovacs RJ, Maldonado G, Azaro A, et al. Cardiac safety of TGF-beta receptor I kinase inhibitor LY2157299 monohydrate in cancer patients in a first-in-human dose study. Cardiovasc Toxicol. Epub 2014 Dec 9; DOI: 10.1007/s12012-014-9297-4. | ||
Azaro A, Rodon J, Carducci M, et al. Case series of cancer patients treated with galunisertib, a transforming growth factor-beta receptor I kinase inhibitor in a first-in-human dose study. J Med Cases. 2014;5(11):603–609. | ||
Rodon J, Carducci M, Sepulveda-Sanchez JM, et al. Pharmacokinetic, pharmacodynamic and biomarker evaluation of transforming growth factor-beta receptor I kinase inhibitor, galunisertib, in phase 1 study in patients with advanced cancer. Invest New Drugs. 2015;33(2):357–370. | ||
Rodon J, Carducci MA, Sepulveda-Sanchez JM, et al. First-in-human dose study of the novel transforming growth factor-beta receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res. 2015;21(3):553–560. | ||
Sepulveda-Sanchez J, Ramos A, Hilario A, et al. Assessment of changes in brain perfusion and permeability in patients with advanced, refractory glioblastoma treated with the combination of lomustine and the oral transforming growth factor-beta (TGF-β) receptor I kinase inhibitor LY2157299 monohydrate. Oncology Lett. 2015;9(6):2442–2448. | ||
Grassian AR, Lin F, Barrett R, et al. Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/microRNA (miR)-200-dependent epithelial-mesenchymal transition (EMT). J Biol Chem. 2012;287(50):42180–42194. | ||
Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. | ||
Anido J, Saez-Borderias A, Gonzalez-Junca A, et al. TGF-beta receptor inhibitors target the CD44 (high)/Id1 (high) glioma-initiating cell population in human glioblastoma. Cancer Cell. 2010;18(6):655–668. | ||
Bruna A, Darken RS, Rojo F, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11(2):147–160. | ||
Holzer TR, Gustavson M, Pinard R, et al. Development and analytical validation of a quantitative tissue-based assay for phospho-Smad2 (Ser465/467) in glioblastoma multiforme: a potential biomarker of TGF-β pathway activation. Presented at: 100th Annual Meeting; 2011; San Antonio, TX. | ||
Kuczynski EA, Patten SG, Coomber BL. VEGFR2 expression and TGF-beta signaling in initial and recurrent high-grade human glioma. Oncology. 2011;81(2):126–134. | ||
Loh JK, Lieu AS, Su YF, et al. Plasma levels of transforming growth factor-beta 1 before and after removal of low- and high-grade astrocytomas. Cytokine. 2013;61(2):413–418. | ||
Penuelas S, Anido J, Prieto-Sanchez RM, et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009;15(4):315–327. | ||
Hardee ME, Marciscano AE, Medina-Ramirez CM, et al. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res. 2012;72(16):4119–4129. | ||
Zhang M, Herion TW, Timke C, et al. Trimodal glioblastoma treatment consisting of concurrent radiotherapy, temozolomide, and the novel TGF-beta receptor I kinase inhibitor LY2109761. Neoplasia. 2011;13(6):537–549. | ||
Zhang M, Kleber S, Rohrich M, et al. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011;71(23):7155–7167. | ||
Parsons S, Sawyer S, Yan L, et al. The combination of the small molecule TGFβR1 inhibitor, LY2157299 monohydrate, with CCNU substantially blocks SMAD phosphorylation and significantly suppresses human glioblastoma xenograft growth. Presented at: Proceedings of the 2011 AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; November 12–16; 2011; San Francisco, CA. Mol Cancer Ther. Abstract C201. | ||
US Food and Drug Administration. Search Orphan Drug Designations and Approvals. Silver Spring (MD): US Food and Drug Administration; 2013 [updated April 1, 2013]. Available from: http://www.accessdata.fda.gov/scripts/opdlisting/oopd/OOPD_Results_2.cfm?Index_Number=378712. Accessed March 3, 2015. | ||
Eli Lilly and Company. A phase 2 study of LY2157299 monohydrate monotherapy or LY2157299 monohydrate plus lomustine therapy compared to lomustine monotherapy in patients with recurrent glioblastoma; 2012 [updated April 19, 2012]. Available from: https://clinicaltrials.gov/ct2/show/NCT01582269?term=NCT01582269&rank=1. NLM identifier: NCT01582269. Accessed March 3, 2015. | ||
Eli Lilly and Company. Phase 1b/2a study combining LY2157299 with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Available from: https://clinicaltrials.gov/ct2/show/NCT01220271?term=NCT01220271&rank=1. NLM identifier: NCT01220271. Accessed March 3, 2015. | ||
Carpentier AF, Brandes AA, Kesari S, et al. Safety interim data from a three-arm phase II study evaluating safety and pharmacokinetics of the oral transforming growth factor-beta (TGF-β) receptor I kinase inhibitor LY2157299 monohydrate in patients with glioblastoma at first progression. In: Annual Meeting of the American Society of Clinical Oncology; 2013; Chicago, IL. Abstract Number 2061. | ||
Wick W, Suarez C, Rodon J, et al. Phase 1b/2a study evaluating safety, pharmacokinetics (PK) and preliminary pharmacodynamics responses of the oral transforming growth factor-beta (TGF-β) receptor I kinase inhibitor LY2157299 monohydrate (LY) when combined with chemoradiotherapy in newly diagnosed malignant gliomas. Poster presented at: 18th Annual SNO Scientific Meeting and Education Day; November 22; 2013;2013; Society of Neurooncology: San Francisco, CA. | ||
Abou-Shady M, Baer HU, Friess H, et al. Transforming growth factor betas and their signaling receptors in human hepatocellular carcinoma. Am J Surg. 1999;177(3):209–215. | ||
Matsuzaki K, Date M, Furukawa F, et al. Regulatory mechanisms for transforming growth factor beta as an autocrine inhibitor in human hepatocellular carcinoma: implications for roles of smads in its growth. Hepatology. 2000;32(2):218–227. | ||
Sacco R, Leuci D, Tortorella C, et al. Transforming growth factor beta1 and soluble Fas serum levels in hepatocellular carcinoma. Cytokine. 2000;12(6):811–814. | ||
Giannelli G, Bergamini C, Fransvea E, Sgarra C, Antonaci S. Laminin-5 with transforming growth factor-beta1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005;129(5):1375–1383. | ||
Giannelli G, Villa E, Lahn M. Transforming growth factor-beta as a therapeutic target in hepatocellular carcinoma. Cancer Res. 2014;74(7):1890–1894. | ||
Fransvea E, Angelotti U, Antonaci S, Giannelli G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology. 2008;47(5):1557–1566. | ||
Fransvea E, Mazzocca A, Antonaci S, Giannelli G. Targeting transforming growth factor (TGF)-betaRI inhibits activation of beta1 integrin and blocks vascular invasion in hepatocellular carcinoma. Hepatology. 2009;49(3):839–850. | ||
Mazzocca A, Fransvea E, Lavezzari G, Antonaci S, Giannelli G. Inhibition of transforming growth factor beta receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology. 2009;50(4):1140–1151. | ||
Garbay D, Serova M, Serrate C, et al. Specific TGF-beta receptor-I inhibition using LY364947 impairs signaling, motility and invasion in parental and multikinase inhibitor-resistant hepatocarcinoma cells. Presented at: 22nd EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics; 2010; Berlin, Germany. | ||
Mazzocca A, Fransvea E, Dituri F, Lupo L, Antonaci S, Giannelli G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology. 2010;51(2):523–534. | ||
Fransvea E, Mazzocca A, Santamato A, Azzariti A, Antonaci S, Giannelli G. Kinase activation profile associated with TGF-beta-dependent migration of HCC cells: a preclinical study. Cancer Chemother Pharmacol. 2011;68(1):79–86. | ||
Giannelli G, Mazzocca A, Fransvea E, Lahn M, Antonaci S. Inhibiting TGF-beta signaling in hepatocellular carcinoma. Biochim Biophys Acta. 2011;1815(2):214–223. | ||
Serova M, Tijeras-Raballand A, Dos Santos C, et al. Effects of TGF-beta signaling inhibition with LY2157299 in hepatocarcinoma models and in ex vivo whole tumor tissue samples from patient specimen. Presented at: 104th annual meeting of the American Association for Cancer Research; April 6–10; 2013; Washington, DC, Philadelphia (PA). Abstract 2094. | ||
Tijeras-Raballand A, Serova M, Neuzillet C, et al. LY2157299 w/wo sorafenib in hepatocarcinomas yields antitumor effects in transgenic mice and xenografts, also inhibiting Tgf-Β signalling in freshly-grown sliced tumor specimens. Presented at: International Liver Cancer Association (ILCA) Annual Conference, 2014; Kyoto; Japan. Abstract P-005. | ||
Wang Q, Yu T, Yuan Y, et al. Sorafenib reduces hepatic infiltrated regulatory T cells in hepatocellular carcinoma patients by suppressing TGF-beta signal. J Surg Oncol. 2013;107(4):422–427. | ||
Serova M, Garbay D, Riveiro ME, et al. Targeting of TGF-beta signaling results in decreased motility and invasion in parental and mulitkinase inhibitor-insensitive hepatocacinoma cells. Presented at: International Liver Cancer Association (ILCA) Annual Conference; 2011; Brussels, Belgium. | ||
Serova M, Riveiro ME, de Gramont A, et al. TGF-β-RI kinase inhibitors, LY2157299 and LY364947, decrease motility and invasion in hepatocarcinoma (HCC) cells developing resistance to VEGFR/PDGFR kinase inhibitors. Presented at: AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; November 12–16; 2011; San Francisco, CA. Mol Cancer Ther. Abstract Number B175. | ||
Eli Lilly and Company. Phase 2 study of LY2157299 in patients with hepatocellular carcinoma. Available from: https://clinicaltrials.gov/ct2/show/NCT01246986?term=NCT01246986&rank=1. NLM identifier: NCT01246986. Accessed March 3, 2015. | ||
Giannelli G, Santoro A, Kelley RK, et al. Phase 2 study of the oral transforming growth factor-beta (TGF-beta) receptor I kinase inhibitor LY2157299. Presented at: 7th ILCA (International Liver Cancer Association) Annual Conference; 2013; Washington, DC, USA. | ||
Faivre S, Santoro A, Kelley RK, et al. A Phase 2 study of a novel transforming growth factor-beta (TGF-β) receptor I kinase inhibitor, LY2157299 monohydrate, in patients with advanced hepatocellular carcinoma (HCC). Presented at: Gastrointestinal Cancers Symposium; 2014; San Francisco, CA. | ||
Truty MJ, Urrutia R. Basics of TGF-beta and pancreatic cancer. Pancreatology. 2007;7(5–6):423–435. | ||
Melisi D, Ishiyama S, Sclabas GM, et al. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther. 2008;7(4):829–840. | ||
Li Z, Chang Z, Chiao LJ, et al. TrkBT1 induces liver metastasis of pancreatic cancer cells by sequestering Rho GDP dissociation inhibitor and promoting RhoA activation. Cancer Res. 2009;69(19):7851–7859. | ||
Lonardo E, Hermann PC, Mueller MT, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 2011;9(5):433–446. | ||
Ostapoff KT, Cenik BK, Wang M, et al. Neutralizing murine TGFbetaR2 promotes a differentiated tumor cell phenotype and inhibits pancreatic cancer metastasis. Cancer Res. 2014;74(18):4996–5007. | ||
Kozloff M, Carbonero R, Nadal T, et al. Phase Ib study evaluating safety and pharmacokinetics (PK) of the oral transforming growth factor-beta (TGF-β) receptor I kinase inhibitor LY2157299 monohydrate (LY) when combined with gemcitabine in patients with advanced cancer. Presented at: ASCO Annual Meeting; 2013; Chicago, IL. Abstract 2563. | ||
Zhou L, Nguyen AN, Sohal D, et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood. 2008;112(8):3434–3443. | ||
Zhou L, McMahon C, Bhagat T, et al. Reduced SMAD7 leads to overactivation of TGF-beta signaling in MDS that can be reversed by a specific inhibitor of TGF-beta receptor I kinase. Cancer Res. 2011;71(3):955–963. | ||
Eli Lilly and Company. Phase 2/3 study of monotherapy LY2157299 monohydrate in very low-, low-, and intermediate-risk patients with myelodysplastic syndromes. Available from: https://clinicaltrials.gov/ct2/show/NCT02008318?term=NCT02008318&rank=1. NLM identifier: NCT02008318. Accessed March 3, 2015. | ||
Xu Y, Tabe Y, Jin L, Watt J, et al. TGF-beta receptor kinase inhibitor LY2109761 reverses the anti-apoptotic effects of TGF-beta1 in myelo-monocytic leukaemic cells co-cultured with stromal cells. Br J Haematol. 2008;142(2):192–201. | ||
Brenet F, Kermani P, Spektor R, Rafii S, Scandura JM. TGFbeta restores hematopoietic homeostasis after myelosuppressive chemotherapy. J Exp Med. 2013;210(3):623–639. | ||
Ge R, Kang Y, McPherson J, Yingling J, Reiss M. Transforming growth factor β pathway antagonists inhibit human breast cancer metastases to lung and bone. Presented at: AACR Meeting; 2007; Los Angeles, CA. Abstract 2231. | ||
Serizawa M, Takahashi T, Yamamoto N, Koh Y. Combined treatment with erlotinib and a transforming growth factor-beta type I receptor inhibitor effectively suppresses the enhanced motility of erlotinib-resistant non-small-cell lung cancer cells. J Thorac Oncol. 2013;8(3):259–269. | ||
Vazquez PF, Carlini MJ, Daroqui MC, et al. TGF-beta specifically enhances the metastatic attributes of murine lung adenocarcinoma: implications for human non-small cell lung cancer. Clin Exp Metastasis. 2013;30(8):993–1007. | ||
Roepman P, Schlicker A, Tabernero J, et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int J Cancer. 2014;134(3):552–562. | ||
Calon A, Espinet E, Palomo-Ponce S, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22(5):571–584. | ||
Arteaga CL, Tandon AK, Von Hoff DD, Osborne CK. Transforming growth factor beta: potential autocrine growth inhibitor of estrogen receptor-negative human breast cancer cells. Cancer Res. 1988;48(14):3898–3904. | ||
Arteaga CL, Hurd SD, Winnier AR, Johnson MD, Fendly BM, Forbes JT. Anti-transforming growth factor (TGF)-beta antibodies inhibit breast cancer cell tumorigenicity and increase mouse spleen natural killer cell activity. Implications for a possible role of tumor cell/host TGF-beta interactions in human breast cancer progression. J Clin Invest. 1993;92(6):2569–2576. | ||
Reiss M, Barcellos-Hoff MH. Transforming growth factor-beta in breast cancer: a working hypothesis. Breast Cancer Res Treat. 1997;45(1):81–95. | ||
Korpal M, Yan J, Lu X, Xu S, Lerit DA, Kang Y. Imaging transforming growth factor-beta signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med. 2009;15(8):960–966. | ||
Lacher MD, Tiirikainen MI, Saunier EF, et al. Transforming growth factor-beta receptor inhibition enhances adenoviral infectability of carcinoma cells via up-regulation of coxsackie and adenovirus receptor in conjunction with reversal of epithelial-mesenchymal transition. Cancer Res. 2006;66(3):1648–1657. | ||
Tan AR, Alexe G, Reiss M. Transforming growth factor-beta signaling: emerging stem cell target in metastatic breast cancer? Breast Cancer Res Treat. 2009;115(3):453–495. | ||
Ganapathy V, Ge R, Grazioli A, et al. Targeting the transforming growth factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol Cancer. 2010;9:122. | ||
Wang E, Xiang B, Olivares M, Chung C, Arteaga C. Transforming growth factor beta engages TACE/ADAM17 and ErbB3 to activate PI3K/Akt in HER2-overexpressing breast cancer and desensitizes cells to trastuzumab. Presented at: CTRC-AACR San Antonio Breast Cancer Symposium; San Antonio, CA. Cancer Res. 2008 | ||
Wang SE, Yu Y, Criswell TL, et al. Oncogenic mutations regulate tumor microenvironment through induction of growth factors and angiogenic mediators. Oncogene. 2010;29(23):3335–3348. | ||
Wang K, Feng H, Ren W, et al. BMP9 inhibits the proliferation and invasiveness of breast cancer cells MDA-MB-231. J Cancer Res Clin Oncol. 2011;137(11):1687–1696. | ||
Bhola NE, Balko J, Arteaga C. Inhibition of the TGFβ pathway inhibits chemotherapy-induced enrichment of breast cancer stem cells in triple-negative breast cancer. Poster presented at: AACR Annual Meeting; 2012; Chicago, IL. Abstract Number 3475. | ||
Connolly EC, Saunier EF, Quigley D, et al. Outgrowth of drug-resistant carcinomas expressing markers of tumor aggression after long-term TβRI/II kinase inhibition with LY2109761. Cancer Res. 2011;71(6):2339–2349. | ||
Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, Slawin KM. Plasma levels of interleukin-6 and its soluble receptor are associated with prostate cancer progression and metastasis. Urology. 2001;58(6):1008–1015. | ||
Shariat SF, Kattan MW, Traxel E, et al. Association of pre- and postoperative plasma levels of transforming growth factor beta(1) and interleukin 6 and its soluble receptor with prostate cancer progression. Clin Cancer Res. 2004;10(6):1992–1999. | ||
Shariat SF, Semjonow A, Lilja H, Savage C, Vickers AJ, Bjartell A. Tumor markers in prostate cancer I: blood-based markers. Acta Oncol. 2011;50 Suppl 1:61–75. | ||
Wan X, Li ZG, Yingling JM, Yang J, et al. Effect of transforming growth factor beta (TGF-beta) receptor I kinase inhibitor on prostate cancer bone growth. Bone. 2012;50(3):695–703. | ||
Cebon JS, Anaka M, Hudson C, et al. Role of cytokine-induced plasticity in melanoma on invasive, therapy-resistant cells that express EMT-related genes and pathways. Poster presented at ASCO Annual Meeting; 2012; Chicago. IL. J Clin Oncol. Abstract 8542. | ||
Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508(7494):118–122. | ||
Hanks BA, Holtzhausen A, Evans K, Heid M, Blobe GC. Combinatorial TGF-beta signaling blockade and anti-CTLA4 antibody immunotherapy in a murine BRAFV600E-PTEN−/− transgenic model of melanoma. Presented at: ASCO Annual Meeting; 2014; Chicago, IL. J Clin Oncol. Abstract 3011. | ||
DeLong P, Carroll RG, Henry AC, et al. Regulatory T cells and cytokines in malignant pleural effusions secondary to mesothelioma and carcinoma. Cancer Biol Ther. 2005;4(3):342–346. | ||
Biswas S, Guix M, Rinehart C, et al. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117(5):1305–1313. | ||
Anscher MS, Kong FM, Andrews K, et al. Plasma transforming growth factor beta1 as a predictor of radiation pneumonitis. Int J Radiat Oncol Biol Phys. 1998;41(5):1029–1035. | ||
Laping N, Huet S. TGF-β receptor kinase inhibitors for the treatment of fibrosis. In: Dijke P, Heldin C-H, editors. Smad Signal Transduction: Smads in Proliferation, Differentiation and Disease. the Netherlands: Springer Verlag; 2006:443–459. | ||
Flechsig P, Dadrich M, Bickelhaupt S, et al. LY2109761 attenuates radiation-induced pulmonary murine fibrosis via reversal of TGF-beta and BMP-associated proinflammatory and proangiogenic signals. Clin Cancer Res. 2012;18(13):3616–3627. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.