")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Cathepsin G cleaves and activates IL-36γ and promotes the inflammation of psoriasis
Authors Guo J, Tu J, Hu YY, Song GX, Yin ZQ
Received 15 November 2018
Accepted for publication 16 January 2019
Published 8 February 2019 Volume 2019:13 Pages 581—588
DOI https://doi.org/10.2147/DDDT.S194765
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Jing Guo,1,* Jie Tu,1,* YingYing Hu,1,2,* GuoXin Song,3 ZhiQiang Yin1
1Department of Dermatology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China; 2Department of Medical Cosmetology, Wuqing People’s Hospital, Wuqing, Tianjin, China; 3Department of Pathology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China
*These authors contributed equally to this work
Background: IL-36γ is considered to be a valuable biomarker in psoriatic patients, which is expressed as an inactive precursor that needs to be proteolytically processed and activated, and neutrophil-derived proteases seemed to be potent activating enzymes of IL-36γ.
Objectives: This study aims to investigate the activation of IL-36γ by cathepsin G (CG) and neutrophil elastase (NE).
Materials and methods: We used inactive recombinant full-length (FL)-IL-36γ with different doses of NE or CG to stimulate HaCaT cells; neutrophil extracellular traps (NETs) were prepared to act on FL-IL-36γ and then stimulate HaCaT cells. Real-time quantitative PCR and ELISA were performed to detect CXCL-1 and CXCL-8 expression. We developed imiquimod-induced psoriasis-like mouse model to evaluate the effect of hypodermic injection of neutrophil-derived protease or its inhibitor. Histopathology and Western blotting were conducted for effect assessment.
Results: Purified CG cleaved and activated recombinant human FL-IL-36γ to promote CXCL-1 and CXCL-8 expression by human keratinocytes, and NETs activated FL-IL-36γ and the activation was inhibited by serpin A3. CG induced expression of a more truncated IL-36γ in psoriasiform lesion of mice and aggravated the psoriasis-like lesion induced by imiquimod, whereas recombinant serpin A3 alleviated the severity of the psoriasis-like mouse mode.
Conclusion: CG has the ability to cleave and activate IL-36γ and aggravate imiquimod-induced mouse psoriasiform lesion. Thus, CG-specific inhibitors might be promising therapeutic drugs for psoriasis.
Keywords: psoriasis, IL-36, neutrophil, cathepsin, elastase
Introduction
The vital role of IL-36 in the pathogenesis of psoriasis including psoriasis vulgaris (PV) and pustular psoriasis, especially generalized pustular psoriasis (GPP), has been extensively elucidated in recent years.1,2 IL-36 cytokines include three receptor agonists (IL-36α, IL-36β, IL-36γ) and a receptor antagonist (IL-36Ra), which use the same receptor complex IL-36R (IL-1Rrp2)/IL-1RAcP. In skin, IL-36 is predominantly expressed in epidermal keratinocytes,3 and IL-36R is widely expressed by keratinocytes and antigen presenting cells (APCs).4,5 IL-36 induces the expression of proinflammatory cytokines and chemokines by keratinocytes and drives APC activation to promote psoriatic inflammation.5
Both mRNA and protein expressions of all IL-36 cytokines were significantly higher in human PV lesions compared with healthy skin, where the expression of IL-36β was far weaker than IL-36α and IL-36γ in PV.6,7 Low-level IL-36β and increased expression of IL-36α, IL-36γ, and IL-36Ra were also seen in GPP lesions, and significantly greater expression of IL-36α and IL-36γ were found in GPP compared with PV lesions.2
As an IL-36R agonist, IL-36γ is now considered to be a valuable biomarker in psoriatic patients, which correlates closely with disease activity.8 Similar to IL-36α and IL-36β, IL-36γ is expressed as an inactive precursor, which needs to be proteolytically processed and activated, and neutrophil-derived proteases seemed to be potent activating enzymes of IL-36γ.9 Henry et al reported neutrophil elastase (NE) could cleave recombinant full-length (FL)-IL-36γ and activate the protein;10 however, Johnston et al found cathepsin G (CG) but not NE had the ability to activate IL-36γ in vitro, and NE could cleave FL-IL-36α.2
This study aims to further investigate the potential of CG and NE to activate IL-36γ in vitro and vivo.
Materials and methods
Study design
Human keratinocyte line HaCaT cells were purchased from KeyGEN BioTECH (Nanjing, China) and cultured as previously described11 and seeded in 6-well culture plates and divided into 15 groups as follows: control, 100 ng/mL recombinant human FL-IL-36γ (2320-IL-025, R&D Systems, Inc., Minneapolis, MN, USA), 100 ng/mL recombinant human truncated (T)-IL-36γ (6835-IL-010, R&D Systems, Inc.), 10 ng/mL recombinant human NE (R&D Systems, Inc.), 10 ng/mL NE + FL-IL-36γ, 100 ng/mL NE, 100 ng/mL NE + FL-IL-36γ, 1 μg/mL NE, 1 μg/mL NE + FL-IL-36γ, 10 ng/mL purified human CG (Enzo Life Sciences, Inc., Ann Arbor, MI, USA), 10 ng/mL CG + FL-IL-36γ, 100 ng/mL CG, 100 ng/mL CG + FL-IL-36γ, 1 μg/mL CG, 1 μg/mL CG + FL-IL-36γ. The cells were incubated for 24 hours, and then prepared for real-time quantitative PCR and ELISA. Experiments were repeated independently at least three times. Recombinant FL-IL-36γ was incubated with a neutrophil-derived protease in PBS for 1 hour at 37°C and then prepared for Western blotting.
Neutrophils were isolated from peripheral blood from healthy volunteers using ficoll and 3% dextran as described.12 Neutrophil extracellular traps (NETs) were generated by stimulating one million neutrophils in 0.5 mL RPMI without FBS with 20 nmol/L phorbol 12-myristate 13-acetate for 3 hours at 37°C.2 After washing with PBS for three times, 1 mL of 100 ng/mL FL-IL-36γ and/or 1 μg/mL recombinant serpin A3 (Enzo Life Sciences, Inc.) in PBS were incubated with the NETs for 1 hour at 37°C. The processed solutions were added to cultured HaCaT cells in 12-well culture plates for 24 hours. T-IL-36γ at 100 ng/mL was used as positive control. Real-time quantitative PCR was subsequently performed.
Thirty female BALB/c mice (6–8 weeks old) were purchased from animal core facility of Nanjing Medical University and kept under specific pathogen-free conditions. The mice were fed with standard laboratory water and food ad libitum and kept on a 12-hour light–dark cycle. All mice were randomly divided into six groups of five mice each as follow: control, imiquimod (Aldara, 3M Health Care Limited, Loughborough, UK), imiquimod + CG, imiquimod + serpin A3, CG, and serpin A3. Topical imiquimod 5% (31.25 mg) was applied on the right dorsal skin, and/or multipoint hypodermic injection with 0.1 mL PBS containing CG (200 ng) or serpin A3 (500 ng) was administered on the right dorsal skin daily. After being treated for 6 days, the mice were killed by cervical dislocation at Day 7, and right dorsal skin from each mouse was collected for Western blotting, and formalin fixed and paraffin embedded for H&E staining. The thickness of mouse epidermis was measured using Olympus cellSens, version 1.16.
Real-time quantitative PCR
After 24 hours of incubation, the supernatant was collected and prepared for ELISA, and the HaCaT cells attached to the culture plates were washed three times using PBS and then lysed in TRIzol (Zoonbio Biotechnology, Nanjing, Jiangsu, China), and total RNA was isolated. First-strand cDNA was synthesized from 2 μg of total RNA using Prime Script II First Strand cDNA Synthesis Kit (Takara Biotechnology (Dalian) Co. Ltd., Dalian, Liaoning, China) using Oligo dT Primer. PCR amplification was performed in a total volume of 20 μL containing 1 μL template cDNA and 10 μL SYBR Premix Ex Taq (SYBR Green) (Takara Biotechnology (Dalian) Co. Ltd.), and transcripts were quantified using StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA).13 All values were normalized to the expression of β-actin. Primer sequences are shown in Table 1.
 | Table 1 Primers for the target genes in real-time quantitative PCR |
ELISA
Measurement of secretory protein in supernatant was performed using CXCL-1 ELISA Kit (CUSABIO, Wuhan, Hubei, China) and CXCL-8 ELISA Kit (Proteintech, Wuhan, Hubei, China). This assay employs the quantitative sandwich enzyme immunoassay technique.
Western blotting
We incubated 20 μg recombinant human FL-IL-36γ with 20 μg CG in 5 mL PBS for 1 hour at 37°C, and then protein concentration in PBS was measured using the Bradford method, and SDS-PAGE performed. The primary antibody was anti-IL-36γ antibody (R&D Systems, Inc.).
The collected mouse dorsal skin was homogenized in cold lysis buffer containing protease inhibitor. Centrifugal separation was conducted at 4°C, at 14,000 rpm for 15 minutes. The upper layer of the solution was tested for protein as above mentioned. The primary antibody was added as below: anti-IL-36γ antibody (Cloud-Clone Corp., Wuhan, Hubei, China) and anti-β-actin antibody (Boster Biological Technology Co., Ltd, Wuhan, Hubei, China), following the manufacturer’s instructions. Gel-Pro 32 (Media Cybernetics, Rockville, MD, USA) was used to detect protein expression.
Statistical analyses
All data were analyzed using GraphPad Prism for Windows (GraphPad Software, San Diego, CA, USA) and presented as mean ± SD. Statistical significance was calculated using a Student’s t-test, Mann–Whitney U-test, or Friedman’s test, as appropriate.13 P<0.05 was defined as statistical significance.
Ethics statement
This study was carried out in accordance with the recommendations of institutional guidelines and Local Ethics Committee of the First Affiliated Hospital of Nanjing Medical University. Healthy volunteers were recruited for blood draws for neutrophil isolation and all subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocols including animal experiment were approved by the Local Ethics Committee of the First Affiliated Hospital of Nanjing Medical University. The institutional guidelines of the Animal Care and Use of Nanjing Medical University were followed for the welfare of the animals.
Results
Purified CG cleaves and activates recombinant human full-length-IL-36γ
We incubated recombinant FL-IL-36γ with different doses of purified CG or recombinant NE to stimulate HaCaT cells, and we found 100 ng/mL FL-IL-36γ alone had low activity to stimulate HaCaT cells, whereas 100 ng/mL CG used with FL-IL-36γ had significant synergistic effect on CXCL-1 and CXCL-8 mRNA expression in HaCaT cells (P<0.05; Figure 1A), which was confirmed at the protein level by ELISA analysis of supernatant (P<0.001; Figure 1B). T-IL-36γ had significantly higher activity compared with FL-IL-36γ (P<0.05). Either CG or NE alone activated HaCaT cells to varying degrees (Figure 1A). Western blot showed purified CG could cleave FL-IL-36γ from 18.7 to 17 KDa (Figure 1C).
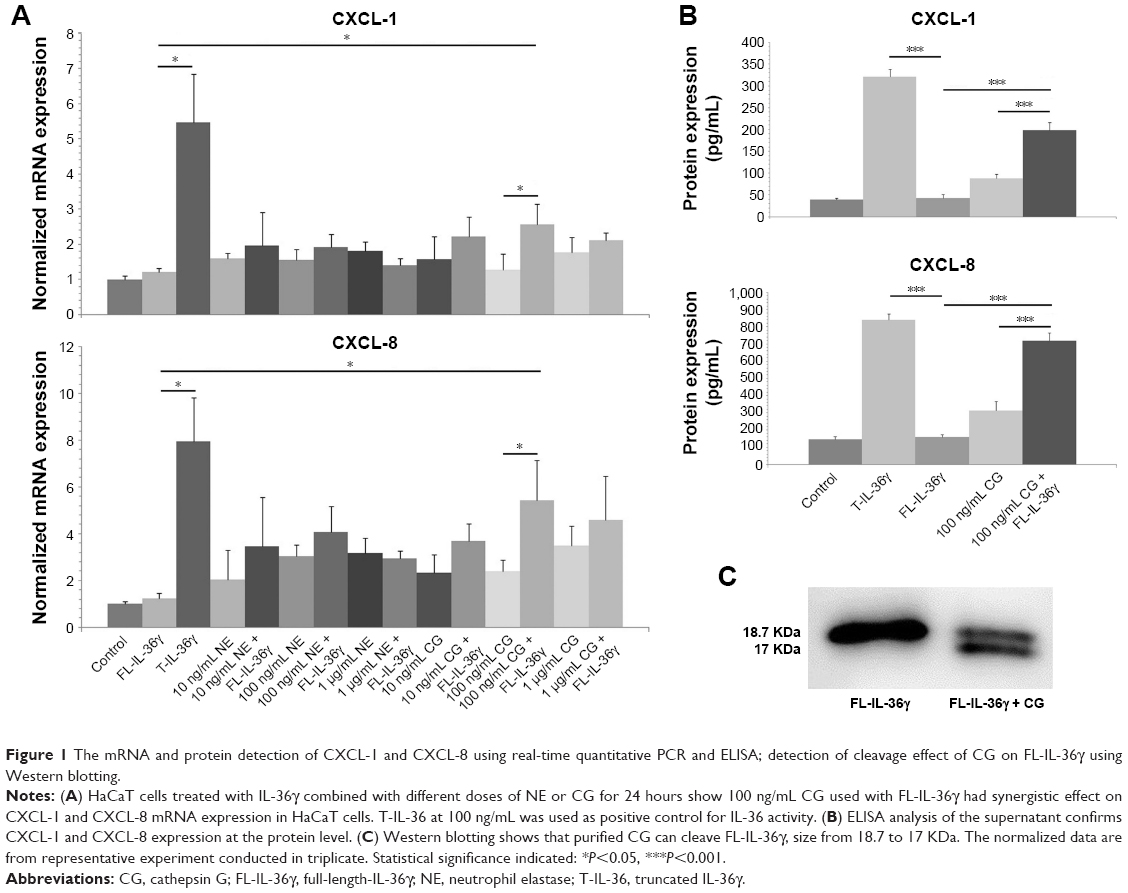 | Figure 1 The mRNA and protein detection of CXCL-1 and CXCL-8 using real-time quantitative PCR and ELISA; detection of cleavage effect of CG on FL-IL-36γ using Western blotting. |
NETs activate full-length-IL-36γ and the activation is inhibited by serpin A3
The DAPI staining of DNA confirmed the formation of NETs under fluorescent microscopy (Figure 2A and B). Exposure to NETs for 1 hour significantly increased the activity of FL-IL-36γ to induce CXCL-1 and CXCL-8 mRNA expression by HaCaT cells in comparison with untreated FL-IL-36γ (P<0.01). Serpin A3, a specific inhibitor of CG, inhibited significantly the action of NETs on FL-IL-36γ (P<0.01; Figure 2C).
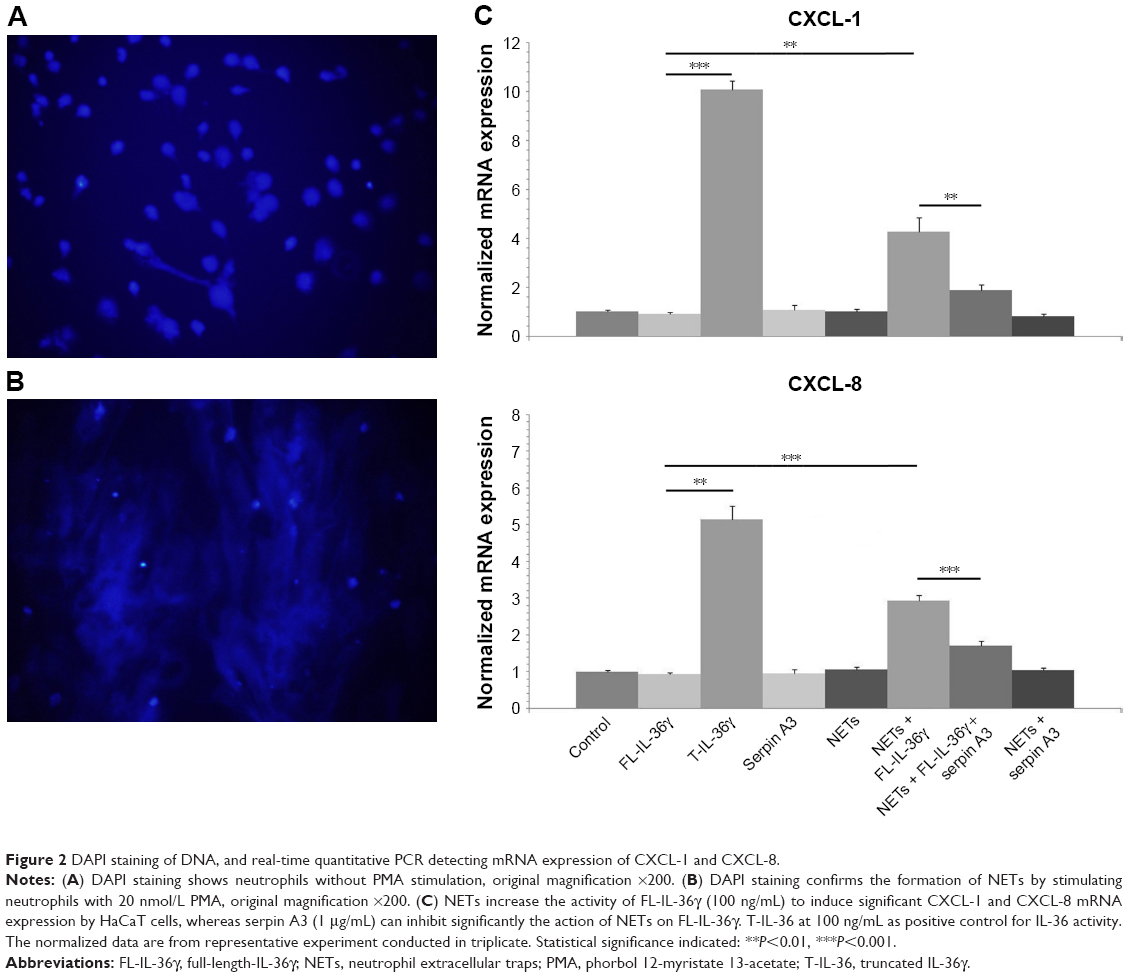 | Figure 2 DAPI staining of DNA, and real-time quantitative PCR detecting mRNA expression of CXCL-1 and CXCL-8. |
Purified CG aggravates psoriasis-like lesion induced by imiquimod whereas recombinant serpin A3 alleviates the severity of psoriasis-like mouse mode
Topical imiquimod successfully induced psoriasis-like mouse model, and hypodermic injection of purified CG or recombinant serpin A3 alone had no effect on skin appearance of mice (Figure 3A). Imiquimod used with CG induced severer psoriasis-like lesion compared with imiquimod alone, whereas serpin A3 relieved the psoriasiform lesion induced by imiquimod (Figure 3A).
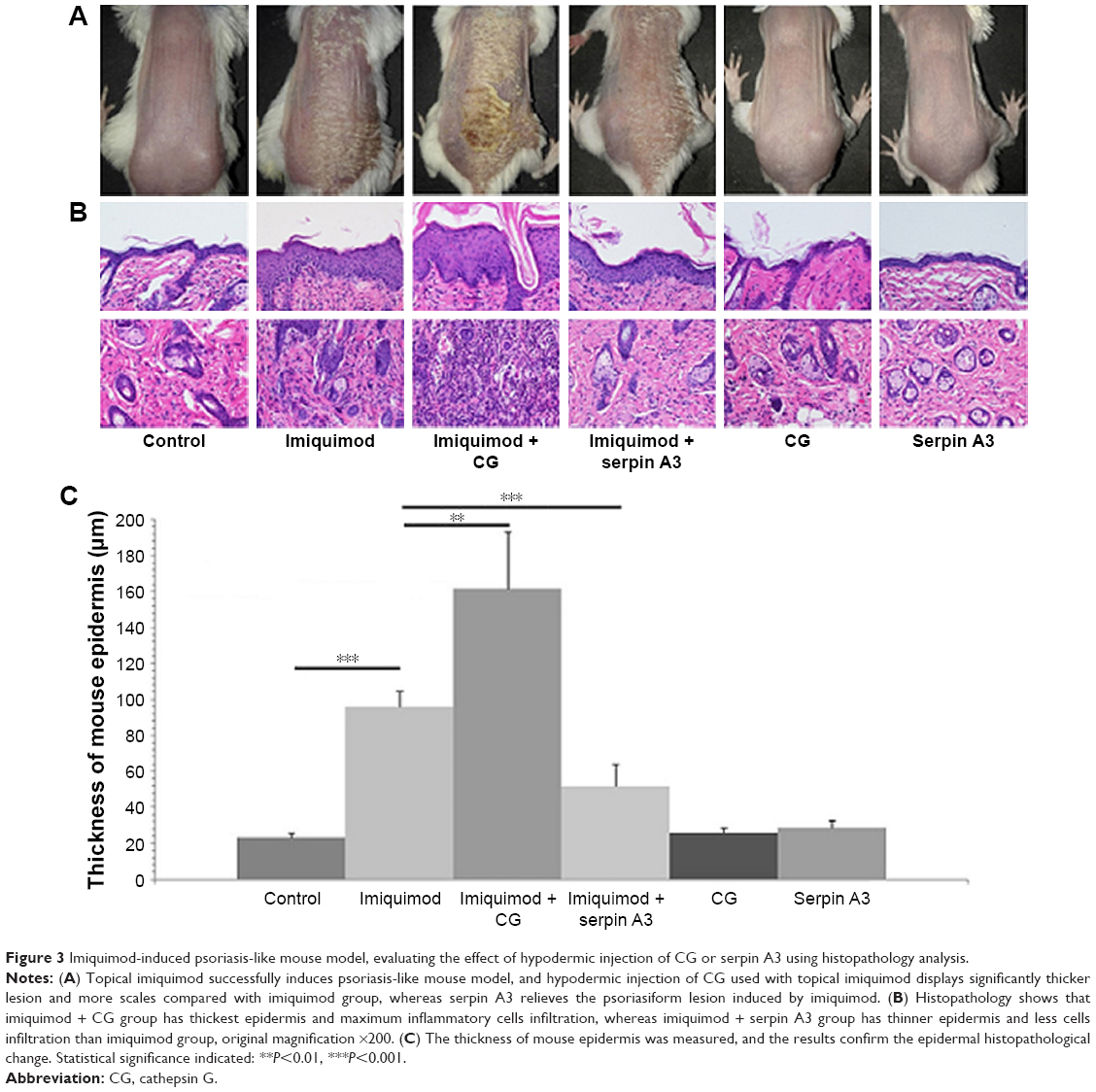 | Figure 3 Imiquimod-induced psoriasis-like mouse model, evaluating the effect of hypodermic injection of CG or serpin A3 using histopathology analysis. |
Imiquimod-induced psoriasis-like lesion had thicker epidermis and more dermal inflammatory cells infiltration than control group, whereas imiquimod used with CG induced more obvious change in both epidermis thickness and dermal cell infiltration compared to the imiquimod group; by contrast, serpin A3 alleviated the imiquimod-induced skin pathological changes in mice (Figure 3B and C).
Western blotting showed that CG promoted but serpin A3 inhibited the expression of truncated IL-36γ (16 KDa) in imiquimod-induced psoriasis-like lesion in mice (P<0.05; Figure 4A and B).
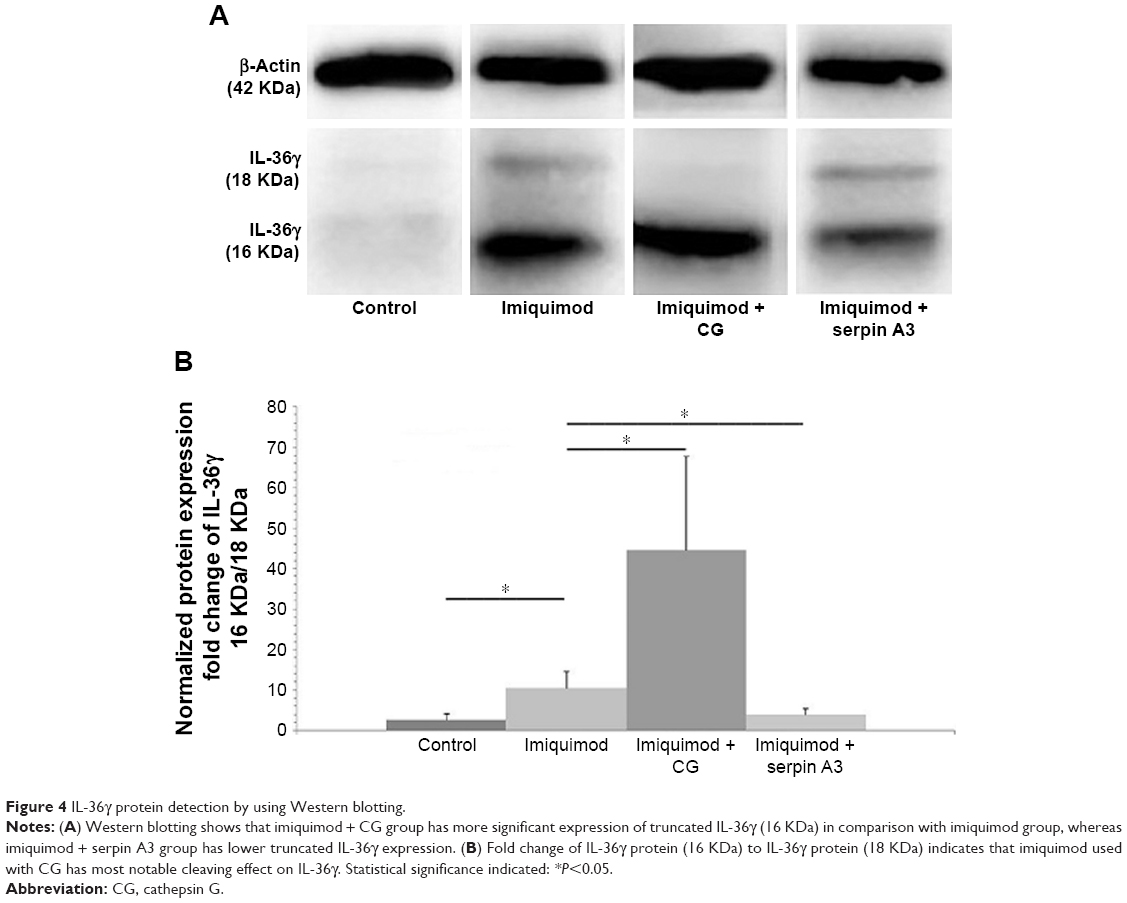 | Figure 4 IL-36γ protein detection by using Western blotting. |
Discussion
The IL-36 cytokines are involved in the pathogenesis of psoriasis where IL-36 can act on keratinocytes and immune cells to induce a powerful inflammatory response.14 IL-36 cytokines are important amplifiers of Th17 signaling and promising therapeutic targets for psoriasis.15 An amplification cycle is initiated by IL-17A, involving IL-36 cytokines and immunocyte-derived proteases; this results in formation of activated IL-36 cytokines that synergize with IL-17A, and this cycle might be related to the persistent state of psoriasis.16
IL-36γ is highly expressed in psoriasis lesions and plays a pivotal role in inflammation response and cell differentiation. IL-36γ induces inflammation and inhibits differentiation of keratinocytes through Wnt signaling pathway.17 IL-36γ signals through IL-36R and IL-1RAcP to activate the pathway leading to NF-κB and MAPKs.18 IL-36γ immunohistochemistry was found to be a useful marker in the histological differential diagnosis between psoriasis and eczema,8 and detection of IL-36γ protein through noninvasive tape stripping of lesion credibly differentiates psoriasis from atopic eczema.19
Keratinocytes secretes IL-36γ in an inactive form that needs to be processed by specific proteases to become bioactive.20 Cathepsin S was identified as the main IL-36γ-activating protease expressed by epithelial cells.21 Neutrophil-derived proteases, NE and CG, have been reported to have the ability to cleave and activate inactive IL-36γ.2,10,22 The diverse results from different studies are considered to possibly be concerned with dissimilar experiment design and condition. Our study has confirmed purified CG could cleave and activate inactive recombinant human FL-IL-36γ to induce significant CXCL-1 and CXCL-8 expression in HaCaT cells at the mRNA and protein levels, and CXCL-1 and CXCL-8 have strong neutrophil chemoattractant activity.
NETs are detected in both PV and GPP lesions, resulting from the programmed death of neutrophils that expose the NE, CG, and proteinase 3 to the extracellular milieu.2 Our study showed NETs could activate FL-IL-36γ to induce significant CXCL-1 and CXCL-8 mRNA expression by HaCaT cells, whereas the activation was inhibited by serpin A3, a specific inhibitor of CG, which suggests that the inhibitor of IL-36γ processing could be considered as a potential anti-inflammatory agent via suppressing activation of the latter cytokines.
Most studies of imiquimod-induced lesions are performed using female BALB/c or C57BL/6 (B6) mice, and expression of IL-36α, IL-36β, and IL-36γ was elevated in imiquimod-treated B6 mice and female BALB/c mice,23,24 which are both suitable for our study on protease processing of IL-36. Swindell et al reported that imiquimod increased IL-17 cytokine expression in B6 mice but not BALB/c mice;23 however, other studies showed that IL-17A expression was induced by imiquimod with a faster response in BALB/c mice than B6 mice, and BALB/c lesions develop more rapidly than B6 lesions.25,26 Milora et al reported that unprocessed IL-36α could cooperate with IL-1α to promote psoriasis-like disease, using an imiquimod-induced mouse model on B6 background.27 Our study showed hypodermic injection of purified CG aggravated imiquimod-induced psoriasis-like lesion in female BALB/c mice, which displayed thicker lesion and more scales in appearance and presented thicker epidermis and more dermal inflammatory cells infiltration in histopathology. Western blotting confirmed that CG could induce more truncated IL-36γ expression in psoriasiform lesion. Conversely, hypodermic injection of serpin A3 significantly relieved the severity of imiquimod-induced mice lesion.
Conclusion
Taken together, our study has confirmed that CG has the ability to cleave and activate IL-36γ to promote CXCL-1 and CXCL-8 expression by human keratinocytes, and CG can aggravate imiquimod-induced mouse psoriasiform lesion, and contrarily serpin A3 alleviates the severity of the mouse model. Neutrophil-derived CG proteolytically processes inactive IL-36γ and then induces neutrophil chemokines secretion from keratinocytes that further recruits surrounding neutrophils, which is a typical amplification cycle very likely involved in the pathogenesis of psoriasis. The specific inhibitors of CG might be promising therapeutic drugs for psoriasis.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81673062), the Youth Medical Talent of Empowering Medicine through Science and Education Program, and the Professionals from Six-Pronged Top-Talent Program (LGY2018052).
Disclosure
The authors report no conflicts of interest in this work.
References
Baliwag J, Barnes DH, Johnston A. Cytokines in psoriasis. Cytokine. 2015;73(2):342–350. | ||
Johnston A, Xing X, Wolterink L, et al. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol. 2017;140(1):109–120. | ||
Gresnigt MS, van de Veerdonk FL. Biology of IL-36 cytokines and their role in disease. Semin Immunol. 2013;25(6):458–465. | ||
Kumar S, McDonnell PC, Lehr R, et al. Identification and initial characterization of four novel members of the interleukin-1 family. J Biol Chem. 2000;275(14):10308–10314. | ||
Foster AM, Baliwag J, Chen CS, et al. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J Immunol. 2014;192(12):6053–6061. | ||
Johnston A, Xing X, Guzman AM, et al. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol. 2011;186(4):2613–2622. | ||
Li B, Tsoi LC, Swindell WR, et al. Transcriptome analysis of psoriasis in a large case-control sample: RNA-seq provides insights into disease mechanisms. J Invest Dermatol. 2014;134(7):1828–1838. | ||
D’Erme AM, Wilsmann-Theis D, Wagenpfeil J, et al. IL-36γ (IL-1F9) is a biomarker for psoriasis skin lesions. J Invest Dermatol. 2015;135(4):1025–1032. | ||
Sullivan GP, Henry CM, Clancy DM, et al. Suppressing IL-36-driven inflammation using peptide pseudosubstrates for neutrophil proteases. Cell Death Dis. 2018;9(3):378. | ||
Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep. 2016;14(4):708–722. | ||
Zhou BR, Zhang JA, Zhang Q, et al. Palmitic acid induces production of proinflammatory cytokines interleukin-6, interleukin-1β, and tumor necrosis factor-α via a NF-κB-dependent mechanism in HaCaT keratinocytes. Mediators Inflamm. 2013;2013:530429. | ||
Clark RA, Nauseef WM. Isolation and functional analysis of neutrophils. Curr Protoc Immunol. 2001. | ||
Yin L, Hu Y, Xu J, Guo J, Tu J, Yin Z. Ultraviolet B inhibits IL-17A/TNF-α-Stimulated activation of human dermal fibroblasts by decreasing the expression of IL-17RA and IL-17RC on fibroblasts. Front Immunol. 2017;8:91. | ||
Bassoy EY, Towne JE, Gabay C. Regulation and function of interleukin-36 cytokines. Immunol Rev. 2018;281(1):169–178. | ||
Capon F. The genetic basis of psoriasis. Int J Mol Sci. 2017;18(12):2526. | ||
Pfaff CM, Marquardt Y, Fietkau K, Baron JM, Lüscher B. The psoriasis-associated IL-17A induces and cooperates with IL-36 cytokines to control keratinocyte differentiation and function. Sci Rep. 2017;7(1):15631. | ||
Wang W, Yu X, Wu C, Jin H. IL-36γ inhibits differentiation and induces inflammation of keratinocyte via Wnt signaling pathway in psoriasis. Int J Med Sci. 2017;14(10):1002–1007. | ||
Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem. 2004;279(14):13677–13688. | ||
Berekméri A, Latzko A, Alase A, Macleod T, Ainscough JS, Laws P. Detection of IL-36y via non-invasive tape stripping reliably discriminates psoriasis from atopic eczema. J Allergy Clin Immunol. 2018;142(3):988–991. | ||
Towne JE, Renshaw BR, Douangpanya J, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J Biol Chem. 2011;286(49):42594–42602. | ||
Ainscough JS, Macleod T, McGonagle D, et al. Cathepsin S is the major activator of the psoriasis-associated proinflammatory cytokine IL-36γ. Proc Natl Acad Sci U S A. 2017;114(13):E2748–E2757. | ||
Sullivan GP, Davidovich PB, Sura-Trueba S, et al. Identification of small-molecule elastase inhibitors as antagonists of IL-36 cytokine activation. FEBS Open Bio. 2018;8(5):751–763. | ||
Swindell WR, Michaels KA, Sutter AJ, et al. Imiquimod has strain-dependent effects in mice and does not uniquely model human psoriasis. Genome Med. 2017;9(1):24. | ||
Tortola L, Rosenwald E, Abel B, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest. 2012;122(11):3965–3976. | ||
van der Fits L, Mourits S, Voerman JS, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182(9):5836–5845. | ||
Flutter B, Nestle FO. TLRs to cytokines: mechanistic insights from the imiquimod mouse model of psoriasis. Eur J Immunol. 2013;43(12):3138–3146. | ||
Milora KA, Fu H, Dubaz O, Jensen LE. Unprocessed Interleukin-36α regulates psoriasis-like skin inflammation in cooperation with interleukin-1. J Invest Dermatol. 2015;135(12):2992–3000. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.