")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Cell-penetrating peptide conjugates of gambogic acid enhance the antitumor effect on human bladder cancer EJ cells through ROS-mediated apoptosis
Authors Lyu L , Huang LQ, Huang T, Xiang W, Yuan JD, Zhang CH
Received 7 January 2018
Accepted for publication 17 February 2018
Published 5 April 2018 Volume 2018:12 Pages 743—756
DOI https://doi.org/10.2147/DDDT.S161821
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Lei Lyu,1,* Lu-qi Huang,2,* Tao Huang,1 Wei Xiang,1 Jing-dong Yuan,1 Chuan-hua Zhang1
1Department of Urology, Wuhan No 1 Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 2Department of Neurology, Wuhan No 1 Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Background: Gambogic acid (GA) is the main active ingredient of resin gamboges and possesses anti-cancer activity toward various human cancer cells. However, clinical application of GA has been limited by its poor aqueous solubility and dose-limiting toxicities. Cell-penetrating peptides (CPPs) are widely used to deliver anti-cancer drugs into cancer cells and to enhance the water solubility of drugs.
Purpose: The object of this study was to synthesize peptide-drug conjugates in which the cell-penetrating peptide TAT (trans-activator of transcription) was conjugated to GA and evaluated the anti-cancer activity of this GA-CPP conjugate (GA-TAT) in EJ bladder cancer cells.
Methods: GA is built onto the TAT, and the GA-TAT conjugates are cleaved from the solid support and purified via HPLC. The equilibrium solubility of GA-TAT was measured using the shake-flask method. The effects of GA-TAT on EJ cell viability and proliferation were determined by MTT assay, Edu assay and colony formation assay, respectively. After treated with 1.0 μM GA-TAT for 24 h, the apoptosis rate of EJ cells were detected by Acridine orange/ethidium bromide (AO/EB) assay and flow cytometry assay. The proteins of caspase-3 (processing), caspase-9 (processing), Bcl-2 and Bax were analyzed by Western blotting, and the intracellular reactive oxygen species (ROS) production was evaluated by a reactive oxygen species assay.
Results: In contrast to free GA, the solubility of GA-TAT in water was significantly improved. Meanwhile, GA-TAT significantly increased EJ cellular uptake, toxicity and apoptosis. Mechanistic analysis revealed that GA-TAT enhanced the anti-cancer effect of GA against EJ cells through ROS-mediated apoptosis. The results were demonstrated that GA-TAT increased the ROS level in EJ cells, and N-acetyl-l-cysteine (NAC; a well-known ROS scavenger) inhibited GA-TAT-induced ROS generation and apoptosis. Additionally, GA-TAT activated caspase-3 and caspase-9 and down-regulated the Bcl-2/Bax ratio, but these effects were largely rescued by NAC.
Conclusion: GA-TAT has outstanding potential for promoting tumor apoptosis and exhibits promise for use in bladder cancer therapy.
Keywords: gambogic acid, cell-penetrating peptides, apoptosis, bladder cancer
Introduction
Bladder cancer is the most common type of malignant tumor in the urinary system. Bladder cancer – with an increasing incidence and mortality – is the leading cause of death in the elderly populations of China.1 Currently, surgery combined with postoperative chemotherapy is the most important treatment for bladder cancer.2 Because bladder cancer has unique characteristics, such as a multi-centric tumor origin, drug resistance, and a tendency to recur, the postoperative recurrence rate in patients with primary bladder cancer is still as high as 40%. Moreover, the degree of tumor malignancy increases significantly with tumor recurrence, which may develop into a metastatic tumor.3 Although chemotherapy is an effective method for improving the survival rate of postoperative patients and those with advanced bladder cancer, the clinical application of chemotherapy is limited by drug resistance and the side effects of anticancer drugs.4 Therefore, identification and development of new anticancer drugs for bladder cancer would significantly improve treatment efficacy.
Natural plant ingredients have always been a major source of chemotherapy drugs, and may reduce the adverse effects of tumor therapies.5 Gambogic acid (GA) is a compound extracted from natural resin gamboges and exhibits anti-proliferative and pro-apoptotic effects on some tumors, such as breast, prostate, and lung cancer cells.6–8 Moreover, selective apoptosis induction by GA in cancer cells, compared with normal cells, suggests that GA might be an effective anticancer drug candidate with low toxicity to normal cells.9 Although GA has good anti-neoplastic activity, the clinical application of this compound has been limited because of its poor cell-penetrating ability, which is a result of its hydrophobicity.10
Cell-penetrating peptides (CPPs) are gaining interest in anticancer drug research because they can be used as very efficient tools for delivering drugs into cancer cells.11 Some antitumor drugs, such as doxorubicin and gemcitabine, have been conjugated to CPPs, thereby enhancing the drug’s anticancer effect.12,13 Most CPPs possess cationic and amphipathic characteristics; conjugating CPPs to drugs not only enhances drug transport into cancer cells but also increases drug solubility in water. The trans-activator of transcription (TAT; CYGRKKRRQRRR) – a part of the HIV-1 virus regulator protein and the first CPP identified – has been widely used as a tool for intracellular delivery of macromolecules.14 Previously, GA was reported to inhibit proliferation and promote apoptosis in bladder cancer cells; however, the anticancer effect of GA-CPP conjugates (GA-TAT) remains unclear.15
In this study, a new GA-TAT conjugate was synthesized for the efficient transduction of GA into bladder cancer EJ cells. GA-TAT conjugates were evaluated for cell toxicity, proliferation inhibition, cell apoptosis induction, and anticancer efficacy, and these parameters were compared with those of GA-treated EJ cells. Furthermore, we explored the potential mechanisms underlying the antitumor effects of GA-TAT in bladder cancer.
Materials and methods
Cell culture
The human bladder cancer EJ cell line was obtained from the Institute of Biochemistry and Cell Biology of Chinese Academy of Sciences (Shanghai, People’s Republic of China). The human immortalized uroepithelium cell line SV-HUC-1 was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). EJ cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin. SV-HUC-1 cells were cultured in F-12K medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained at 37°C in a humidified incubator with 5% CO2.
Design and synthesis of GA-TAT conjugates
GA (Sigma, St Louis, MO, USA) was prepared as a stock solution at a concentration of 25 mmol/L in anhydrous dimethyl sulfoxide (DMSO; Sigma), aliquoted, and then stored at −20°C. GA-TAT conjugates were synthesized by the Qiangyao Biological Technology Company (Shanghai, People’s Republic of China). Briefly, the CPP-TAT (CYGRKKRRQRRR) was mechanically assembled onto the resin, and the GA component was coupled to the N-terminal of the peptides (Figure 1A and B). The GA-TAT constructs were cleaved from the solid phase. The crude GA-TAT conjugate peptide was purified by high-performance liquid chromatography (HPLC), and the purified compounds were analyzed (Figure 1C and D) by HPLC and mass spectrometry (MS). The purity of these peptides was >95%.
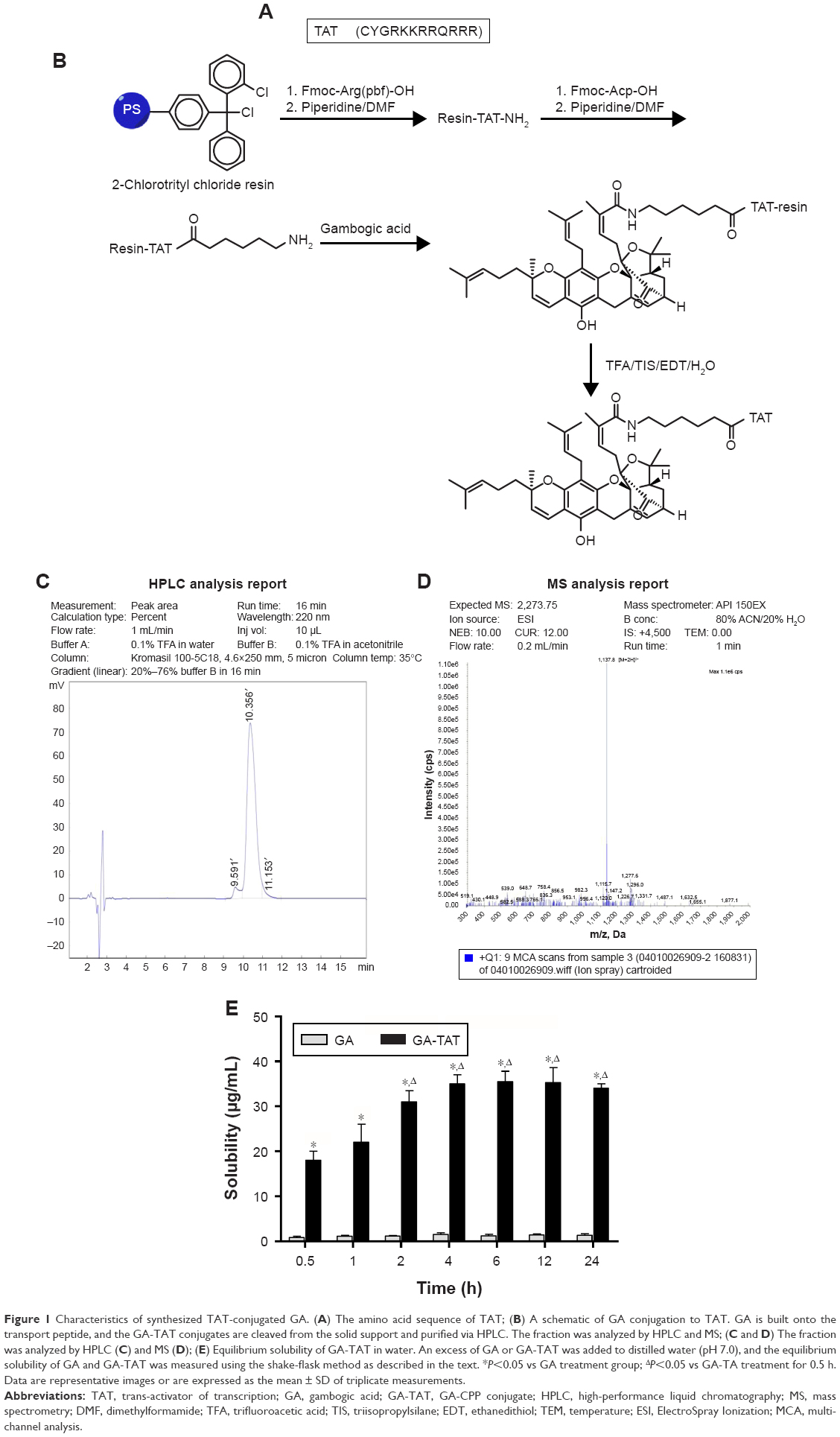 | Figure 1 Characteristics of synthesized TAT-conjugated GA. (A) The amino acid sequence of TAT; (B) A schematic of GA conjugation to TAT. GA is built onto the transport peptide, and the GA-TAT conjugates are cleaved from the solid support and purified via HPLC. The fraction was analyzed by HPLC and MS; (C and D) The fraction was analyzed by HPLC (C) and MS (D); (E) Equilibrium solubility of GA-TAT in water. An excess of GA or GA-TAT was added to distilled water (pH 7.0), and the equilibrium solubility of GA and GA-TAT was measured using the shake-flask method as described in the text. *P<0.05 vs GA treatment group; ΔP<0.05 vs GA-TA treatment for 0.5 h. Data are representative images or are expressed as the mean ± SD of triplicate measurements. |
Equilibrium solubility assay
The equilibrium solubility of GA-TAT was measured using the shake-flask method. Briefly, 10 mL buffer solution (distilled water, pH 7.0) and an excess of GA or GA-TAT were mixed in a flask. Then, the samples were maintained in an incubator shaker at 37°C and stirred at the indicated time (0.5, 1, 2, 4, 6, 12, and 24 h) until a heterogeneous system was obtained (comprising a saturated liquid and undissolved drug solid). The saturated solution and precipitate were separated by sedimentation and centrifugation. The supernatant was collected, and the concentration of samples was determined by UV spectrophotometry.
Cellular uptake assay
EJ and SV-HUC-1 cells, at a concentration of 1×106 cells per well, were seeded in six well plates and were incubated overnight. After exposure to different concentrations of GA or GA-TAT for 2 h, the cells were washed three times with PBS. Then, the cells were scraped from the wells, transferred to sterile Eppendorf tubes, and sonicated using a probe sonicator (OMNI Sonic Ruptor 400, Kennesaw, GA, USA). The cell solutions were centrifuged for 15 min at 8,000×g, and the supernatant was removed and tested via HPLC. To further test the effect of incubation time on cellular uptake, cells were treated with 2.5 μM GA or GA-TAT conjugates for different durations, followed by the other experimental procedures as described earlier.
Cell viability assay
Cell viability was measured with an MTT assay (Beyotime Institute Biotechnology, Shanghai, People’s Republic of China), according to the manufacturer’s instructions. Briefly, EJ and SV-HUC-1 cells at a concentration of 5×103 cells per well were seeded in a 96-well plate and treated with various concentrations of GA or GA-TAT. After treatment for the indicated times, 20 μL MTT reagent (5 mg/mL) was added to each well and was incubated at 37°C for an additional 4 h. Then, the cell supernatants were discarded. MTT crystals were dissolved in DMSO, and the absorbance values were detected at 570 nm by using a microplate reader (TECAN ULTRA, Research Triangle Park, NC, USA). The relative cell viability (%) was defined as the absorbance of the treated samples versus that of the untreated control, and the untreated control cells were designated as having 100% cell viability. All experiments were repeated at least three times.
EdU incorporation assay
Cell proliferation was assessed by a 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay by using an EdU assay kit (Ribobio, Guangzhou, People’s Republic of China) according to the manufacturer’s instructions. Briefly, 5×103 EJ cells per well were seeded into 96 well plates and treated with 1.0 μM of either GA or GA-TAT for 24 h. Then, the cells were washed twice with PBS, and 50 μM EdU was added for 4 h at 37°C in the dark. The cells were fixed with 4% formaldehyde for 15 min at room temperature, and then permeabilized with 0.5% Triton X-100 for 20 min. After being washed with PBS three times, the cells were treated with 100 μL Apollo reaction cocktail for 30 min. Subsequently, the DNA contents of the cells were stained with 100 μL Hoechst 33342 for 30 min and visualized under a fluorescence microscope.
Colony-formation assay
Cells at 1×106 cells per well were seeded into six well plates and treated with 1.0 μM of either GA or GA-TAT for 24 h. Then, the EJ cells were harvested and were cultured at 5×102 cells per well in six well plates, and the medium was replaced with fresh medium containing 10% FBS. Colonies were allowed to grow for 10 days. Subsequently, colonies were fixed, stained with 1% crystal violet (Sigma, St Louis, MO, USA) in 10% methanol, and counted under a microscope (Olympus, Tokyo, Japan).
Morphological assessment of apoptotic cells
Acridine orange/ethidium bromide (AO/EB; Sigma, St Louis, MO, USA) dual staining was done to detect the change in cell morphology. After treatment with 1.0 μM of either GA or GA-TAT for 24 h, the EJ cells were collected by trypsinization and centrifugation. The cells were washed with PBS and stained with 100 μg/mL AO and 100 μg/mL EB at 4°C in the dark for 10 min. Morphological changes, including nuclear condensation and cell shrinkage, were assessed using a fluorescence microscope (Olympus, Tokyo, Japan). The percentage of apoptotic cells was calculated by the following formula: apoptotic rate (%) = number of apoptotic cells/the total number of cells counted.
Quantitation of cell apoptosis rates by flow cytometry
The apoptosis rates of cells were determined by Annexin V-FITC and propidium iodide (PI) staining and flow cytometry, using the Annexin V-FITC/PI Apoptosis Detection Kit (KeyGen BioTECH, Nanjing, People’s Republic of China) according to the manufacturer’s instructions. Briefly, EJ cells at 1×106 cells per well were seeded into six well plates, incubated overnight, and treated with 1.0 μM of either GA or GA-TAT for 24 h. The cells were harvested and stained with Annexin V-FITC (V-FITC) and PI. The apoptotic cells were assessed using a FACScan flow cytometer (Becton Dickinson, NJ, USA) and Cell Quest analysis software.
Western blotting analysis
EJ cells were harvested and lysed with RIPA buffer containing protease inhibitor (Thermo Fisher Scientific, Waltham, MA, USA). Proteins were quantified using a bicinchoninic acid (BCA) protein assay kit (Beyotime Institute Biotechnology, Shanghai, People’s Republic of China) according to the manufacturer’s instructions. Equal amounts of protein lysate (30 μg) were resolved by SDS-PAGE on 10%–12% polyacrylamide gels and electrotransferred to polyvinylidene difluoride (PVDF) membranes (Millipore Bedford, MA, USA). After being blocked with Tris-buffered saline (TBS) containing 0.1% Triton X-100 and 5% skimmed milk at room temperature for 30 min, the membranes were incubated with primary antibodies against caspase-3, caspase-9, Bcl-2, Bax (Cell Signaling Technology, Beverly, MA, USA), and GAPDH (Santa Cruz Biotechnology, CA, USA) overnight at 4°C. After being washed, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology, CA, USA) at room temperature for 1 h. Protein bands were detected using a chemiluminescence method (ECL; Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instruction.
Caspase activity measurement
Caspase-3 and caspase-9 activities were analyzed using the caspase-3 and caspase-9 activity assay kit (Beyotime Institute Biotechnology, Shanghai, People’s Republic of China) according to the manufacturer’s instructions and the acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) and acetyl-Leu-Glu-His-Asp p-nitroanilide (Ac-LEHD-pNA) substrates, respectively. Briefly, after treatment with 1.0 μM of either GA or GA-TAT for 24 h (in some cases, cells were pretreated for 2 h with a caspase inhibitor [Ac-DEVD-CHO or Z-LEHD-FMK]). Cell lysates (10 μL) were prepared and mixed with buffer (80 μL) containing the caspase substrate (10 μL, 2 mM) attached to pNA. Then, the lysates were incubated at 37°C for 4 h in the dark. The release of pNA was measured with a fluorescence plate reader at an absorbance of 405 nm. Caspase activities were expressed as the ratio of treated to non-manipulated control cells.
Intracellular reactive oxygen species detection
Intracellular reactive oxygen species (ROS) production was evaluated by a ROS assay with 2′7′-dichlorofluorescin diacetate (DCFH-DA; Beyotime Institute Biotechnology, Shanghai, People’s Republic of China). Briefly, after treatment with 1.0 μM of either GA or GA-TAT for 24 h, EJ cells were washed with PBS twice and incubated with 10 μM DCFH-DA for 30 min at 37°C. After washing with PBS three times, cells were collected and observed under a fluorescence microscope (Olympus, Tokyo, Japan). The qualitative analysis of ROS production was conducted using a fluorescence microplate reader (ReadMax 1800Plus, Flash Spectrum Biological Technology, Shanghai, People’s Republic of China) with excitation at 485 nm and emission at 520 nm. The values were determined as the mean absorbance normalized to the percentage of the control.
Statistical analysis
All statistical analyses were conducted using SPSS 18.0 (SPSS Inc., Chicago, IL, USA). Data are presented as the mean ± SD of at least three independent experiments. Statistical significance (P<0.05) was determined by one-way ANOVA or the Student–Newman–Keuls (SNK)-q test.
Results
Equilibrium solubility of GA-TAT
The equilibrium solubility of GA-TAT was tested using the shake-flask method. As shown in Figure 1E, the measured solubility of GA-TAT increased with increased stirring time. This increased solubility of GA-TAT reached its peak at 4 h after stirring, and stayed at this level for 6–24 h after stirring. The results were consistent with a previous report that stated GA was almost completely insoluble in water.10 Although the solubility of GA-TAT in water was improved as compared with GA, the solubility values of GA-TAT were much lower than that of the standard sample, which was dissolved with DMSO and diluted with water. Thus, both GA and GA-TAT stock solutions were prepared in DMSO (25 mmol/L) and diluted with serum-free medium or PBS for experiments.
Cellular uptake assay
Either GA or GA-TAT was added at a final concentration of 0.25, 1.0, 2.5, or 5 μM to a six well plate, and the GA cellular uptake was evaluated in EJ and SV-HUC-1 cells. The results revealed that, as compared with GA-treated cells, the EJ and SV-HUC-1 cells incubated with GA-TAT exhibited higher cellular uptake of GA as compared with the GA-treated cells (P<0.05), regardless of the concentration (0.25, 1.0, 2.5, or 5 μM) (Figure 2A). Furthermore, the cellular GA and GA-TAT uptake was increased in EJ and SV-HUC-1 cells with a prolonged incubation time. However, more GA-TAT was internalized than GA (Figure 2B). These results demonstrated the efficacy of peptide-mediated GA-TAT internalization in EJ and SV-HUC-1 cells.
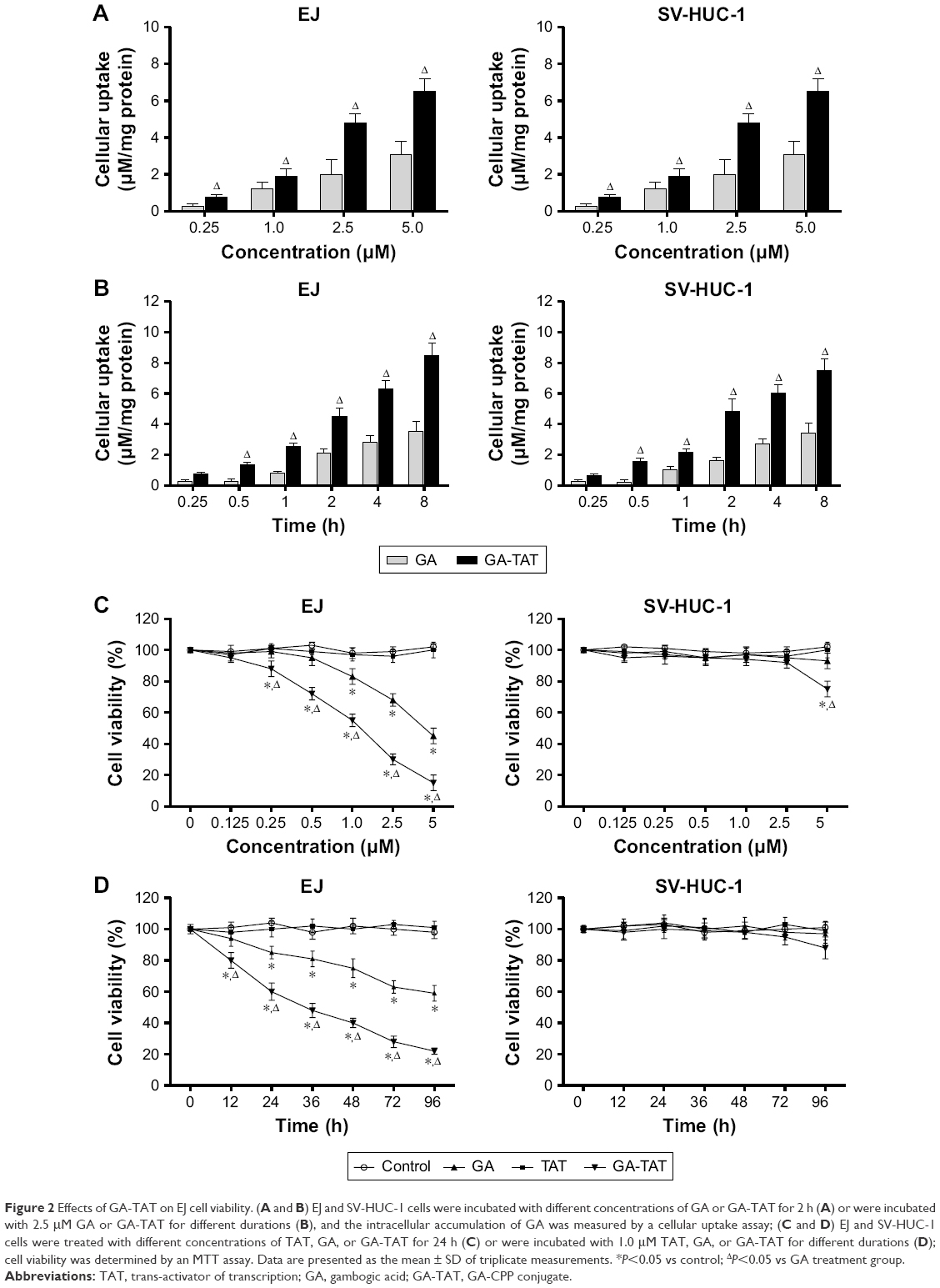 | Figure 2 Effects of GA-TAT on EJ cell viability. (A and B) EJ and SV-HUC-1 cells were incubated with different concentrations of GA or GA-TAT for 2 h (A) or were incubated with 2.5 μM GA or GA-TAT for different durations (B), and the intracellular accumulation of GA was measured by a cellular uptake assay; (C and D) EJ and SV-HUC-1 cells were treated with different concentrations of TAT, GA, or GA-TAT for 24 h (C) or were incubated with 1.0 μM TAT, GA, or GA-TAT for different durations (D); cell viability was determined by an MTT assay. Data are presented as the mean ± SD of triplicate measurements. *P<0.05 vs control; ΔP<0.05 vs GA treatment group. |
GA-TAT cytotoxicity on bladder cancer cells
The effect of a series of GA and GA-TAT concentrations on cell viability was tested using the MTT assay. Moreover, TAT peptides were used at the same concentration as GA and GA-TAT to test CPP toxicity. As shown in Figure 2C and D, the TAT peptide had no effect on the viability of EJ and SV-HUC-1 cells. GA inhibited EJ cell viability in a dose- and time-dependent manner, and the inhibitory effect of GA-TAT conjugate compounds on cell viability was significantly greater than that of the GA treatment. A low GA-TAT (0.25 μM) concentration did induce clear cytotoxic effects on EJ cells, whereas the equivalent dose of GA alone was insufficient. GA had to be used at 1.0 μM to attain a comparable effect. The 50% inhibitory concentration (IC50) of GA-TAT at 24 h was 1.24 μM, which was lower than that of the GA treatment (4.95 μM). Furthermore, the viability of the cultured SV-HUC-1 cells was nearly unaffected by GA and GA-TAT, which was consistent with previous evidence demonstrating that normal cells are resistant to GA-mediated cytotoxicity.9
GA-TAT inhibited EJ cell proliferation
To compare the inhibitory functions of GA and GA-TAT on EJ cell proliferation, we used the EdU incorporation assay – a sensitive and specific method – in this study. The results revealed that the number of EdU+ cells was significantly reduced in the 1.0 μM GAT or GA-TAT–treated groups as compared with the TAT-treated and control groups (Figure 3A). More importantly, the number of EdU+ cells was markedly less in the GA-TAT–treated groups than in the GA-treated group. These results indicated that GA-TAT inhibited EJ cell proliferation in vitro and that the inhibitory effect of GA-TAT was much greater than that of GA alone. To further determine whether GA-TAT enhances the long-term inhibitory effect of GA on proliferation, a clonogenic assay was conducted. Colony growth was significantly decreased after the 1.0 μM GA treatment, and was further reduced following the GA-TAT treatment (Figure 3B). These results further demonstrated that GA-TAT conjugate compounds are a promising candidate to sensitize tumor cells to bladder cancer treatment.
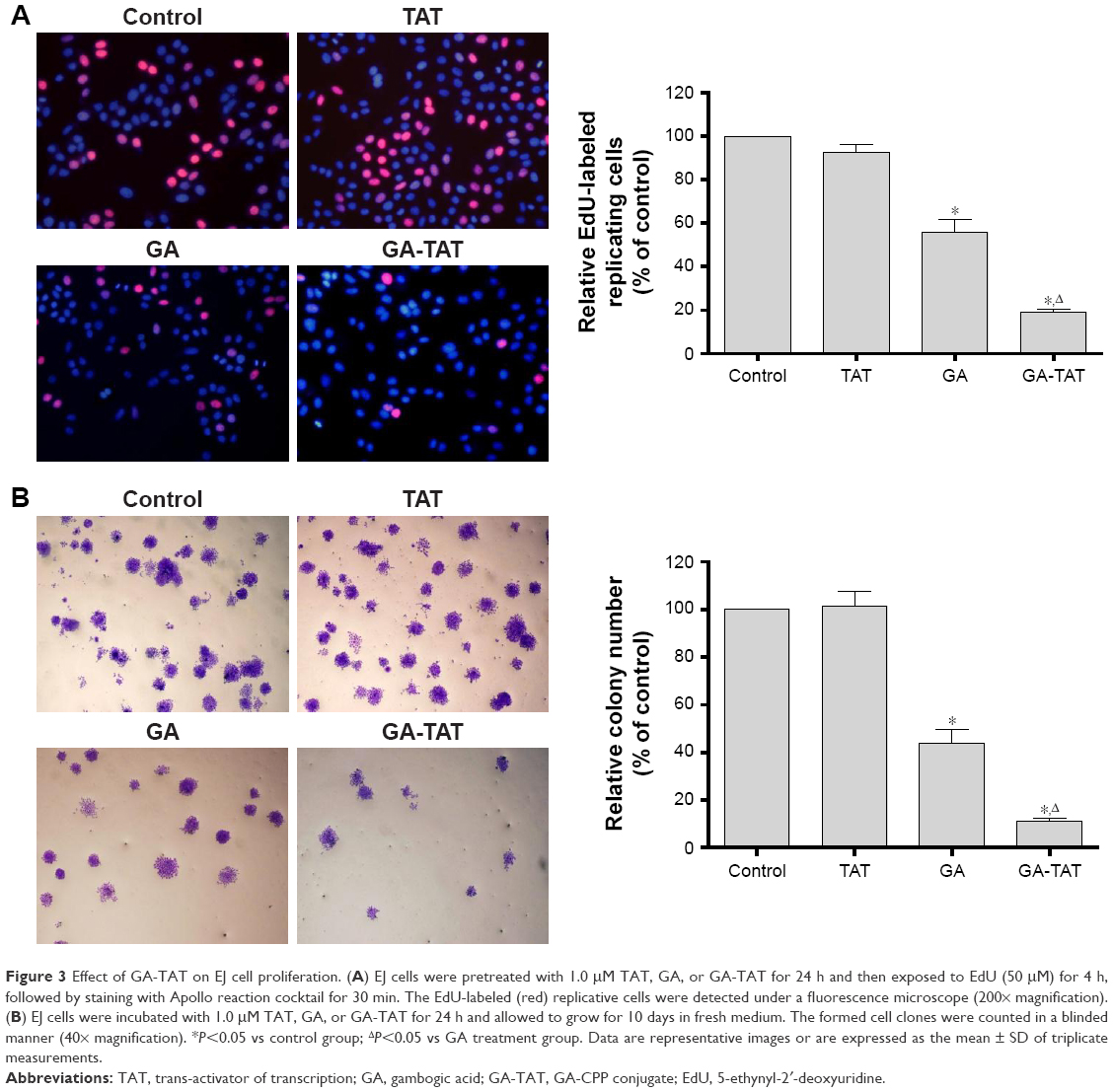 | Figure 3 Effect of GA-TAT on EJ cell proliferation. (A) EJ cells were pretreated with 1.0 μM TAT, GA, or GA-TAT for 24 h and then exposed to EdU (50 μM) for 4 h, followed by staining with Apollo reaction cocktail for 30 min. The EdU-labeled (red) replicative cells were detected under a fluorescence microscope (200× magnification). (B) EJ cells were incubated with 1.0 μM TAT, GA, or GA-TAT for 24 h and allowed to grow for 10 days in fresh medium. The formed cell clones were counted in a blinded manner (40× magnification). *P<0.05 vs control group; ΔP<0.05 vs GA treatment group. Data are representative images or are expressed as the mean ± SD of triplicate measurements. |
GA-TAT induced EJ cell apoptosis
To investigate whether GA-TAT toxicity was due to the induction of apoptosis, we examined the induced apoptosis by the AO/EB dual staining assay and V-FITC/PI staining flow cytometric method. First, cell apoptosis was analyzed using AO/EB dual detection wherein apoptotic cells present with orange chromatin fragments. As shown in Figure 4A, compared with the control and TAT-treated cells, GA- and GA-TAT–treated cells presented with characteristics of apoptosis. However, treating cells with GA-TAT further enhanced apoptosis. The percentage of apoptotic cells in the GA-TAT group was much greater than that in the GA treatment group (42.15%±5.23% vs 19.18%±4.35%). The V-FITC/PI flow cytometric staining was used to further quantify the number of apoptotic cells. As shown in Figure 4B, GA induced apoptosis in only 21.3%±2.27% of EJ cells. However, after treatment with GA-TAT, the percentage of apoptotic cells was greatly increased to 46.4%±4.86%. The apoptosis-inducing effect of GA-TAT on EJ cells was greater than that of GA alone.
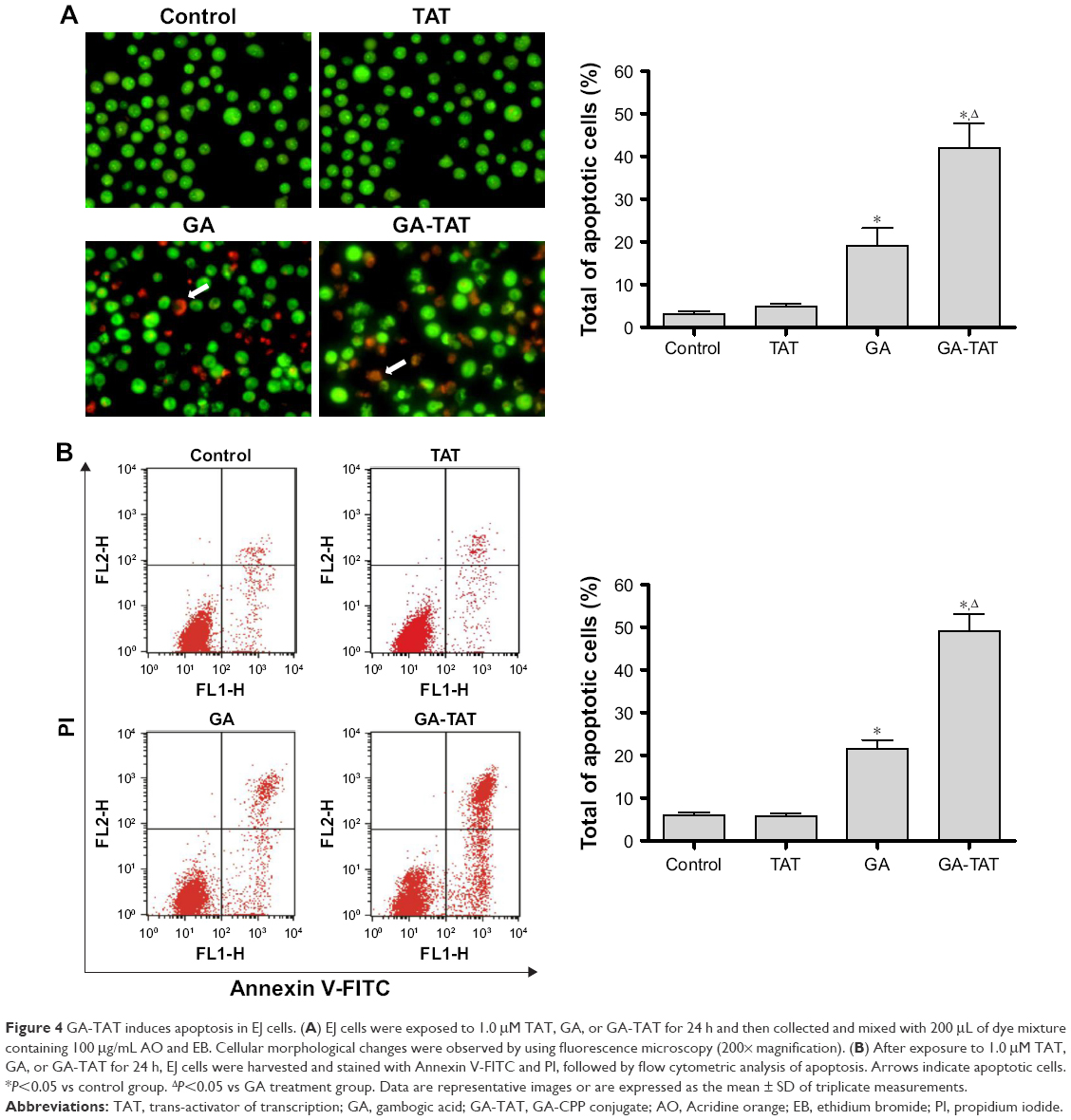 | Figure 4 GA-TAT induces apoptosis in EJ cells. (A) EJ cells were exposed to 1.0 μM TAT, GA, or GA-TAT for 24 h and then collected and mixed with 200 μL of dye mixture containing 100 μg/mL AO and EB. Cellular morphological changes were observed by using fluorescence microscopy (200× magnification). (B) After exposure to 1.0 μM TAT, GA, or GA-TAT for 24 h, EJ cells were harvested and stained with Annexin V-FITC and PI, followed by flow cytometric analysis of apoptosis. Arrows indicate apoptotic cells. *P<0.05 vs control group. ΔP<0.05 vs GA treatment group. Data are representative images or are expressed as the mean ± SD of triplicate measurements. |
Caspase activation in response to GA-TAT treatment in EJ cells
To determine the association between cell apoptosis and GA-TAT treatment in bladder cancer EJ cells, we examined the proteolytic processing and activities of caspase-3 and caspase-9, which are crucial for apoptosis signal transduction via the caspase cascade. As shown in Figure 5A, caspase-9 and caspase-3 activities were increased in GA-treated cells as compared with control and TAT-treated cells. Cells treated with GA-TAT presented with a further enhanced processing and activation of caspase-9 and caspase-3.
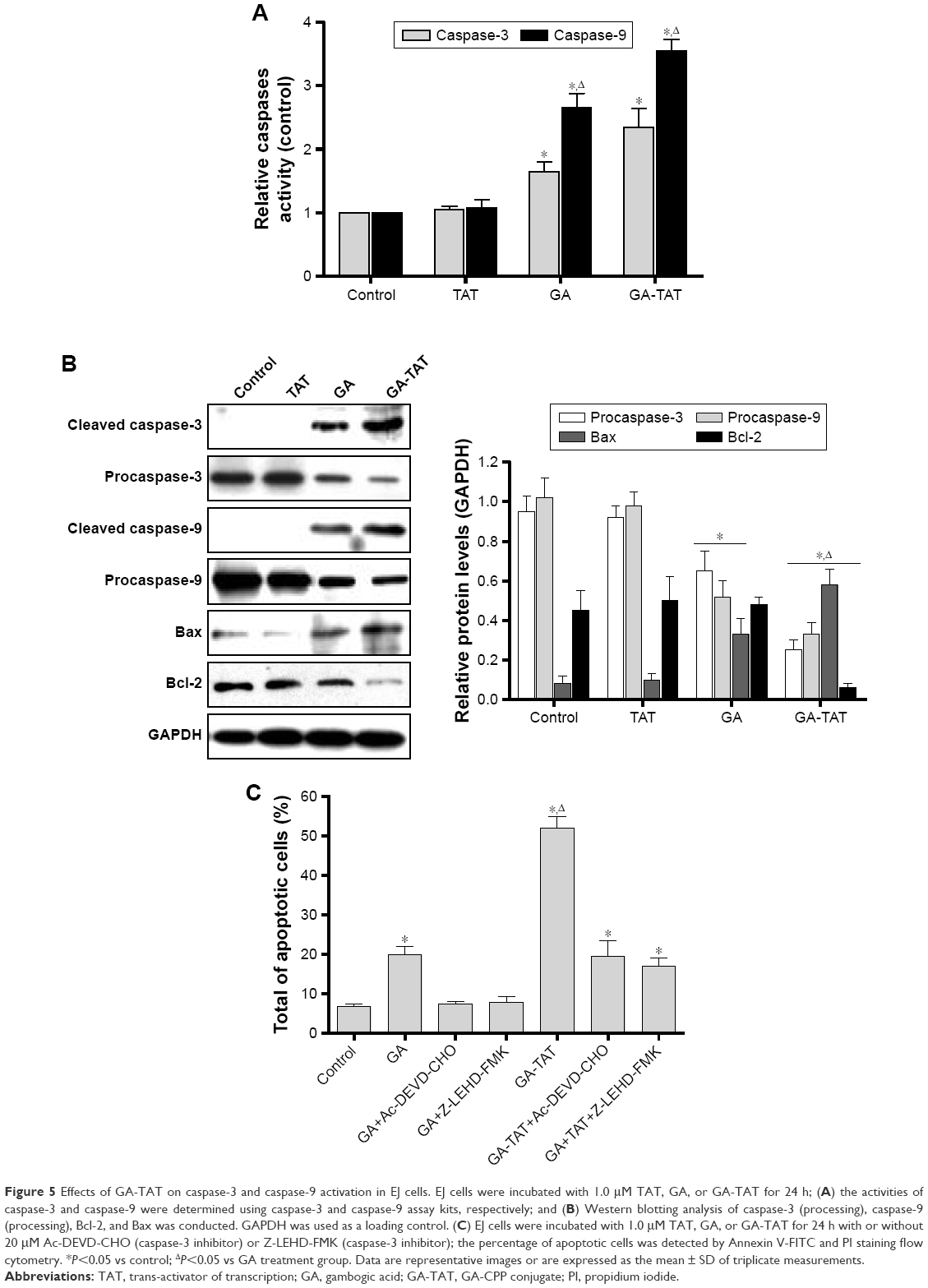 | Figure 5 Effects of GA-TAT on caspase-3 and caspase-9 activation in EJ cells. EJ cells were incubated with 1.0 μM TAT, GA, or GA-TAT for 24 h; (A) the activities of caspase-3 and caspase-9 were determined using caspase-3 and caspase-9 assay kits, respectively; and (B) Western blotting analysis of caspase-3 (processing), caspase-9 (processing), Bcl-2, and Bax was conducted. GAPDH was used as a loading control. (C) EJ cells were incubated with 1.0 μM TAT, GA, or GA-TAT for 24 h with or without 20 μM Ac-DEVD-CHO (caspase-3 inhibitor) or Z-LEHD-FMK (caspase-3 inhibitor); the percentage of apoptotic cells was detected by Annexin V-FITC and PI staining flow cytometry. *P<0.05 vs control; ΔP<0.05 vs GA treatment group. Data are representative images or are expressed as the mean ± SD of triplicate measurements. |
These results were further confirmed by Western blotting. In contrast to GA treatment, GA-TAT substantially enhanced the cleaved caspase-3, caspase-9, and Bax upregulation. However, the Bcl-2 protein level was not affected by GA but was downregulated after treatment with GA-TAT (Figure 5B). We further determined the apoptotic rates of EJ cells after the addition of a caspase inhibitor. The caspase dependence of the apoptosis induced by GA or GA-TAT treatment was confirmed with the use of a caspase-9–specific inhibitor Z-LEHD-FMK and a caspase-3–specific inhibitor z-DEVD-fmk. As shown in Figure 5C, Z-LEHD-FMK and z-DEVD-fmk completely suppressed apoptosis in GA-treated cells but partially inhibited GA-TAT–induced apoptosis.
Role of ROS in GA-TAT–induced apoptosis
To determine the mechanism involved in GA-TAT–induced apoptosis, we examined ROS production in EJ cells. As shown in Figure 6A, ROS production was substantially increased in GA and GA-TAT–treated EJ cells compared with control and TAT-treated cells and was significantly higher in GA-TAT–treated cells than in cells treated with GA (P<0.05). To further investigate the role of ROS in GA-TAT–treated EJ cell apoptosis, an ROS inhibitor was used. ROS accumulation was markedly abrogated when cells were pretreated with the ROS scavenger N-acetyl-L-cysteine (NAC), demonstrating the effective elimination of GA and GA-TAT–induced ROS production by NAC (Figure 6A). Moreover, inhibiting ROS generation by adding NAC significantly blocked GA-TAT–induced caspase-3 and caspase-9 activities. Meanwhile, NAC also attenuated the GA-TAT–mediated upregulation of Bax protein expression and downregulation of Bcl-2 protein expression (Figure 6B and C). Importantly, when cells were pretreated with NAC for 2 h, followed by incubation with GA or GA-TAT for an additional 24 h, the percentage of apoptotic EJ cells was markedly reduced to 8.3%±0.8% or 9.0%±1.3%, respectively. Thus, NAC treatment alone – in the absence of GA and GA-TAT – did not significantly affect apoptosis (Figure 6D).
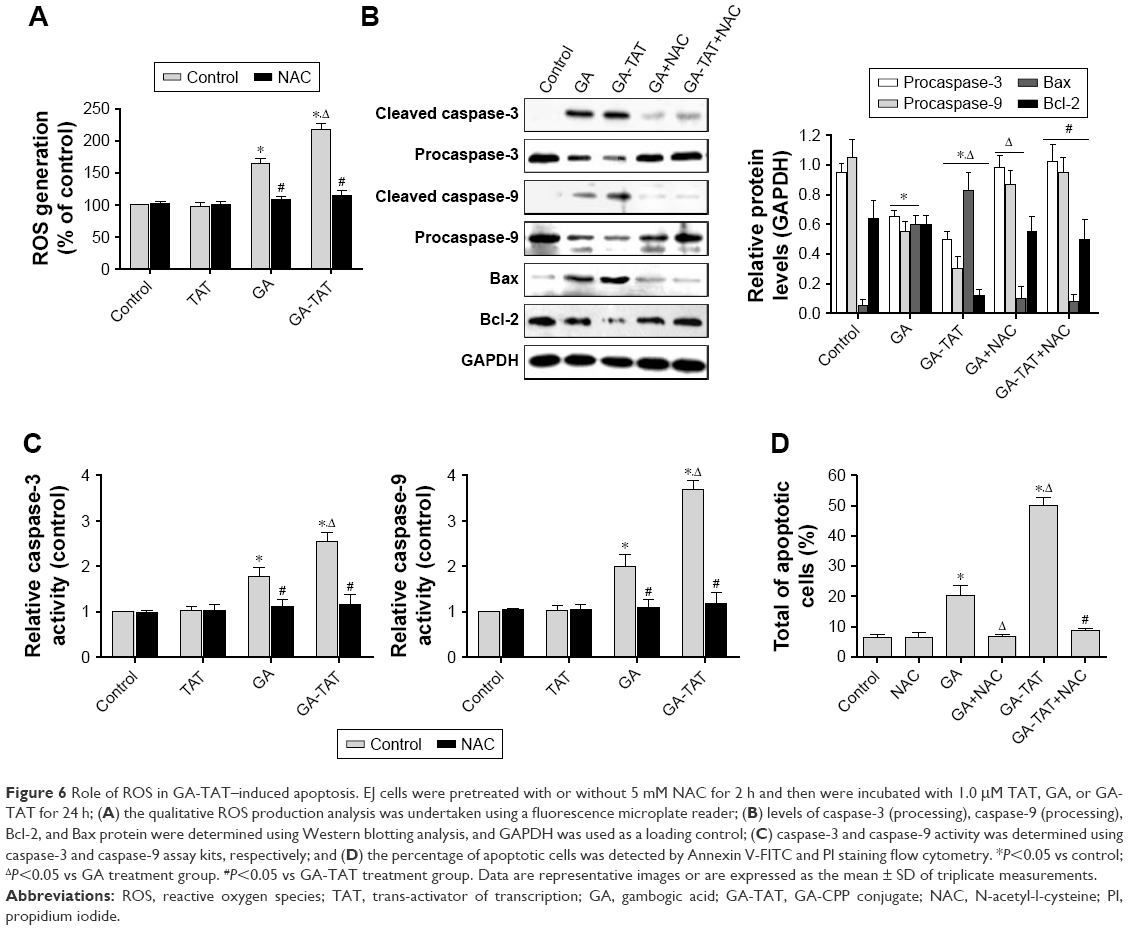 | Figure 6 Role of ROS in GA-TAT–induced apoptosis. EJ cells were pretreated with or without 5 mM NAC for 2 h and then were incubated with 1.0 μM TAT, GA, or GA-TAT for 24 h; (A) the qualitative ROS production analysis was undertaken using a fluorescence microplate reader; (B) levels of caspase-3 (processing), caspase-9 (processing), Bcl-2, and Bax protein were determined using Western blotting analysis, and GAPDH was used as a loading control; (C) caspase-3 and caspase-9 activity was determined using caspase-3 and caspase-9 assay kits, respectively; and (D) the percentage of apoptotic cells was detected by Annexin V-FITC and PI staining flow cytometry. *P<0.05 vs control; ΔP<0.05 vs GA treatment group. #P<0.05 vs GA-TAT treatment group. Data are representative images or are expressed as the mean ± SD of triplicate measurements. |
Discussion
The novel anticancer agent GA has significant anticancer activity against human cancers. GA exhibits anticancer effects via multiple mechanisms, including the promotion of apoptosis, induction of cell-cycle arrest as well as inhibition of telomerases, cell invasion, and angiogenesis.16–18 In this study, we demonstrated that GA presented with dose- and time-dependent anticancer activity in human bladder cancer EJ cells at concentrations starting at 1.0 μM. Furthermore, our study confirmed that cell viability was inhibited via the promotion of apoptosis in bladder cancer EJ cells. These results further indicated that GA is an attractive agent for cancer therapy. However, the major obstacle to the clinical application of GA is its poor cell-penetration ability, which is caused by extremely poor water solubility.19 To achieve an effective concentration of GA intracellularly, a high dose of GA was used to induce apoptosis. However, the relatively high therapeutic dose of GA has inhibited the development of GA for tumor therapy due to potential side effects, such as vascular stimulation, neurotoxicity, hepatotoxicity, nephrotoxicity, and cardiotoxicity, which discourage its clinical application.20
In recent years, an increasing amount of evidence has suggested that CPPs are an improved vehicle for intracellular delivery due to their low toxicity levels and inability to promote an immune response. The greatest qualities of CPPs are their ability to efficiently pass through cellular membranes with low cytotoxicity, to carry a large variety of cargo into the cell, and to enhance the water solubility of drugs.21 With regard to research on CPP application, several CPP drugs have been introduced in preclinical studies and have shown some advantages in cancer treatment. In this study, we have synthesized a novel CPP-GA conjugate using the well-known CPP sequence, TAT – a short peptide with amphipathic and cationic characteristics. TAT not only enhances the water solubility of drugs but also shows a high level of cellular uptake. TAT contains more than five positively charged amino acids (arginine) that can create hydrogen bonds with polar lipid groups, and the location of arginines in the TAT sequence have been associated with better uptake efficiency.22 In addition, macropinocytosis has been shown to be involved in TAT uptake.23 TAT has been reported to have the ability to transport macromolecules across the cell membrane of numerous cancer cells, such as breast, lung, and liver cancer cells.24–26 The results of the present study demonstrated that the solubility of GA-TAT in water was improved as compared with that of GA. In addition, our cellular uptake assays data showed that GA-TAT enhanced the accumulation of GA in EJ cells as compared with unconjugated GA, which confirmed that TAT facilitates cargo uptake. More importantly, in EJ cells, GA-TAT cytotoxicity was detectable by MTT assay at lower treatment concentrations than those required during treatment with unconjugated GA. The results demonstrated that the IC50 value of GA-TAT (1.24 μM), as obtained by an MTT assay, was lower than that of GA (4.95 μM). Meanwhile, GA-TAT treatment alone did not induce cell death in the normal human uroepithelium cell line SV-HUC-1. These results implied that GA-TAT may have therapeutic potential in patients with bladder cancer if the lower doses can be achieved clinically, thereby potentially reducing nephrotoxicity and cardiotoxicity which are the main side effects of chemotherapeutics. Next, we evaluated the effect of GA-TAT on EJ cell proliferation by using EdU and colony-formation assays. We found that, compared with GA alone, the GA-TAT conjugates had a more potent inhibitory effect on EJ cell proliferation in vitro. These results are supported by a fewer number of EdU+ cells and cell clones being observed in the GA-TAT–treated groups than in the GA treatment group.
The potential mechanisms underlying the anticancer activity of GA-TAT in EJ cells were characterized by apoptosis induction. Apoptosis is characterized by a series of typical morphological changes.27 In this work, we used AO/EB dual staining to detect the morphological changes of EJ cells. After 24 h of treatment with GA, EJ cells presented with typical apoptotic morphological changes: cell shrinkage, chromatin condensation, nuclear fragmentation, and cell size and shape alterations. Moreover, more of these morphological changes were detected in the GA-TAT–treated group than in the GA alone–treated group, indicating that treating cells with GA-TAT further enhanced apoptosis. The V-FITC/PI staining flow cytometric assay further confirmed our results that, in EJ cells, the apoptosis-inducing effect of GA-TAT was greater than that of GA alone.
The caspase family contains 12 cysteine aspartate-specific proteases that play essential roles in cell apoptosis.28 Activated caspase-9 works as an initiating caspase by cleaving and, thereby, activating the downstream executioner caspases, such as caspase-3, that then trigger the rest of the caspase cascade.29 In the present study, we have demonstrated that GA-TAT significantly increased apoptosis in bladder cancer EJ cells, which was accompanied by increased caspase-9 and caspase-3 processing. However, the caspase-9 inhibitor Z-LEHD-FMK and caspase-3 inhibitor Ac-DEVD-CHO completely inhibited GA-induced apoptosis but only partially suppressed the apoptosis in GA-TAT–treated cells. These findings indicated that, in EJ cells, GA-induced apoptosis occurred mainly through the caspase-dependent pathway; however, other mechanisms are probably involved in GA-TAT–induced apoptosis.
Intracellular ROS generation is an important cause of apoptotic cell death and is also involved in the development of bladder cancer.30 ROS has been shown to undertake certain functions in early stages of apoptosis and to induce mitochondrial membrane depolarization.31 We, therefore, investigated whether GA-TAT induced ROS production. Our results demonstrated that GA or GA-TAT treatment greatly increased ROS production, which was blocked by NAC – an ROS scavenger. Interestingly, NAC also completely inhibited GA-TAT–induced caspase-3 and caspase-9 activation and suppressed cell apoptosis. These findings suggest that GA-TAT–induced EJ cell apoptosis is mediated via ROS production. These results are consistent with a previous study that ROS generation triggered apoptosis via caspase-dependent pathway activation.32
ROS have been shown to regulate the expression of Bcl-2 family members to activate the intrinsic apoptotic pathway.33 Therefore, we next focused on two members of this family: pro-death protein Bax and pro-survival protein Bcl-2. Bcl-2 and Bax protein form homo- and heterodimers that regulate apoptosis susceptibility.34 An imbalance between Bax/Bcl-2 leads to cytochrome C release, caspase activation, and subsequent cell apoptosis.35 In the present study, GA or GA-TAT treatment in EJ cells upregulated the Bax expression level. However, the Bcl-2 protein level was not affected by GA alone, but rather, was downregulated after GA-TAT treatment. These results indicated that, compared with GA treatment, GA-TAT treatment induced a greater decrease in the Bcl-2/Bax ratio, leading to increased EJ cell apoptosis. This finding was consistent with previous studies wherein a lower Bcl-2/Bax ratio was associated with increased caspase-3 levels and, consequently, cell apoptosis susceptibility.36 Furthermore, the GA-TAT–mediated upregulation of Bax, downregulation of Bcl-2, and induction of apoptosis were also attenuated by NAC treatment, suggesting that GA-TAT enhances the pro-apoptotic effect of human bladder cancer EJ cells partly through a ROS-mediated decrease in the Bcl-2/Bax ratio.
In summary, we successfully synthesized a new conjugate – GA-TAT – which is more efficient and induces greater cell apoptosis than GA alone in bladder cancer EJ cells. We offer the first evidence that the conjugation of GA with a CPP enhances the pro-apoptotic effect. Our study demonstrated that GA-TAT enhanced the pro-apoptotic effect via increasing caspase-3 and caspase-9 processing and activities and decreasing the Bcl-2/Bax ratio, which were regulated by intracellular ROS. The results of this study highlight the potential of GA-TAT as a promising clinical therapeutic for the treatment of bladder cancer.
Acknowledgment
This study was supported by the National Natural Science Foundation of China (grant no 81502204), Hubei province Nature Science Foundation of China (grant no 2014CFB399), and Wuhan Clinical Medical Research of China (grant no WX14A01).
Disclosure
The authors report no conflicts of interest in this work.
References
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Carneiro BA, Meeks JJ, Kuzel TM, Scaranti M, Abdulkadir SA, Giles FJ. Emerging therapeutic targets in bladder cancer. Cancer Treat Rev. 2015;41(2):170–178. | ||
Sfakianos JP, Kim PH, Hakimi AA, Herr HW. The effect of restaging transurethral resection on recurrence and progression rates in patients with nonmuscle invasive bladder cancer treated with intravesical bacillus Calmette-Guerin. J Urol. 2014;191(2):341–345. | ||
Brausi M, Oddens J, Sylvester R, et al. Side effects of Bacillus Calmette-Guerin (BCG) in the treatment of intermediate- and high-risk Ta, T1 papillary carcinoma of the bladder: results of the EORTC genito-urinary cancers group randomised phase 3 study comparing one-third dose with full dose and 1 year with 3 years of maintenance BCG. Eur Urol. 2014;65(1):69–76. | ||
Ouyang L, Luo Y, Tian M, et al. Plant natural products: from traditional compounds to new emerging drugs in cancer therapy. Cell Prolif. 2014;47(6):506–515. | ||
Wang S, Wang L, Chen M, Wang Y. Gambogic acid sensitizes resistant breast cancer cells to doxorubicin through inhibiting P-glycoprotein and suppressing survivin expression. Chem Biol Interact. 2015;235:276–284. | ||
Lü L, Tang D, Wang L, et al. Gambogic acid inhibits TNF-α-induced invasion of human prostate cancer PC3 cells in vitro through PI3K/Akt and NF-κB signaling pathways. Acta Pharmacol Sin. 2012;33(4):531–541. | ||
Qi Q, Lu N, Li C, et al. Involvement of RECK in gambogic acid induced anti-invasive effect in A549 human lung carcinoma cells. Mol Carcinog. 2015;54 (Suppl 1):E13–E25. | ||
Yang Y, Yang L, You QD, et al. Differential apoptotic induction of gambogic acid, a novel anticancer natural product, on hepatoma cells and normal hepatocytes. Cancer Lett. 2007;256(2):259–266. | ||
Doddapaneni R, Patel K, Owaid IH, Singh M. Tumor neovasculature-targeted cationic PEGylated liposomes of gambogic acid for the treatment of triple-negative breast cancer. Drug Deliv. 2016;23(4):1232–1241. | ||
Ruoslahti E. Tumor penetrating peptides for improved drug delivery. Adv Drug Deliv Rev. 2017;110–111:3–12. | ||
Aroui S, Brahim S, De Waard M, Bréard J, Kenani A. Efficient induction of apoptosis by doxorubicin coupled to cell-penetrating peptides compared to unconjugated doxorubicin in the human breast cancer cell line MDA-MB 231. Cancer Lett. 2009;285(1):28–38. | ||
Vale N, Ferreira A, Fernandes I, et al. Gemcitabine anti-proliferative activity significantly enhanced upon conjugation with cell-penetrating peptides. Bioorg Med Chem Lett. 2017;27(13):2898–2901. | ||
Rosenkranz AA, Ulasov AV, Slastnikova TA, Khramtsov YV, Sobolev AS. Use of intracellular transport processes for targeted drug delivery into a specified cellular compartment. Biochemistry (Mosc). 2014;79(9):928–946. | ||
Wang Y, Xiang W, Wang M, et al. Methyl jasmonate sensitizes human bladder cancer cells to gambogic acid-induced apoptosis through down-regulation of EZH2 expression by miR-101. Br J Pharmacol. 2014;171(3):618–635. | ||
Zhao W, You CC, Zhuang JP, et al. Viability inhibition effect of gambogic acid combined with cisplatin on osteosarcoma cells via mitochondria-independent apoptotic pathway. Mol Cell Biochem. 2013;382(1–2):243–252. | ||
Xu P, Li J, Shi L, Selke M, Chen B, Wang X. Synergetic effect of functional cadmium-tellurium quantum dots conjugated with gambogic acid for HepG2 cell-labeling and proliferation inhibition. Int J Nanomedicine. 2013;8:3729–3736. | ||
Yan X, Yang Y, He L, Peng D, Yin D. Gambogic acid grafted low molecular weight heparin micelles for targeted treatment in a hepatocellular carcinoma model with an enhanced anti-angiogenesis effect. Int J Pharm. 2017;522(1–2):110–118. | ||
Liu YT, Hao K, Liu XQ, Wang GJ. Metabolism and metabolic inhibition of gambogic acid in rat liver microsomes. Acta Pharmacol Sin. 2006;27(9):1253–1258. | ||
Qi Q, Gu H, Yang Y, et al. Involvement of matrix metalloproteinase 2 and 9 in gambogic acid induced suppression of MDA-MB-435 human breast carcinoma cell lung metastasis. J Mol Med (Berl). 2008;86(12):1367–1377. | ||
Vhora I, Patil S, Bhatt P, Misra A. Protein- and peptide-drug conjugates: an emerging drug delivery technology. Adv Protein Chem Struct Biol. 2015;98:1–55. | ||
Pooga M, Langel Ü. Classes of cell-penetrating peptides. Methods Mol Biol. 2015;1324:3–28. | ||
Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10(3):310–315. | ||
Diao Y, Liu J, Ma Y, Su M, Zhang H, Hao X. A specific aptamer-cell penetrating peptides complex delivered siRNA efficiently and suppressed prostate tumor growth in vivo. Cancer Biol Ther. 2016;17(5):498–506. | ||
Morshed RA, Muroski ME, Dai Q, et al. Cell-penetrating peptide-modified gold nanoparticles for the delivery of doxorubicin to brain metastatic breast cancer. Mol Pharm. 2016;13(6):1843–1854. | ||
Liu L, Dong X, Zhu D, Song L, Zhang H, Leng XG. TAT-LHRH conjugated low molecular weight chitosan as a gene carrier specific for hepatocellular carcinoma cells. Int J Nanomedicine. 2014;9:2879–2889. | ||
Vanden Berghe T, Grootjans S, Goossens V, et al. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods. 2013;61(2):117–129. | ||
Murray J, Renslo AR. Modulating caspase activity: beyond the active site. Curr Opin Struct Biol. 2013;23(6):812–819. | ||
Julien O, Wells JA. Caspases and their substrates. Cell Death Differ. 2017;24(8):1380–1389. | ||
Park BH, Lim JE, Jeon HG, et al. Curcumin potentiates antitumor activity of cisplatin in bladder cancer cell lines via ROS-mediated activation of ERK1/2. Oncotarget. 2016;7(39):63870–63886. | ||
Tokarz P, Blasiak J. Role of mitochondria in carcinogenesis. Acta Biochim Pol. 2014;61(4):671–678. | ||
Zhang M, Harashima N, Moritani T, Huang W, Harada M. The roles of ROS and caspases in TRAIL-induced apoptosis and necroptosis in human pancreatic cancer cells. PLoS One. 2015;10(5):e0127386. | ||
Um HD. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: a review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget. 2016;7(5):5193–5203. | ||
Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5(4):a008714. | ||
Xu CF, Wu AR, Zhu H, et al. Melatonin is involved in the apoptosis and necrosis of pancreatic cancer cell line SW-1990 via modulating of Bcl-2/Bax balance. Biomed Pharmacother. 2013;67(2):133–139. | ||
Xu G, Kuang G, Jiang WG, Jiang R, Jiang DM. Polydatin promotes apoptosis through upregulation the ratio of Bax/Bcl-2 and inhibits proliferation by attenuating the β-catenin signaling in human osteosarcoma cells. Am J Transl Res. 2016;8(2):922–931. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.