")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Baicalin provides neuroprotection in traumatic brain injury mice model through Akt/Nrf2 pathway
Authors Fang J, Wang H , Zhou J, Dai W, Zhu Y, Zhou Y, Wang X, Zhou M
Received 28 January 2018
Accepted for publication 30 May 2018
Published 10 August 2018 Volume 2018:12 Pages 2497—2508
DOI https://doi.org/10.2147/DDDT.S163951
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Jiang Fang,1 Handong Wang,1 Jian Zhou,2 Wei Dai,2 Yihao Zhu,3 Yuan Zhou,2 Xiaoliang Wang,3 Mengliang Zhou3
1Department of Neurosurgery, Jinling Hospital, School of Medicine, Southeast University, Nanjing, China; 2Department of Neurosurgery, Jinling Hospital, Jinling Clinical Medical College, Nanjing Medical University, Nanjing, China; 3Department of Neurosurgery, Jinling Hospital, School of Medicine, Nanjing University, Nanjing, China
Background: The neuroprotective effects of Baicalin have been confirmed in several central nervous system (CNS) diseases. However, its possible effect on traumatic brain injury (TBI) model is still not clear. The present study is aimed to investigate the role and the underling mechanisms of 7-d-glucuronic acid-5,6-dihydroxyflavone (Baicalin) on TBI model.
Methods: The weight-drop model of TBI in Institute of Cancer Research mice was treated with Baicalin intraperitoneally at 30 minutes after TBI. LY294002 (LY) (a commonly used PI3K/Akt pathway inhibitor) was injected into the left ventricle at 30 minutes before TBI. All mice were euthanized at 24 hours after TBI to collect the brain tissue for a series of tests except for neurological function, which was measured at 2 hours and 1 and 3 days post-TBI.
Results: Baicalin administration significantly improved neurobehavioral function, alleviated brain edema, and reduced apoptosis-positive cells by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay accompanied with the upregulation of B-cell lymphoma 2 (Bcl-2) and downregulation of Bcl-2-associated X protein (Bax) and cleaved-caspase 3 by Western blot. Besides, TBI-induced oxidant stress status was also restored in the Baicalin group by measuring malondialdehyde (MDA) content, glutathione peroxidase (GPx), and superoxide dismutase (SOD) levels in the injured brain cortex. Furthermore, translocation of Nrf2 to the nucleus was dramatically enhanced by Baicalin verified by immunofluorescence and Western blot analyses. Accordingly, its downstream antioxidative enzymes nicotinamide adenine dinucleotide phosphate:quinine oxidoreductase 1 (NQO-1) and heme oxygenase 1 (HO-1) were also activated by Baicalin confirmed by quantitative reverse transcription polymerase chain reaction (RT-qPCR) and Western blot. However, cotreatment with Baicalin and LY could partly abolish Baicalin-induced activation of Nrf2 and its neuroprotective effects in TBI.
Conclusion: This study demonstrates that Baicalin provides a neuroprotective effect in TBI mice model via activating the Akt/Nrf2 pathway.
Keywords: Baicalin, traumatic brain injury, Akt, nuclear factor erythroid 2-related factor 2, apoptosis, oxidative stress
Introduction
Traumatic brain injury (TBI) has been a major health and socioeconomic problem with high morbidity and mortality.1,2 Secondary brain injury was triggered after the irreversible primary brain injury following TBI, including glutamate excitotoxicity, calcium overload, oxidative stress, and inflammatory response, leading to the worse outcome for TBI patients.3–5 Among these processes, oxidative stress is a major contributor to the pathogenesis of the secondary brain injury.6 The excessive production of reactive oxygen species (ROS), resulted from exhaustion of antioxidant enzymes, could damage cellular components such as proteins, lipids, and DNA, exacerbating neuronal apoptosis.7 Therefore, it is vital to find an efficient approach to alleviate oxidative stress caused by TBI in clinical patients.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a member of the basic leucine zipper (bZIP) family of transcription factors.8 It regulates a series of antioxidant and cytoprotective factors in response to oxidative stress.9 Under normal conditions, Nrf2 locates in the cytoplasm and binds to its inhibitor, Kelch-like ECH-associated protein 1 (Keap1), which is associated with actin cytoskeleton and promotes Nrf2 to the proteasomal degradation. However, once activated, Nrf2 is released from Keap1 and translocates into the nucleus.10 By binding to the antioxidant response element (ARE) in the promoter of antioxidant genes, Nrf2 exhibits a powerful antioxidative effect via enhancing the transcription of antioxidative Phase II enzymes such as nicotinamide adenine dinucleotide phosphate:quinine oxidoreductase 1 (NQO-1) and heme oxygenase 1 (HO-1).11,12
The phosphatidylinositol 3-kinase (PI3K)/Akt is a well-known pathway in promoting neuronal survival.13,14 Activation of Akt pathway has been proved to be an effective strategy to provide therapeutic benefits against TBI.15,16 Besides, the activated Akt protein could phosphorylate several prosurvival factors including Nrf2 by facilitating its translocation to the nucleus.17
7-D-Glucuronic acid-5,6-dihydroxyflavone (Baicalin) is a major bioactive flavone isolated from the radix of Scutellaria baicalensis, which is widely used in traditional Chinese medicine for clinical treatment.18 It can freely cross the blood–brain barrier19 and provide neuroprotection in a variety of central nervous system (CNS) diseases according to previous studies, including cerebral ischemia,20 spinal cord injury,21 and subarachnoid hemorrhage.22 More importantly, recent studies have demonstrated that Baicalin could protect the brain from ketamine-induced neurotoxicity in rats through the activation of PI3K/Akt23 and attenuates subarachnoid hemorrhagic brain injury through Nrf2 pathway in mice.22 However, no literature has investigated its role on Nrf2 and Akt pathways in TBI model. This inspired the authors to explore whether Baicalin could ameliorate brain injury after TBI through protective pathways of Nrf2 and Akt.
Methods
Animals
Male Institute of Cancer Research mice (6–8 weeks, 28–32 g) were housed under controlled environmental conditions with a 12-hour light/dark cycle and were given free access to food and water. The animals were purchased from Animal Experimental Centre of Nanjing Medical University, Jiangsu, China. All procedures involving animals were approved by the Animal Care and Use Committee of Southeast University, Nanjing, China, and conformed to the Guide for Care and Use of Animals of Laboratory by the National Institutes of Health (NIH).
Model of TBI
The TBI model was induced using a modified weight-drop model previously described by Flierl et al.24 Briefly, mice were anesthetized with 10% chloral hydrate (4 mL/kg, ip) and then placed onto the platform of weight-drop apparatus. A longitudinal midline scalp incision (2.0–2.5 cm) was made to expose the bony skull. The area to be impacted lies on the left frontal skull (1.5 mm lateral to the midline on the mid-coronal plane). After confirming the correct impact location again, the skull was impacted by a 200 g of weight releasing from the height of 2.5 cm. Then, the scalp was closed and the mice were returned to quondam cages. Mice in the Sham group were subjected to the same procedures except the weight drop.
Groups and drug administration
All mice were randomly assigned to the following groups: sham, TBI, TBI + vehicle (V), and TBI + Baicalin (involving three subgroups: 50, 100, and 150 mg/kg). The dose of Baicalin injection used in this study was based on previous studies performed on the cerebral ischemia models.20,25 Baicalin (Sigma-Aldrich Co., St Louis, MO, USA) or equivalent volumes of saline were administered intraperitoneally in the TBI + Baicalin or TBI + V groups for 30 minutes after TBI. The mice used for Neurological Severity Score (NSS) tests were injected with Baicalin or equivalent volumes of saline 30 minutes and every 24 hours after TBI. To further confirm the role of Akt pathway in the neuroprotection of Baicalin on TBI, it was attempted to apply commonly used PI3K/Akt inhibitor, LY294002 (LY) (Sigma-Aldrich Co.). Groups were set as follows: sham, TBI + V, TBI + Baicalin (100 mg/kg), TBI + Baicalin + LY (500 nmol), and TBI + LY (500 nmol). A 5 μL solution of LY was injected into the left ventricle (bregma; 1.0 mm lateral, 0.3 mm posterior, and 2.6 mm deep) for 30 minutes before TBI according to previous studies.26,27 All the mice of each group were euthanized at 24 hours post-TBI except neurobehavioral evaluation.
Neurobehavioral evaluation
The neurological impairment was evaluated using the NSS at 2 hours and 1 and 3 days after TBI as previously described.7,24 This method consists of 10 separated tasks on motor function, alertness, and physiological behavior. One point is awarded for inability to perform each task, no point for succeeding. The higher score represents the more serious neurological impairment. All mice were trained the tasks at 1 day before TBI. Evaluation of tasks was carried out by an investigator who blinded about animal groups.
Brain water content
A dry–wet weight method was used to measure the brain water content.28 The brain was removed from anesthetized mice at 24 hours after TBI. The brain stem, cerebellum, and right cerebral hemispheres were removed, and the remaining left cerebral hemispheres were weighed immediately to obtain the wet weight (WW). Then, the hemispheres were dried at 72°C for 72 hours and the dry weight (DW) was measured. The brain water content was calculated as: (WW − DW)/WW ×100%.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) immunofluorescence
TUNEL assay was performed to detect apoptosis-positive cells using a TUNEL staining kit (Hoffman-La Roche Ltd., Basel, Switzerland) as previously described.29 Briefly, all brain samples were sliced (6 μm) and incubated with TUNEL reaction mixture for 2 hours at room temperature in the dark, then washed with PBS prior to be incubated with DAPI for 5 minutes. All slides were covered by microscopic cover glass with antifluorescence quenching sealant after three washes in PBS. The area surrounding the injury epicenter was detected, and the positive cells were identified and counted by an investigator who blinded to the animal groups in random six microscopic fields (400× magnification) in each section. The apoptotic index was defined as the ratio of the number of TUNEL-positive cells to DAPI positive cells. Four sections of each mouse were used to calculate the final average of the apoptotic index, which was regarded as the data of each sample.
Western blot analysis
Ipsilateral-injured cerebral cortex was collected at 24 hours after TBI. Nuclear and cytoplasmic proteins were isolated by the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology, Shanghai, China) according to manufacturer’s instructions. Total protein extraction was acquired by the Total Protein Extraction Kit (Beyotime Biotechnology) according to kit instructions. The proteins were dissolved in the loading buffer (Beyotime Biotechnology) and subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by transfer to polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked in blocking buffer (5% freshly prepared milk–Tris-buffered saline with Tween 20) for 2 hours at indoor temperature and then incubated overnight at 4°C with the following primary antibodies: Nrf2 (1:1,000; Abcam, Cambridge, MA, USA), H3 (1:1,000; Bioworld Technology, St Louis, MO, USA), Bcl-2 (1:200; Santa Cruz Biotechnology Inc., Dallas, TX, USA), Bax (1:200; Santa Cruz Biotechnology Inc.), cleaved-caspase 3 (1:1,000; Cell Signaling Technology, Beverly, MA, USA), GPx1 (1:1,000; Abcam), SOD1 (1:200; Santa Cruz Biotechnology Inc.), NQO-1 (1:1,000; Abcam), HO-1 (1:200; Santa Cruz Biotechnology Inc.), β-actin (1:5,000; Bioworld Technology), p-Akt(Ser473) (1:1,000; Cell Signaling Technology), and Akt(pan) (1:1,000; Cell Signaling Technology). After washing the membranes with Tris-buffered saline with Tween 20 three times, they were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (1:6,000; Bioworld Technology) for 1 hour at room temperature. The signal was visualized using the ECL substrate (EMD Millipore). The optical density of protein bands was measured by ImageJ software (NIH), with normalization to β-actin or histone 3.
Malondialdehyde (MDA) level, superoxide dismutase (SOD), and glutathione peroxidase (GPx) activities
MDA content, SOD activity, and GPx activity were determined as previously described.10 The injured cerebral cortex samples were homogenized in 2 mL of PBS (10 mM) solution and then centrifuged at 10,000× g for 25 minutes at 4°C to reduce cell debris. The numerical results were obtained by a spectrophotometer using commercial kits (Nanjing Jiancheng Biology Engineering Institute, Nanjing, China) according to manufacturer’s instructions. Protein concentration was determined via bicinchoninic acid (BCA) assay (Beyotime Biotechnology). The activities of MDA, SOD, and GPx were expressed as nanomole per milligram of protein, unit per milligram of protein, and unit per milligram of protein, respectively.
Immunofluorescence staining
Brain tissue samples were fixed in 4% formalin, embedded in paraffin, and cut into 6 μm sections. The sections were treated with blocking buffer (10% normal goat serum in PBS) for 1 hour at room temperature and then incubated with Nrf2 (1:100; Abcam) overnight at 4°C. After being washed with PBS three times, the sections were incubated with appropriate secondary antibodies (Alexa Fluor 594, 1:200) for 2 hours at room temperature. After three washes, the slides were incubated with NeuN (1:100; EMD Millipore) for 3 hours, then counterstained with DAPI for 5 minutes, rinsed with PBS, and coverslipped with mounting medium. Fluorescence was captured on a Zeiss HB050 inverted microscope system and analyzed by the Image-Pro Plus 6.0 software (Media Cybernetics, Sarasota, FL, USA). The specificity of the immunofluorescence reaction was evaluated by replacement of the primary antibody with PBS.
Quantitative reverse transcription polymerase chain reaction (RT-qPCR)
RT-qPCR was performed as previously decribed.29 Total RNA was isolated from the injury cortex with the TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) at 24 hours after TBI. Primer sequences used in our study were as follows: NQO-1 forward and reverse primers were 5′-ACGACAACGGTCCTTTCCAGA-3′ and 5′-CAGAAACGCAGGATGCCACT-3′; HO-1 forward and reverse primers were 5′-GCTGGTGATGGCTTCCTTGTA-3′ and 5′-ACCTCGTGGAGACGCTTTACAT-3′; β-actin forward and reverse primers were 5′-GTGACGTTGACATCCGTAAAGA-3′ and 5′-GTAACAGTCCGCCTAGAAGCAC-3′.
RT-qPCR analysis was performed using the SYBR Green qRT-PCR kit (Takara Biotechnology, Kyoto, Japan) according to the manufacturer’s instructions. All samples were analyzed in triplicate with normalization to the β-actin.
Statistical analysis
All data are expressed as mean ± SD and evaluated by the one-way ANOVA followed by Tukey’s post hoc tests except the NSS, which was analyzed by two-way ANOVA with repeated measures followed by Bonferroni post-tests. The SPSS 19.0 software (IBM Corporation, Armonk, NY, USA) was used for the statistical analysis. P<0.05 was considered statistically significant.
Results
Baicalin alleviated TBI-induced neurological deficits and brain edema
To assess the effects of Baicalin on TBI mice model and determine the optimum dose of Baicalin, animal groups were set as follows: sham, TBI, TBI + V, TBI + Baicalin (50, 100, and 150 mg/kg) groups. The scores in the sham group were almost zero at different time points, and no difference was detected between the TBI and TBI + V groups. Thus, the data of scores in the sham and TBI groups were not shown. There were no significant differences among all groups at 2 hours after TBI (Figure 1A). However, at 1 and 3 days post-TBI, the mice in the Baicalin (100 mg/kg)-treated group had significant lower NSS than those in the V-treated group (P<0.01 vs <0.001, respectively). Meanwhile, the lower does (50 mg/kg) failed to provide a neuroprotective effect (P>0.05) and the larger dose (150 mg/kg) did not exhibit a better result than 100 mg/kg dose group.
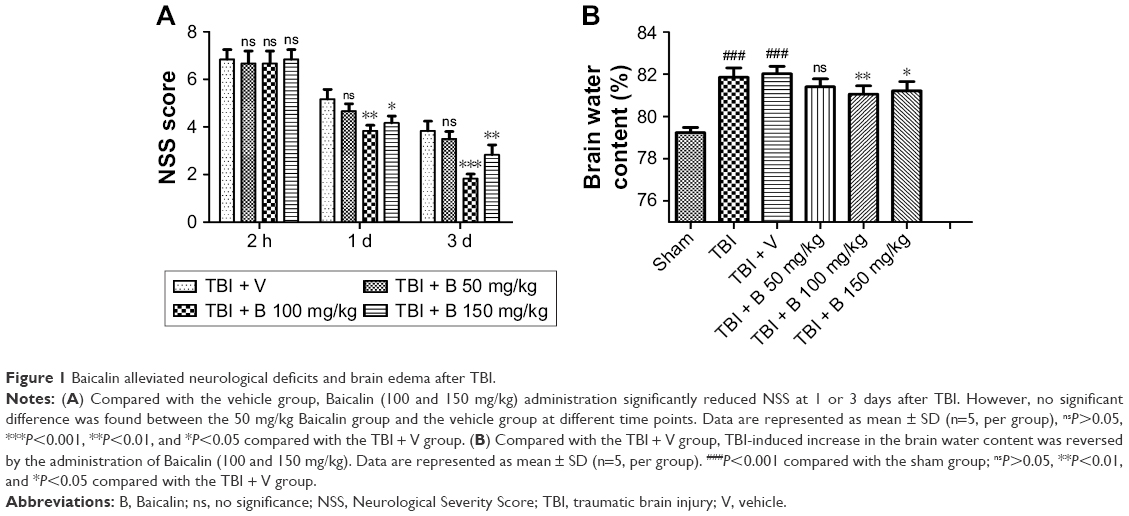 | Figure 1 Baicalin alleviated neurological deficits and brain edema after TBI. |
Then, the brain water content was examined to confirm the protective effect of Baicalin at 24 hours post-TBI. Compared with the sham group, the brain water content was dramatically increased in the TBI and TBI + V groups (P<0.001 and <0.001) as shown in Figure 1B. However, they were significantly reduced by treatment with Baicalin at 100 and 150 mg/kg doses (P<0.01 and <0.05). The lower does (50 mg/kg) failed to alleviate TBI-induced brain edema as before. The results are consistent with the NSS data, indicating that Baicalin was a neuroprotective agent, and the 100 mg/kg dose was appropriate for mice in this study. Therefore, 100 mg/kg dose was used for subsequent studies.
Baicalin suppressed neuronal apoptosis after TBI
To further investigate the potential mechanism of neuroprotective effects by Baicalin, we used TUNEL immunofluorescence to explore the role of Baicalin on neuronal apoptosis at 24 hours after TBI (Figure 2A). The apoptotic index increased significantly in the TBI and TBI + V groups (P<0.001 and <0.001, respectively) as illustrated in Figure 2B. However, it was remarkably decreased after treatment with Baicalin (P<0.001). Afterward, several apoptotic indicators were examined: B-cell lymphoma 2 (Bcl-2) (Figure 2C), Bcl-2-associated X protein (Bax) (Figure 2D), and cleaved-caspase 3 (Figure 2E) to further verify the antiapoptotic effect of Baicalin. The achieved results revealed that proapoptotic proteins (Bax and cleaved-caspase 3) were increased (P<0.01 and <0.001), and antiapoptotic protein (Bcl-2) was decreased (P<0.01) after TBI. However, Baicalin reversed the changes, indicating that protein levels of cleaved-caspase 3 (P<0.05) and Bax (P<0.05) were downregulated and the expression of Bcl-2 was upregulated (P<0.05) compared with the TBI + V group (Figure 2C–E). In other words, Baicalin elevated the ratio of Bcl-2/Bax and decreased cleaved-caspase 3 after TBI. The results indicated that Baicalin possesses the antiapoptotic property in the TBI mice model.
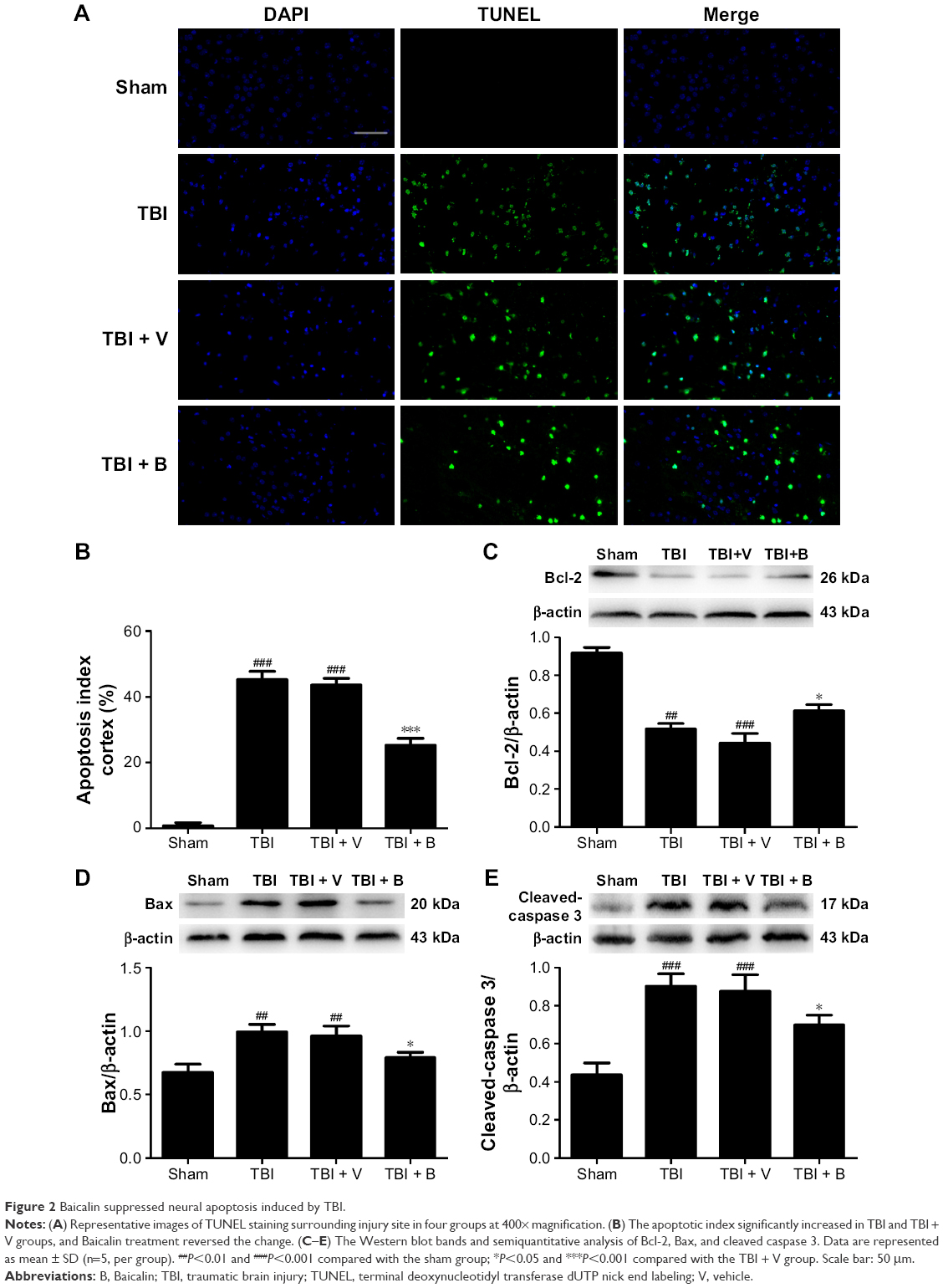 | Figure 2 Baicalin suppressed neural apoptosis induced by TBI. |
Baicalin alleviated oxidative stress in the injured cortex
To determine whether the antiapoptotic property of Baicalin was associated with the ability to restore the oxidative stress, we examined the MDA level as an indicator of lipid peroxidation and the activities of GPx and SOD as the reflections of endogenous antioxidant enzyme activity levels in the injured brain tissues at 24 hours after TBI. Observations showed that a significant increase in MDA was detected in the TBI and TBI + V groups compared with the sham group (P<0.01 and <0.01) as depicted in Figure 3A. However, Baicalin could markedly reduce TBI-induced high MDA level (P<0.05). Inversely, GPx and SOD were decreased after TBI (P<0.001 and <0.001) (Figure 3B and C), while Baicalin could significantly enhance their activities (P<0.01 and <0.05). Besides, we further measured the protein levels of GPx1, an important member of GPx family functions in the detoxification of hydrogen peroxide30 and SOD1, one of the three SODs responsible for reducing free superoxide radicals.31 The changes in their protein levels were consistent with activities of GPx and SOD (Figure 3D and E). These data demonstrated that Baicalin increased antioxidant enzyme activities accompanied with elevated expression of their protein levels. Thus, we infer that Baicalin has antioxidative effects against TBI in the mice model.
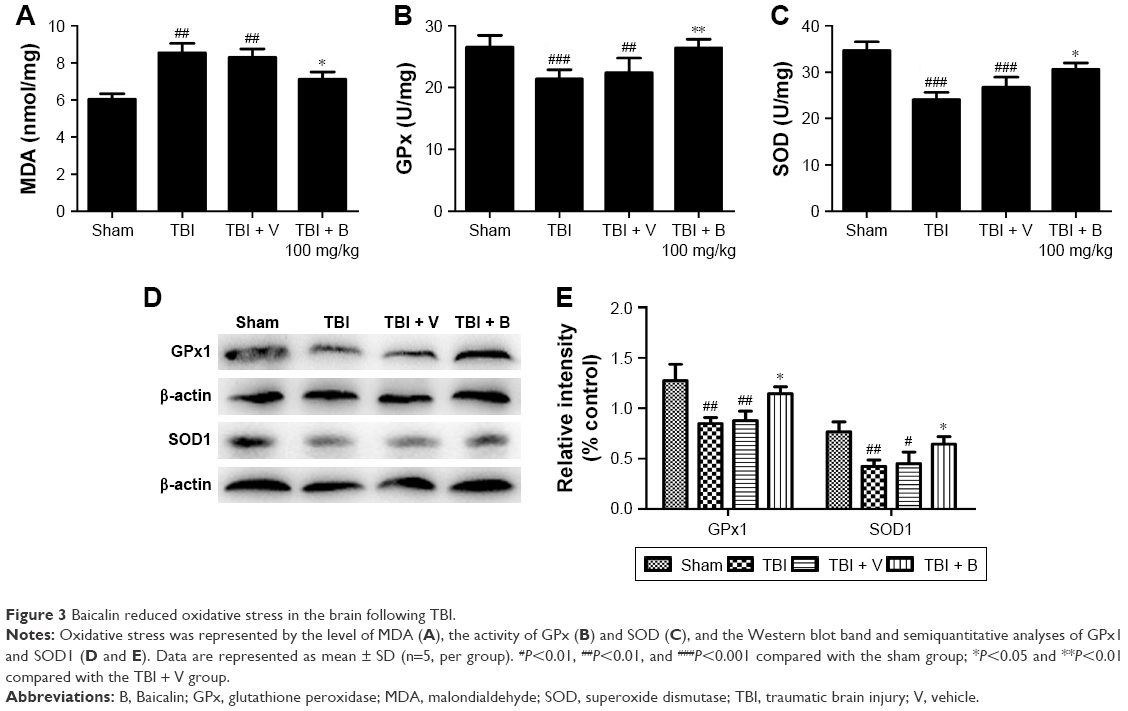 | Figure 3 Baicalin reduced oxidative stress in the brain following TBI. |
Baicalin enhanced the translocation of Nrf2 from cytoplasm to nucleus after TBI
Since Nrf2 is a well-known factor in restoring oxidative stress in the brain,32 we hypothesized that the antioxidative ability of Baicalin might be associated with Nrf2 pathway. Thus, it was attempted to investigate the expression of Nrf2 at 24 hours after TBI by Western blot. The results showed that TBI remarkably increased the nuclear Nrf2 and decreased the cytoplasmic Nrf2 compared with the sham group (Figure 4A and B). Moreover, when compared with the TBI + V group, the Baicalin-treated group increased the nuclear Nrf2 and reduced the cytoplasmic Nrf2 (P<0.01 and <0.05), but no significant difference was observed between the two groups (P>0.05) (Figure 4C), which indicated that Baicalin could further promote Nrf2 nuclear translocation without increase in total Nrf2. These results were also confirmed by the immunofluorescence (Figure 4D). Nrf2 expression in the sham-injured group was predominately detected in the cytoplasm compared with the TBI or V-treated group. The translocation of Nrf2 from the cytoplasm to the nucleus could be obviously observed after TBI insult. Moreover, more Nrf2 immunoreactivity was detected in cell nuclei in the Baicalin-treated group compared with the V group.
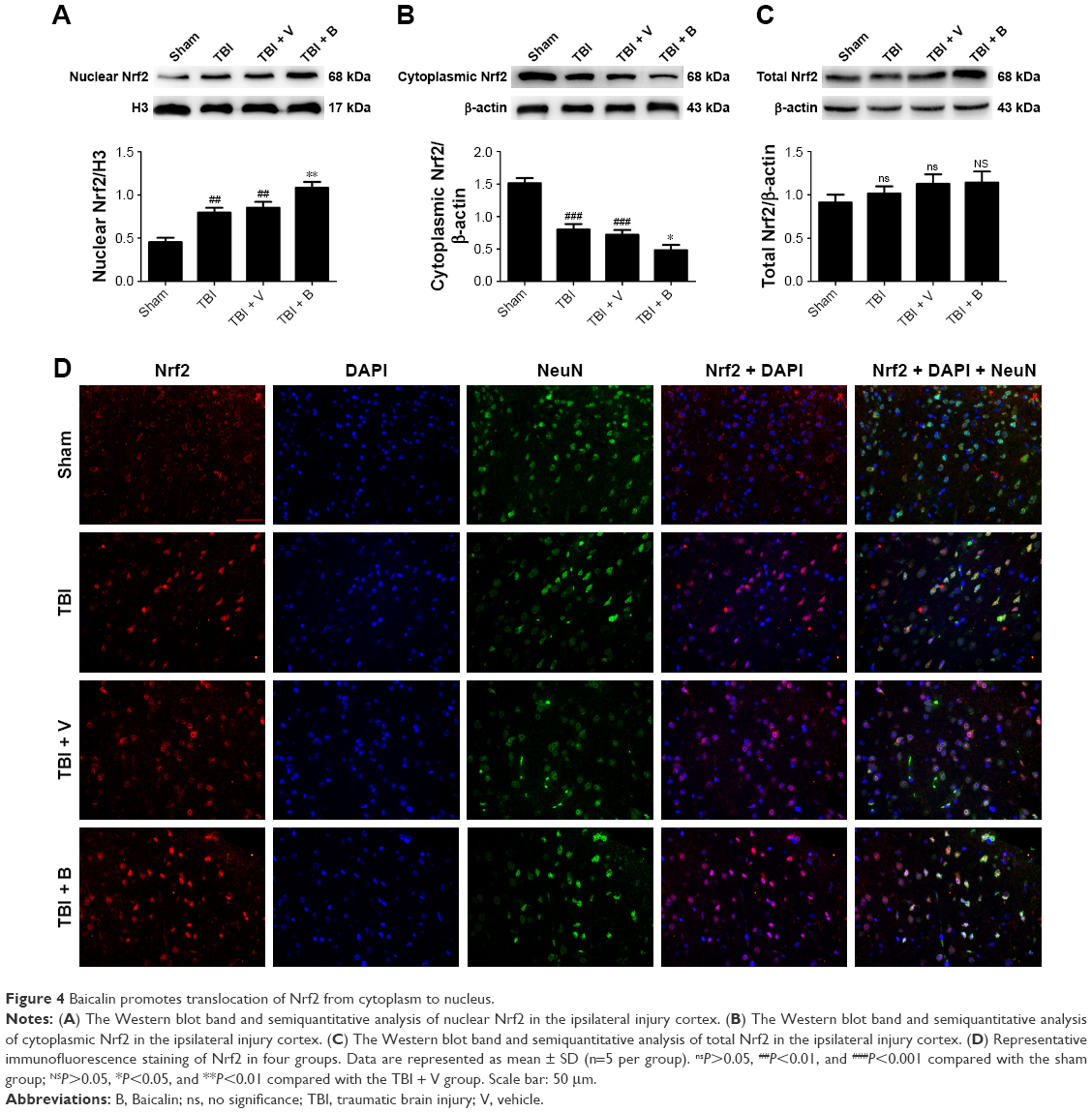 | Figure 4 Baicalin promotes translocation of Nrf2 from cytoplasm to nucleus. |
Baicalin upregulates the expression of Nrf2 downstream proteins
It is clear that the mechanism of Nrf2 to reduce oxidative stress is mostly through transcriptional regulation of its downstream antioxidant genes such as HO-1 and NQO-1.7 Therefore, it was hypothesized that Baicalin could upregulate the two well-known Nrf2 downstream enzymes, thus providing antioxidative neuroprotection. RT-qPCR and Western blot were used to measure mRNA and protein levels of HO-1 and NQO-1. RT-qPCR results showed that both HO-1 and NQO-1 mRNA were increased in the TBI group (P<0.001 and <0.01) (Figure 5A and B). Moreover, Baicalin further enhanced their expressions compared with the TBI + V group (P<0.01 and <0.01). Consistent with mRNA variations, Western blot results also demonstrated that Baicalin could upregulate the expressions of HO-1 and NQO-1 in protein level as compared with the TBI + V group (P<0.05 and <0.01) as shown in Figure 5C and D. These results indicated that Baicalin induced the expression of HO-1 and NQO-1 at transcription and translation levels.
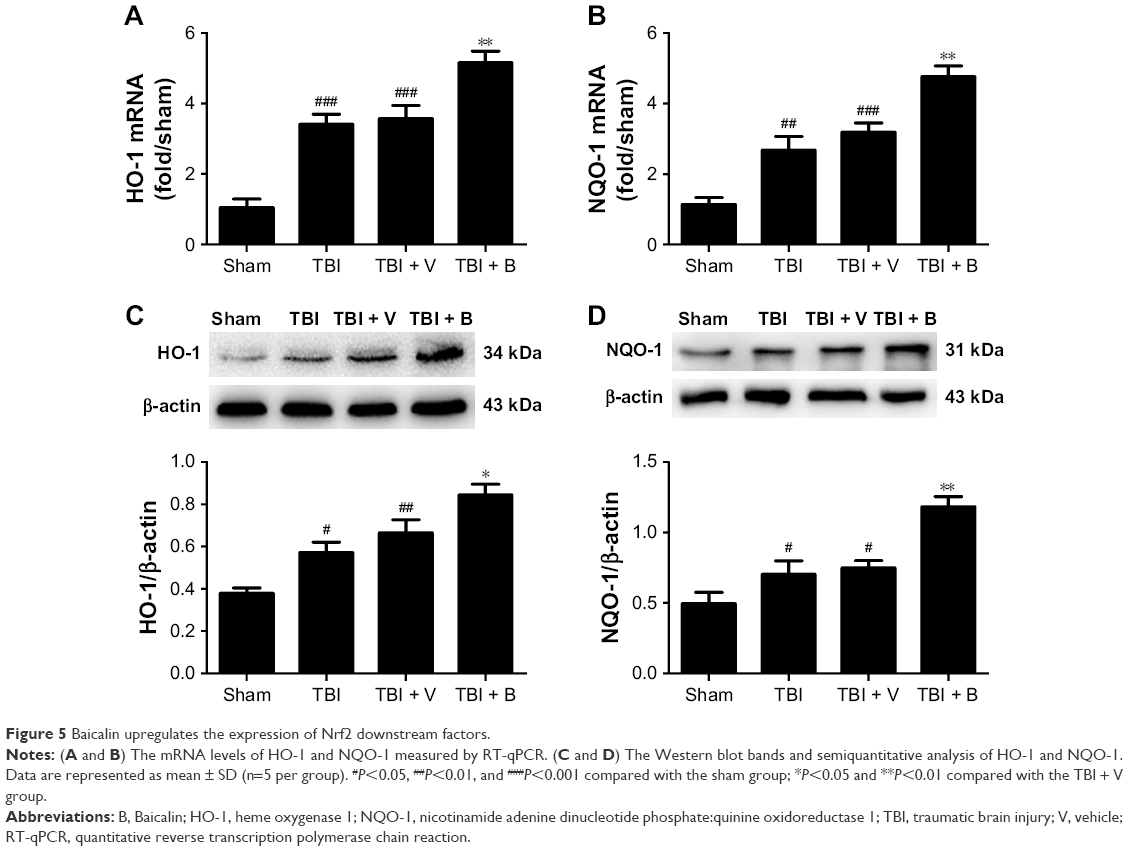 | Figure 5 Baicalin upregulates the expression of Nrf2 downstream factors. |
Baicalin activates Nrf2 pathway and provides neuroprotection through modulation of Akt pathway
Based on the results mentioned earlier, we used a specific PI3K/Akt inhibitor to determine the role of Akt in the activation of Nrf2 pathway by Baicalin. The results showed a significant increase in phosphorylation Akt and nuclear Nrf2 in the Baicalin-treated group compared with the V-treated group (P<0.05 and <0.05) (Figure 6A and B). When combined with LY and Baicalin, the variations were blocked, namely, both expressions of p-Akt and nuclear Nrf2 were decreased (P<0.01 and <0.01). Conversely, Baicalin reduced the cytoplasmic Nrf2 when compared with the V group (P<0.05) (Figure 6A and B), while the changes were also reversed by treatment with LY (P<0.05). The results showed that Baicalin-induced Nrf2 translocation from cytoplasm to nucleus in TBI mice model required activation of Akt pathway.
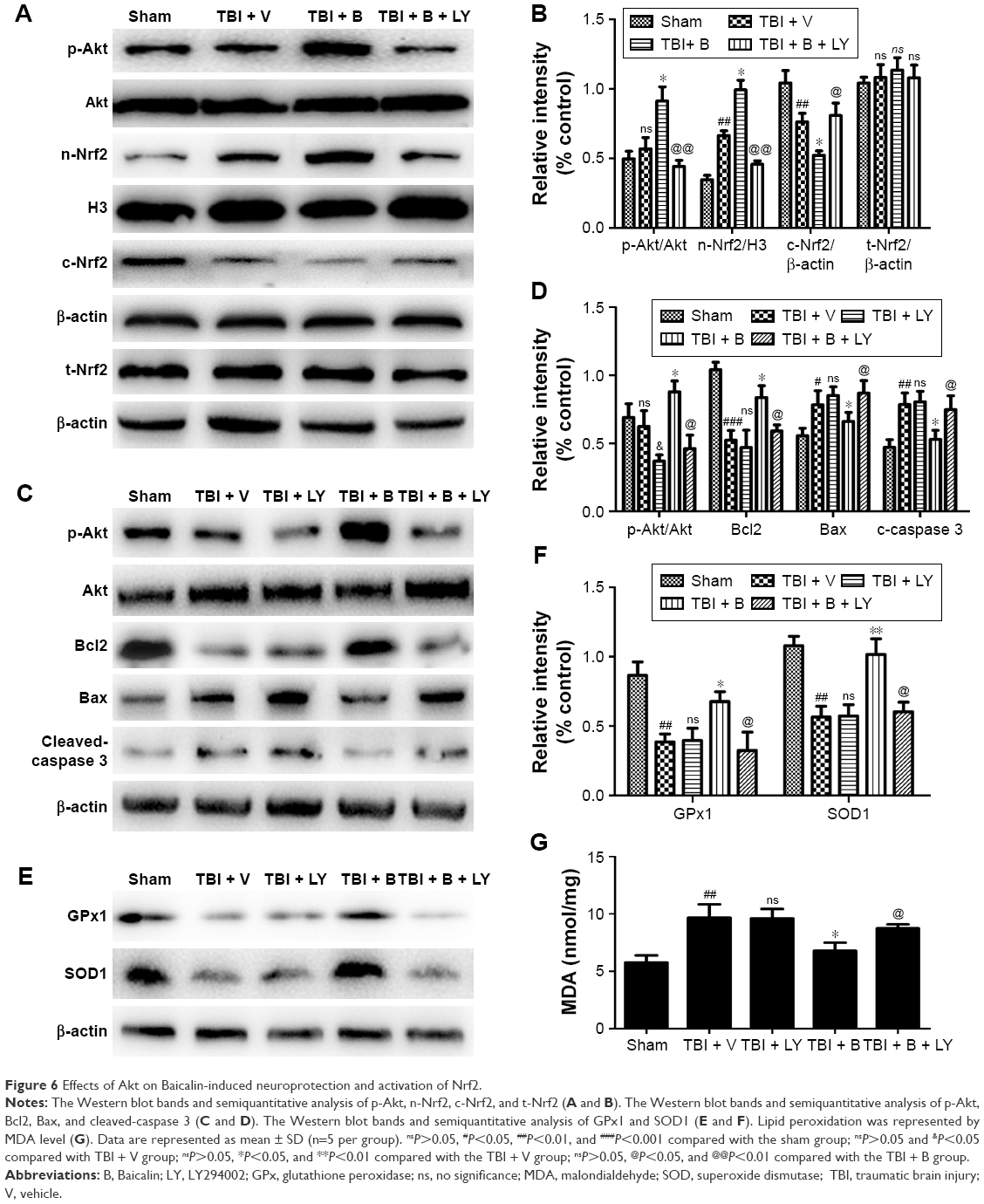 | Figure 6 Effects of Akt on Baicalin-induced neuroprotection and activation of Nrf2. |
In addition, we measured Bcl2, Bax, cleaved-caspase 3, GPx1, SOD1, and MDA levels to determine whether Baicalin-induced antiapoptotic and antioxidative properties through Nrf2 pathway involved with Akt pathway. Figure 6C reveals that Baicalin-induced antiapoptotic properties (including increased Bcl2 level and decreased bax and cleaved-caspase 3 levels) were reversed by co-treatment with Baicalin and LY (P<0.05, <0.05, and <0.05) (Figure 6C and D). Figure 6E demonstrates Baicalin-induced antioxidative properties (including increased SOD1 and GPx1 levels and decreased MDA level) were abrogated by co-treatment with Baicalin and LY (P<0.05, <0.05, and P<0.05) (Figure 6E–G). Besides, LY did not have significant effects on the antiapoptotic and antioxidative indicators when comparing the TBI + V group with the TBI + LY group (Figure 6C–G). Thus, we inferred that LY abrogated Baicalin-induced neuroprotection by the inactivation of Nrf2 and not by decreased p-Akt.
From the above, we demonstrated that Baicalin provides neuroprotection in TBI mice model through the activation of Akt/Nrf2 pathway.
Discussion
To the best of our knowledge, this is the first study exploring the protective effects of Baicalin in TBI mice model. In this study, we confirmed the neuroprotection of Baicalin in mice after TBI and investigated its potential mechanism. The main results are summarized as follows: 1) Baicalin provided neuroprotection after TBI, specifically, it improved neurological function and alleviated cerebral edema; 2) Baicalin inhibited TBI-induced neuronal apoptosis and oxidative stress; 3) Baicalin activated Nrf2 pathway as well as its downstream antioxidants, HO-1 and NQO-1 following TBI; 4) LY abrogated Baicalin-induced activation of Nrf2, antiapoptotic and antioxidative effects in TBI mice. Based on these results, we concluded that Baicalin protected the brain against TBI through Akt/Nrf2 pathway.
Brain edema increased the intracranial pressure and reached peak at 24 hours after TBI, which is a major death cause in the early stage of TBI patients.33 It has been shown that Baicalin could alleviate brain edema in subarachnoid hemorrhage22 and cerebral ischemia model.34 Consistent with the previous studies, we demonstrated that Baicalin treatment dramatically reduced TBI-induced brain edema accompanied with a significant improvement in neurological deficits. These data proved that Baicalin was a protective agent in TBI model. In addition, Baicalin treatment with the dose of 50 or 150 mg/kg did not exhibit a better effect than that of 100 mg/kg. We speculated that the protective dose–response curve of Baicalin in TBI mice model might be U shaped.
Oxidative stress, resulted from the overproduction of ROS and the exhaustion of endogenous antioxidative enzymes such as SOD and GPx, significantly exacerbates neuronal apoptosis in the pathological process of the secondary brain injury following TBI.29 It is not only because the excessive amounts of ROS causing cellular lipid peroxidation, protein nitration, and DNA damages,6,35 but also because the brain is vulnerable to the oxidative damages due to its high content of lipid and consumption of oxygen.36 In this study, we determined MDA content in injury brain tissues as an indicator of lipid peroxidation, it increased immediately following injury and remained high for 24–48 hours.37 SOD and GPx levels, the indicators of the endogenous antioxidative capacities, were also measured at 24 hours after TBI. They were both major intracellular antioxidant enzymes that convert peroxides into nontoxic forms, and their activities underwent a descent and minimum 24 hours postinjury.38,39 In this study, we disclosed that Baicalin treatment remarkably reduced MDA content and increased GPx and SOD activities accompanied with elevated expressions of GPx1 and SOD1 in injury brain tissues compared with the V group at 24 hours post-TBI, reflecting that Baicalin owned an antioxidant ability in the TBI mice model. Besides, the antiapoptotic effects of Baicalin sere also demonstrated with a substantial evidence including lower apoptotic index, elevated ratio of Bcl-2/Bax, and downregulation of cleaved-caspase 3 in the Baicalin-treated group. Although the exact mechanism of antiapoptotic property has not been fully illustrated, it was inferred that it might be related to the ability to restore the oxidative stress, therefore preventing mitochondria dysfunction from TBI.
Nrf2, as a key translate factor in the maintenance of cellular redox balance against oxidative stress in TBI, has been widely studied in the past 10 years.40 Loads of evidence revealed that it is beneficial for TBI when activated in a classical pattern of translocation from cytoplasm to nucleus, thereby inducing its downstream ARE-containing genes such as NQO-1 and HO-1.28 Moreover, recent studies in our laboratory have demonstrated that some flavonoid compounds such as quercetin and luteolin could further enhance the translocation of Nrf2 and provide neuroprotection in the TBI model.41,42 Baicalin is also a bioactive flavone with a powerful antioxidative characteristic in the brain in vivo22,25,43 and in vitro.44 The property has been demonstrated to be associated with the activation of Nrf2 pathway in several experiments, such as Aβ1–42-induced learning and memory deficit,43 a rat ischemic stroke model,45 and a vitro model of 6-OHDA-induced neurotoxicity in PC12 cells.46 Therefore, we hypothesized that the Nrf2 pathway was involved in the antioxidative neuroprotection by Baicalin in the TBI mice model. The findings were consistent with our hypothesis, showing that Baicalin could also enhance the Nrf2 translocation from the cytoplasm to the nucleus as well as its targeting downstream antioxidative enzymes, NQO-1 and HO-1, at transcriptional and translational levels.
It has been shown that Nrf2 could be regulated by the PI3K/Akt pathway.32 Under the oxidative stress status, the activated Akt signaling pathway resulted in the depolymerization of actin microfilaments, thereby facilitating Nrf2 translocation to the nucleus.17 Besides, Nrf2 downstream antioxidative proteins: NQO-1 and HO-1 were also reported to be induced by p-Akt in a previous study.47 In addition, recently, reports have demonstrated that Baicalin could activate Akt pathway in rats against ketamine-induced neurotoxicity23 and hypoxic–ischemic injury.48 Therefore, it was attempted to investigate the p-Akt to determine its variations with Baicalin treatment in TBI and whether it was contributed to the activation of Nrf2. Interestingly, the achieved data were in line with previous studies, demonstrating that both p-Akt and nuclear Nrf2 were upregulated by Baicalin after TBI. However, when combined with LY, Baicalin-induced activation of Nrf2, the antiapoptotic and antioxidative properties were abrogated. According to these results, we concluded that the neuroprotective effects of Baicalin in TBI through Nrf2 pathway required the activation of Akt.
However, there are some limitations in the present study. First, some other Nrf2 upstream factors (eg, p38MAPK, JNK, and protein kinase C) also have significant impacts on Nrf2-ARE pathway.32 Further study should investigate whether these signaling pathways also involve the Baicalin-induced Nrf2 activation. Second, since Baicalin is a multitarget drug with a variety of protective effects such as antioxidative and anti-inflammatory properties,22,49 we should consider if its neuroprotection in our study is related to its anti-inflammation or other properties. Third, a single injection of Baicalin at 30 minutes after TBI is not practical in the clinical treatment. Whether multiple treatments of Baicalin with different time courses could exhibit a better effect against TBI should be determined in the further study.
Conclusion
This study demonstrated that Baicalin attenuated brain injury against TBI by improving neurological function and reducing brain edema, apoptosis, and oxidative stress with the activation of Akt/Nrf2 pathways. The obtained data in this study make Baicalin as an attractive therapeutic agent against TBI in clinical treatment in the future.
Acknowledgments
This study was grant supported by the National Natural Science Foundation of China (nos 81672503, 81571162, and 81401026), Military Medical Youth Training Program (no 16QNP042), and the Project of Hospital Management of Nanjing General Hospital of Nanjing Military Region (no 2017013).
Disclosure
The authors report no conflicts of interest in this work.
References
Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7(8):728–741. | ||
Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21(5):375–378. | ||
Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab. 2004;24(2):133–150. | ||
Cornelius C, Crupi R, Calabrese V, et al. Traumatic brain injury: oxidative stress and neuroprotection. Antioxid Redox Signal. 2013;19(8):836–853. | ||
Bell JD, Park E, Ai J, Baker AJ. PICK1-mediated GluR2 endocytosis contributes to cellular injury after neuronal trauma. Cell Death Differ. 2009;16(12):1665–1680. | ||
Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med. 2008;45(4):443–452. | ||
Ding H, Wang H, Zhu L, Wei W. Ursolic acid ameliorates early brain injury after experimental traumatic brain injury in mice by activating the Nrf2 pathway. Neurochem Res. 2017;42(2):337–346. | ||
Li W, Jain MR, Chen C, et al. Nrf2 possesses a redox-insensitive nuclear export signal overlapping with the leucine zipper motif. J Biol Chem. 2005;280(31):28430–28438. | ||
Owuor ED, Kong AN. Antioxidants and oxidants regulated signal transduction pathways. Biochem Pharmacol. 2002;64(5–6):765–770. | ||
Ding K, Wang H, Xu J, et al. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. | ||
de Vries HE, Witte M, Hondius D, et al. Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic Biol Med. 2008;45(10):1375–1383. | ||
Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36(10):1208–1213. | ||
Frebel K, Wiese S. Signalling molecules essential for neuronal survival and differentiation. Biochem Soc Trans. 2006;34(Pt 6):1287–1290. | ||
Ali T, Kim T, Rehman SU, et al. Natural dietary supplementation of anthocyanins via PI3K/Akt/Nrf2/HO-1 pathways mitigate oxidative stress, neurodegeneration, and memory impairment in a mouse model of Alzheimer’s disease. Mol Neurobiol. 2018;55(7):6076–6093. | ||
Shen M, Wang S, Wen X, et al. Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed Pharmacother. 2017;95:885–893. | ||
Gao Y, Li J, Wu L, et al. Tetrahydrocurcumin provides neuroprotection in rats after traumatic brain injury: autophagy and the PI3K/AKT pathways as a potential mechanism. J Surg Res. 2016;206(1):67–76. | ||
Kang KW, Lee SJ, Park JW, Kim SG. Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2-related factor 2 through actin rearrangement in response to oxidative stress. Mol Pharmacol. 2002;62(5):1001–1010. | ||
Xi YL, Li HX, Chen C, et al. Baicalin attenuates high fat diet-induced insulin resistance and ectopic fat storage in skeletal muscle, through modulating the protein kinase B/Glycogen synthase kinase 3 beta pathway. Chin J Nat Med. 2016;14(1):48–55. | ||
Tarrago T, Kichik N, Claasen B, Prades R, Teixido M, Giralt E. Baicalin, a prodrug able to reach the CNS, is a prolyl oligopeptidase inhibitor. Bioorg Med Chem. 2008;16(15):7516–7524. | ||
Tu XK, Yang WZ, Shi SS, Wang CH, Chen CM. Neuroprotective effect of baicalin in a rat model of permanent focal cerebral ischemia. Neurochem Res. 2009;34(9):1626–1634. | ||
Cao Y, Li G, Wang YF, et al. Neuroprotective effect of baicalin on compression spinal cord injury in rats. Brain Res. 2010;1357:115–123. | ||
Shi X, Fu Y, Zhang S, Ding H, Chen J. Baicalin attenuates subarachnoid hemorrhagic brain injury by modulating blood-brain barrier disruption, inflammation, and oxidative damage in mice. Oxid Med Cell Longev. 2017;2017:1401790. | ||
Zuo D, Lin L, Liu Y, et al. Baicalin attenuates ketamine-induced neurotoxicity in the developing rats: involvement of PI3K/Akt and CREB/BDNF/Bcl-2 pathways. Neurotox Res. 2016;30(2):159–172. | ||
Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E. Mouse closed head injury model induced by a weight-drop device. Nat Protoc. 2009;4(9):1328–1337. | ||
Cao Y, Mao X, Sun C, et al. Baicalin attenuates global cerebral ischemia/reperfusion injury in gerbils via anti-oxidative and anti-apoptotic pathways. Brain Res Bull. 2011;85(6):396–402. | ||
Tang G, Yang H, Chen J, et al. Metformin ameliorates sepsis-induced brain injury by inhibiting apoptosis, oxidative stress and neuroinflammation via the PI3K/Akt signaling pathway. Oncotarget. 2017;8(58):97977–97989. | ||
Du G, Zhao Z, Chen Y, et al. Quercetin attenuates neuronal autophagy and apoptosis in rat traumatic brain injury model via activation of PI3K/Akt signaling pathway. Neurol Res. Epub 2016 Oct 3:1–8. | ||
Liu Z, Wang H, Shi X, et al. DL-3-n-butylphthalide (NBP) provides neuroprotection in the mice models after traumatic brain injury via Nrf2-ARE signaling pathway. Neurochem Res. 2017;42(5):1375–1386. | ||
Yang Y, Wang H, Li L, et al. Sinomenine provides neuroprotection in model of traumatic brain injury via the Nrf2-ARE pathway. Front Neurosci. 2016;10:580. | ||
Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830(5):3289–3303. | ||
Kojima T, Dogru M, Ibrahim OM, et al. Effects of oxidative stress on the conjunctiva in Cu, Zn-superoxide dismutase-1 (Sod1)-knockout mice. Invest Ophthalmol Vis Sci. 2015;56(13):8382–8391. | ||
Lee JS, Surh YJ. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005;224(2):171–184. | ||
Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22(5):E1. | ||
Tu XK, Yang WZ, Liang RS, et al. Effect of baicalin on matrix metalloproteinase-9 expression and blood-brain barrier permeability following focal cerebral ischemia in rats. Neurochem Res. 2011;36(11):2022–2028. | ||
Adibhatla RM, Hatcher JF. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2010;12(1):125–169. | ||
Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7(1):51–61. | ||
Hou Z, Luo W, Sun X, et al. Hydrogen-rich saline protects against oxidative damage and cognitive deficits after mild traumatic brain injury. Brain Res Bull. 2012;88(6):560–565. | ||
Wang HC, Lin YJ, Shih FY, et al. The role of serial oxidative stress levels in acute traumatic brain injury and as predictors of outcome. World Neurosurg. 2016;87:463–470. | ||
Ansari MA, Roberts KN, Scheff SW. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma. 2008;25(5):513–526. | ||
Yan W, Wang HD, Hu ZG, Wang QF, Yin HX. Activation of Nrf2-ARE pathway in brain after traumatic brain injury. Neurosci Lett. 2008;431(2):150–154. | ||
Li X, Wang H, Gao Y, et al. Protective effects of quercetin on mitochondrial biogenesis in experimental traumatic brain injury via the Nrf2 signaling pathway. PLoS One. 2016;11(10):e0164237. | ||
Xu J, Wang H, Ding K, et al. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic Biol Med. 2014;71:186–195. | ||
Ding H, Wang H, Zhao Y, Sun D, Zhai X. Protective effects of baicalin on abeta(1)(-)(4)(2)-induced learning and memory deficit, oxidative stress, and apoptosis in rat. Cell Mol Neurobiol. 2015;35(5):623–632. | ||
Zheng WX, Wang F, Cao XL, et al. Baicalin protects PC-12 cells from oxidative stress induced by hydrogen peroxide via anti-apoptotic effects. Brain Inj. 2014;28(2):227–234. | ||
Zhang Q, Wang J, Zhang C, et al. The components of Huang-Lian-Jie-Du-Decoction act synergistically to exert protective effects in a rat ischemic stroke model. Oncotarget. 2016;7(49):80872–80887. | ||
Zhang Z, Cui W, Li G, et al. Baicalein protects against 6-OHDA-induced neurotoxicity through activation of Keap1/Nrf2/HO-1 and involving PKCalpha and PI3K/AKT signaling pathways. J Agric Food Chem. 2012;60(33):8171–8182. | ||
Li L, Dong H, Song E, Xu X, Liu L, Song Y. Nrf2/ARE pathway activation, HO-1 and NQO1 induction by polychlorinated biphenyl quinone is associated with reactive oxygen species and PI3K/AKT signaling. Chem Biol Interact. 2014;209:56–67. | ||
Zhou ZQ, Li YL, Ao ZB, et al. Baicalin protects neonatal rat brains against hypoxic-ischemic injury by upregulating glutamate transporter 1 via the phosphoinositide 3-kinase/protein kinase B signaling pathway. Neural Regen Res. 2017;12(10):1625–1631. | ||
He P, Wu Y, Shun J, Liang Y, Cheng M, Wang Y. Baicalin ameliorates liver injury induced by chronic plus binge ethanol feeding by modulating oxidative stress and inflammation via CYP2E1 and NRF2 in mice. Oxid Med Cell Longev. 2017;2017:4820414. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.