")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Autophagy facilitates lung adenocarcinoma resistance to cisplatin treatment by activation of AMPK/mTOR signaling pathway
Authors Wu T , Wang M, Jing L, Liu Z, Guo H, Liu Y, Bai Y, Cheng Y, Nan K, Liang X, Ma X
Received 2 September 2015
Accepted for publication 10 November 2015
Published 14 December 2015 Volume 2015:9 Pages 6421—6431
DOI https://doi.org/10.2147/DDDT.S95606
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Tao Wu,1 Min-Cong Wang,1 Li Jing,1 Zhi-Yan Liu,2 Hui Guo,1 Ying Liu,3 Yi-Yang Bai,1 Yang-Zi Cheng,1 Ke-Jun Nan,1 Xuan Liang1
1Department of Medical Oncology, The First Affiliated Hospital of Xi’an Jiaotong University, 2Department of Respiratory Medicine, Xi’an Central Hospital, 3Department of Medical Oncology, Shaanxi Provincial People’s Hospital, Xi’an, Shaanxi, People’s Republic of China
Abstract: Resistance to cisplatin-based therapy is a major challenge in the control of lung cancer progression. However, the underlying mechanisms remain largely unclear. Autophagy is closely associated with resistance to lung cancer therapy, but the function of autophagy in cisplatin treatment is still controversial. Here, we investigated whether autophagy was involved in lung adenocarcinoma resistance to cisplatin and further elucidated the underlying molecular mechanisms. Cisplatin-refractory lung adenocarcinoma cells increased autophagic vacuole formation detected by monodansylcadaverine staining. When exposed to cisplatin, lung adenocarcinoma cells demonstrated increased levels of autophagy detected by MAP1A/1B LC3B and mammalian homologue of yeast Atg6 (Beclin-1) expression using Western blot analysis. Activation of cisplatin-induced autophagic flux was increased by using chloroquine (CQ), which can accumulate LC3B-II protein and increase punctate distribution of LC3B localization. The combination of cisplatin with CQ was more potent than cisplatin alone in inhibiting lung adenocarcinoma cell growth, which also increased cisplatin-induced apoptosis. Compared to cisplatin treatment alone, the combination of cisplatin and CQ decreased p-AMPK and increased p-mTOR protein expressions, in addition, the AMPK inhibitor Compound C plus cisplatin downregulated p-AMPK and upregulated p-mTOR as well as depressed LC3B cleavage. These findings demonstrate that activation of autophagy is a hallmark of cisplatin exposure in human lung adenocarcinoma cells, and that there is a cisplatin-induced autophagic response via activation of the AMPK/mTOR signaling pathway. We speculate that autophagy can be used as a novel therapeutic target to overcome cisplatin-resistant lung adenocarcinoma.
Keywords: autophagy, lung cancer, A549 cells, A549/DDP cells, chemoresistance, AMPK, chloroquine
Introduction
Lung cancer is a major health problem with a high incidence and mortality rate worldwide.1 Surgery remains the cornerstone of therapy in early-stage lung cancer patients.2 However, the majority of patients have locally advanced or distant metastatic disease at diagnosis and thus do not undergo surgery.3 Thus, for these patients, platinum-based combination chemotherapy is the mainstay of palliation.4 However, the clinical outcome is still disappointing and the median survival for these individuals is only approximately 10 months.5,6
The platinum chemotherapeutic agent, cisplatin, is a major frontline drug to treat various human malignant tumors, including lung cancer.7,8 Nonetheless, clinical usage of this drug is limited because of intrinsic and acquired resistance.9 To date, mounting evidence indicates that cisplatin resistance has a multifactorial nature, and the underlying mechanisms remain obscure; most often targeting one mechanism of resistance fails to fully circumvent chemoresistance.7,10 Accordingly, exploring new agents that can overcome resistance of lung cancer to cisplatin is still urgently needed. Recently, autophagy has been found to be a potential anticancer therapeutic strategy.11 It is a conserved physiological catabolic process that allows portions of cytoplasmic organelles that are engulfed within vesicles with a double membrane structure to be subsequently degraded through fusion with lysosomes.12 In chemotherapy, the role of autophagy following anticancer treatment remains a paradoxical topic. Autophagy functions as a “prosurvival” or “prodeath” mechanism to re-sensitize or augment the anticancer effect of chemotherapeutic drugs.13,14 As for lung cancer, there are also some conflicting views on the involvement of autophagy in cisplatin treatment. One study found that cisplatin exposure inhibited autophagy,15 while another showed that it induced an autophagic response. This could lead to poor treatment outcomes and development of drug resistance,16 interestingly, a recent study found that treatment with low-dose cisplatin induced p53-dependent autophagy.17
Although these findings suggest a connection between cisplatin treatment and autophagy in lung cancer, it is unknown whether autophagy is cytoprotective and what its function is on acquired cisplatin resistance in specific molecular settings. In this study, we utilized A549 cells and cisplatin-refractory A549/DDP cells to explore the effect of autophagy modulators on cisplatin-induced anti-proliferation and acquired resistance. Specifically, we aimed to identify a critical molecular mechanism underlying the autophagic response during cisplatin treatment in these lung adenocarcinoma (ADC) cell lines.
Materials and methods
Reagents and antibodies
Cisplatin was purchased from Qilu Pharmaceutical Co. Ltd. (Jinan, People’s Republic of China) and prepared in stock solution, 1 mg/mL, at room temperature (RT). Chloroquine (CQ), Compound C, and monodansylcadaverine (MDC) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). CQ was administered as a stock water-based solution, 50 mg/mL, and stored at 4°C in darkness. Primary antibodies p-AMPK, AMPK, LC3B, Beclin-1, p-mTOR, mTOR, β-actin, and secondary antibodies were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). Fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG were from Beyotime Company, (Nanjing, People’s Republic of China). Annexin V-FITC/propidium iodide kit was purchased from Join Care Medicine Company (Zhuhai, People’s Republic of China).
Cell culture
The human lung ADC cells, A549 and A549/DDP-cisplatin-resistant clone were purchased from Chinese Academy of Sciences Institute of Life Sciences Cell Resource Center in Shanghai. These were grown in high glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (Hyclone, Logan, UT, USA) containing 10% heat-inactivated fetal bovine serum (Hyclone) and 100 U/mL penicillin and streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) at 37°C with 5% CO2. For drug treatment, cisplatin-refractory A549/DDP cells were maintained in DMEM supplemented with 2 μg/mL cisplatin and withdrawn from treatment 3 days before being used. No ethics approval was required from the Institutional Review Board for the use of these cell lines.
Cell viability assay
Drug effects on cell viability were analyzed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay. A549 cells (5,000/well) and A549/DDP cells (6,000/well) were seeded into 96-well plates overnight, and then treated in triplicate with cisplatin and/or CQ at indicated concentrations. The half maximal inhibitory concentration (IC50) was determined as the concentration resulting in 50% cell growth reduction compared with untreated control cells. There were five duplicate wells for each group with a volume of 200 μL in each well. After drug treatment, cells were cultured in DMEM containing MTT (0.5 mg/mL) for 4 hours at 37°C and formazan crystals were reabsorbed in 150 μL of dimethyl sulfoxide (DMSO) (Sigma-Aldrich Co.) for each well. After mixing, cell viability was determined by measuring absorbance at a wavelength of 490 nm. The ratio of cell viability was evaluated by the following equation: (optical density [OD]490 of treated group/OD490 of untreated group) ×100.
Flow cytometric apoptosis assay
Cells were seeded in six-well plates with 1×105 cells/well. Floating and adherent cells were collected after drug treatment. All cells were washed twice with cold phosphate-buffered saline (PBS) and followed by staining with Annexin V-FITC and PI for 15 minutes at 37°C in darkness. Apoptotic cells were analyzed followed by flow cytometric analysis (FACS Calibur). Apoptosis was calculated as percentage of early and late apoptotic cells.
Detection of autophagic vacuoles
To visualize autophagic response in lung cancer cells, cells were stained with MDC, a specific autophagolysosome marker. Briefly, approximately 1×103 cells were seeded on a coverslip in six-well culture plates and treated with cisplatin and/or CQ at indicated concentrations for 24 hours, then exposed to 0.05 mM MDC in PBS for 1 hour at 37°C. The cells were fixed and then imaged immediately under a fluorescence microscope. Cells treated with CQ were used as autophagy control.
Western blot analysis
After drug treatment, cells were harvested in ice-cold PBS and lysed with radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors, then centrifuged (12,000 rpm, 20 minutes) at 4°C. The protein supernatant was collected and its concentration determined using BCA protein assay reagent (both from Wolsen Company, Xi’an, People’s Republic of China). Each protein sample was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (8%–12%), then transferred onto polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA), blocked with 10% non-fat milk in 0.05% Tris-based saline-Tween 20 for 2 hours at RT. Membranes were labeled with primary antibodies against LC3B, Beclin-1 p-AMPK, AMPK, p-mTOR, mTOR (1:1,000), and β-actin (1:3,000) overnight at 4°C. Subsequently, blots were washed with Tris-based saline-Tween 20 for 30–40 minutes, then further probed with secondary antibody at RT for 1 hour and subjected to reaction with enhanced chemiluminescence solution and visualized using ChemiDoc XRS+ system (BioRad Laboratories Inc., Hercules, CA, USA). For quantification of specific protein bands, films were scanned and analyzed using Labworks software (Mission Viejo, CA, USA).
Immunofluorescence staining
A549 and A549/DDP cells, grown on glass coverslips, were treated with cisplatin and/or CQ at designed dose for 24 hours. Next, slides were fixed with 4% paraformaldehyde for 20 minutes and treated with 0.05% Triton X-100 for 15 minutes at RT. Thereafter, the coverslips were blocked with 10% goat serum for 1 hour, then stained with anti-LC3B antibody (1:200) overnight and, the next day, washed with PBS thrice, followed by incubation with FITC-conjugated goat anti-rabbit secondary antibody (1:500) for 1 hour at 37°C. Next, nuclei were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Invitrogen, Thermo Fisher Scientific) for 1 minute. Images were acquired using Zeiss LSM 710 fluorescence microscope (Carl Zeiss Meditec AG, Jena, Germany).
Statistical analysis
All experiments were performed in triplicate, the data are expressed as means ± standard deviation (SD). Student’s t-test was used to compare differences between groups using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). In each case, a P-value <0.05 was considered statistically significant.
Results
Sensitivity of lung ADC cell lines to cisplatin treatment
To determine the possible involvement of autophagy in cisplatin treatment, we first investigated sensitivity of lung ADC A549 and A549/DDP cells to cisplatin. After the cells were treated with cisplatin at indicated concentrations, viability was measured by MTT assay. As shown in Figure 1A, A549 cell viability was reduced by cisplatin treatment in a time- and dose-dependent manner. The IC50 of cisplatin on A549 cells was 4.07±0.32, 2.26±0.43, and 1.52±0.41 μg/mL, for 24, 48, and 72 hours, respectively. Thus, we used 4, 2, and 1 μg/mL of cisplatin to treat A549 cells in the subsequent experiments. A549/DDP cells were derived from A549 cells by continuous culture in cisplatin-containing medium. Data in Figure 1A and B showed approximately ten-fold resistance to cisplatin compared to A549 cells (IC50 of cisplatin for 24-hour treatment was 42.71±2.21 vs 4.07±0.32 μg/mL).
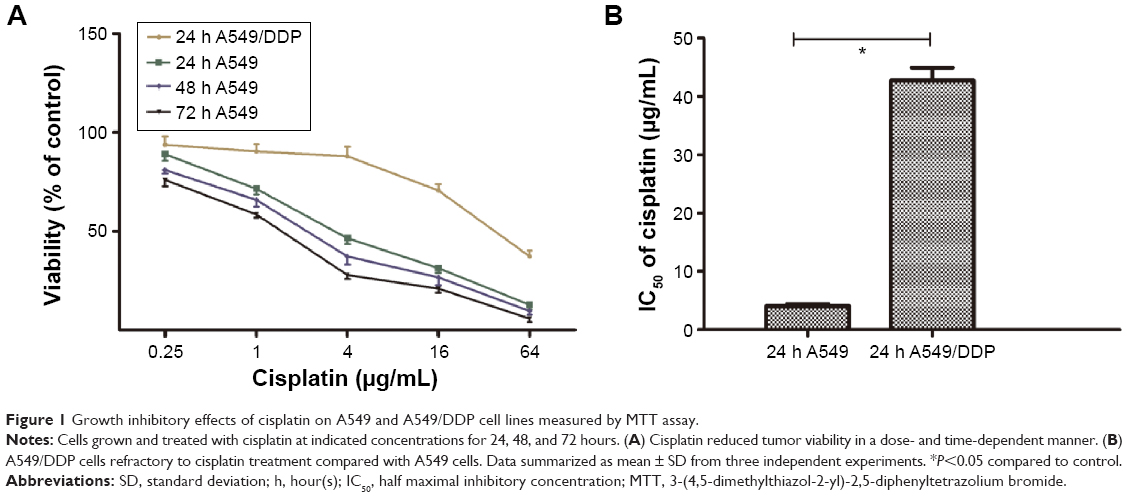 | Figure 1 Growth inhibitory effects of cisplatin on A549 and A549/DDP cell lines measured by MTT assay. |
Cisplatin induced autophagy in lung ADC cell lines
To explore the involvement of cisplatin on regulation of tumor cell autophagy, we detected autophagic response genes. LC3B (MAP1A/1B LC3B) is the first identified mammalian homologue of yeast Atg8 protein, and plays a key role in the process of autophagosome formation; it is a widely used biomarker to monitor autophagy. More specifically, LC3B comprised LC3B-I and LC3B-II. LC3B-I is conjugated to phosphatidylethanolamine and processed to LC3B-II, the hallmark of the autophagic pathway.18 We, therefore, detected LC3B conversion using immunoblot analysis. As shown in Figure 2A and B, cisplatin induced the switch of LC3B-I to LC3B-II for A549cells in time- and dose-dependent manners. Similarly, we found that the baseline conversion in cisplatin-refractory A549/DDP cells was more prominent than in A549 cells.
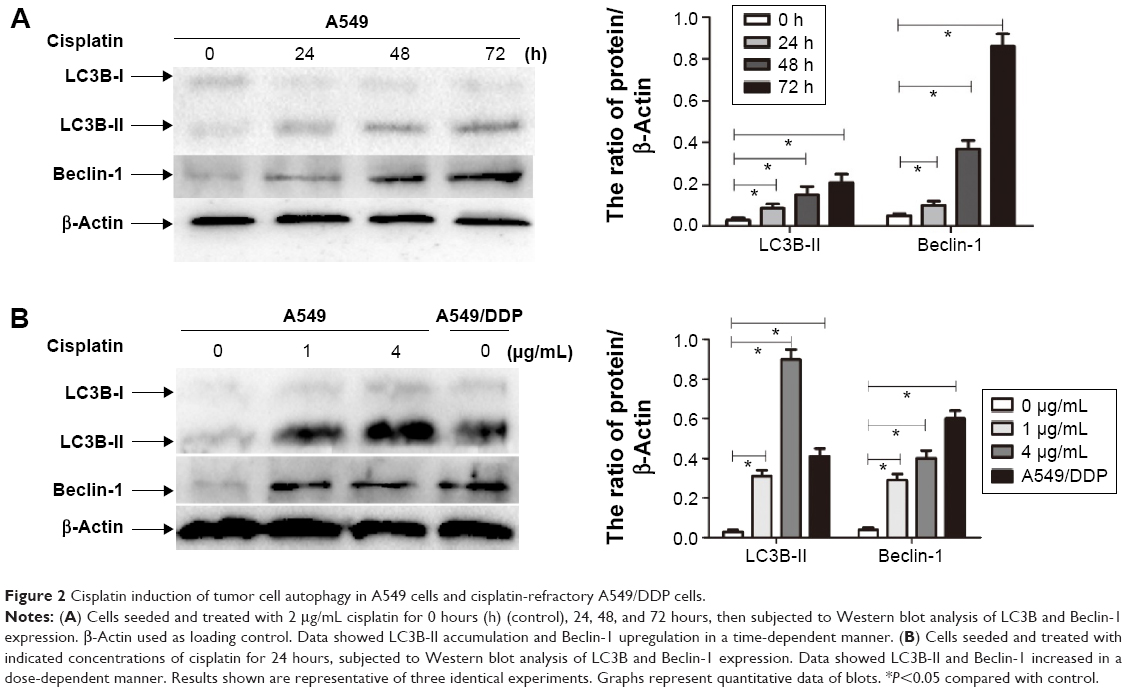 | Figure 2 Cisplatin induction of tumor cell autophagy in A549 cells and cisplatin-refractory A549/DDP cells. |
Beclin-1 was also one of the first identified mammalian autophagy-related proteins, a critical component involved in regulating the development of autophagosomes.19 After cisplatin treatment, we next assessed expression of Beclin-1 and found that Beclin-1 expression was increased in a time- and dose-dependent manner. Likewise, A549/DDP cells showed a high Beclin-1 expression (Figure 2A and B).
Increasing LC3B-II expression could contribute to autophagosomes’ formation or blocking lysosomal degradation.20 To address this issue, we used the lysosome inhibitor, CQ, to study cisplatin-triggered autophagic flux in A549 cells. CQ caused an increase in the formation of autophagosomes due to preventing autophagosome-lysosome fusion, eventually inhibiting late stage autophagy.21 As shown in Figure 3A, cisplatin treatment led to LC3B-II expression, which was upregulated in A549 cells. However, co-treatment of CQ and cisplatin resulted in further LC3B-II accumulation. These data demonstrated cisplatin-induced LC3B-II accumulation was due to increasing autophagosome formation, revealing that cisplatin induced autophagic flux in A549 cells.
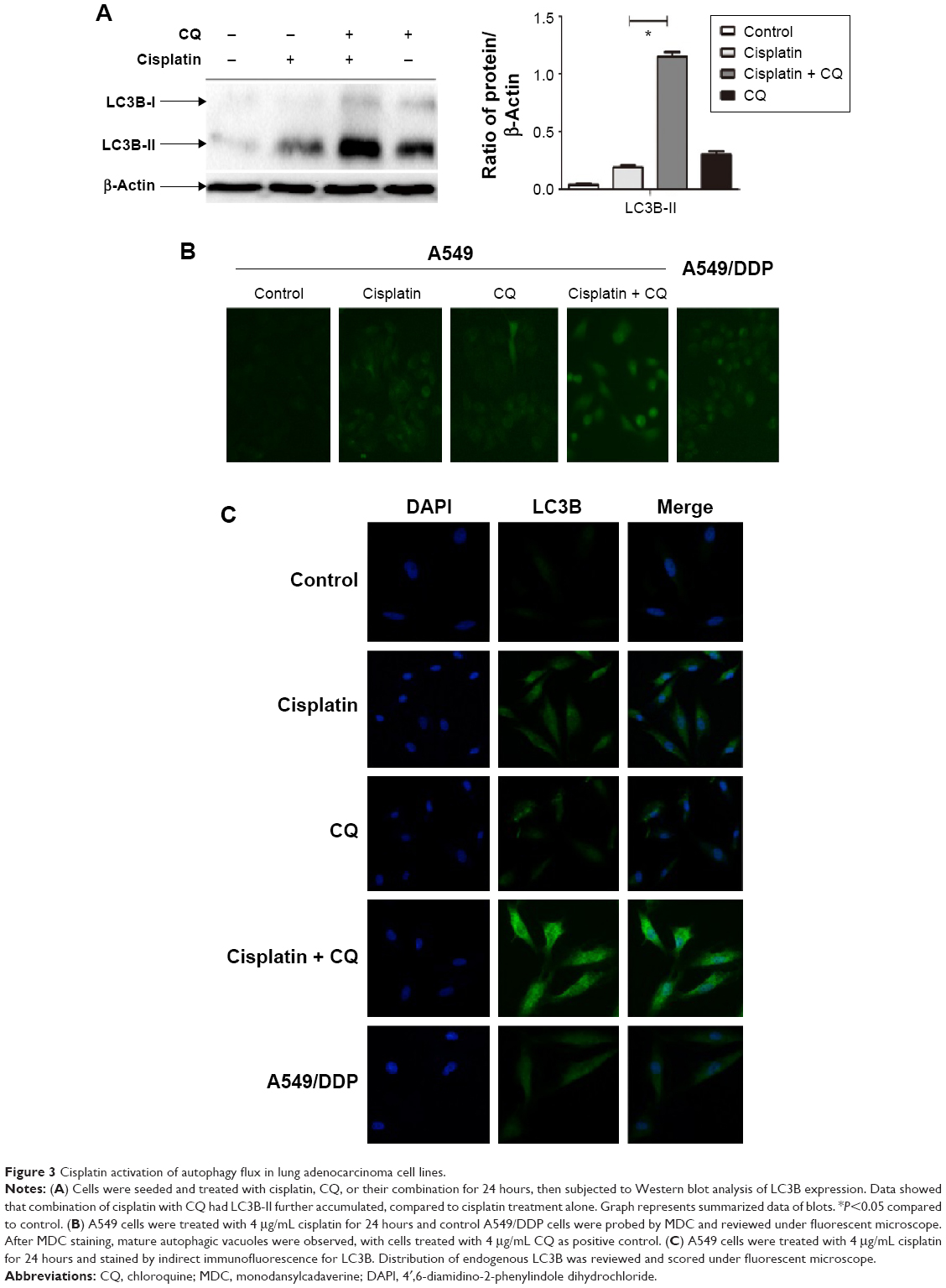 | Figure 3 Cisplatin activation of autophagy flux in lung adenocarcinoma cell lines. |
We also measured the incorporation of MDC, an autofluorescent base that concentrates on autophagic vacuoles, a specific marker for autophagolysosomes.22,23 As shown in Figure 3B, the fluorescent density and formation of MDC-labeled vacuoles increased after 24 hours of cisplatin treatment. CQ, as a positive control, increased autophagosomal number; we observed a similar increase in A549/DDP cells. The findings showed that cisplatin exerts an induced effect on autophagic vacuoles’ formation in lung ADC cells.
To further confirm aforementioned data, we examined the distribution of LC3B localization in lung ADC cells by indirect immunofluorescence assay. As shown in Figure 3C, specific increased punctate distribution occurred in A549 cells after being treated with cisplatin. CQ alone showed an increase in the number of LC3B-positive granules, since it blocked autophagosome-lysosome fusion and induced a defect in autolysosomal degradation. The combination of CQ with cisplatin led to an increase of LC3B-positive accumulation, consistent with the aforementioned data of the immunoblot.
Taken together, these findings demonstrated that cisplatin could induce autophagic response and activate autophagic flux in A549 cells.
Autophagy inhibition attenuated cisplatin-induced drug resistance
The aforementioned findings indicated that lung ADC cells treated with cisplatin resulted in autophagy, which is positively associated with the refractory effect of cisplatin in cisplatin-refractory A549/DDP cells. Similarly, induction of autophagy can be viewed as a prosurvival mechanism and can contribute to drug resistance.14 Hence, we further investigated whether cisplatin-induced drug resistance was mediated by autophagy. We speculated that autophagy suppression could sensitize A549 cells to cisplatin and create a cisplatin-sensitive phenotype in A549/DDP cells. CQ treatment, alone, up to 4 μg/mL, did not significantly affect viability of A549 cells (Figure 4A), whereas treatment with cisplatin with or without CQ decreased viability of A549 cells (Figure 4B and C). Moreover, IC50 of these combined drugs on A549 and A549/DDP lung ADC cells decreased to 2.45±0.45 and 23.68±1.32 μg/mL, respectively. This indicated that CQ caused a prominent sensitization effect to cisplatin by decreasing cisplatin IC50 value by nearly two-fold. Next, we explored whether disruption of autophagy with CQ could promote cell apoptosis. As shown in Figure 4D and E, The apoptotic rates of A549 cells of the control, CQ, cisplatin, co-treatment of cisplatin and CQ groups were 8.0%±0.9%, 10.5%±1.0%, 16.6%±0.8%, and 21.8%±0.9%, respectively. The apoptotic rates of A549/DDP cells of the control, CQ, cisplatin, co-treatment of cisplatin and CQ groups were 2.6%±0.6%, 2.9%±0.8%, 8.8%±0.8%, and 12.9%±0.7%, respectively. These data indicated that cisplatin induced tumor cell apoptosis, although CQ did not; the CQ plus cisplatin had an increased percentage of apoptotic cells, as compared with the cisplatin alone.
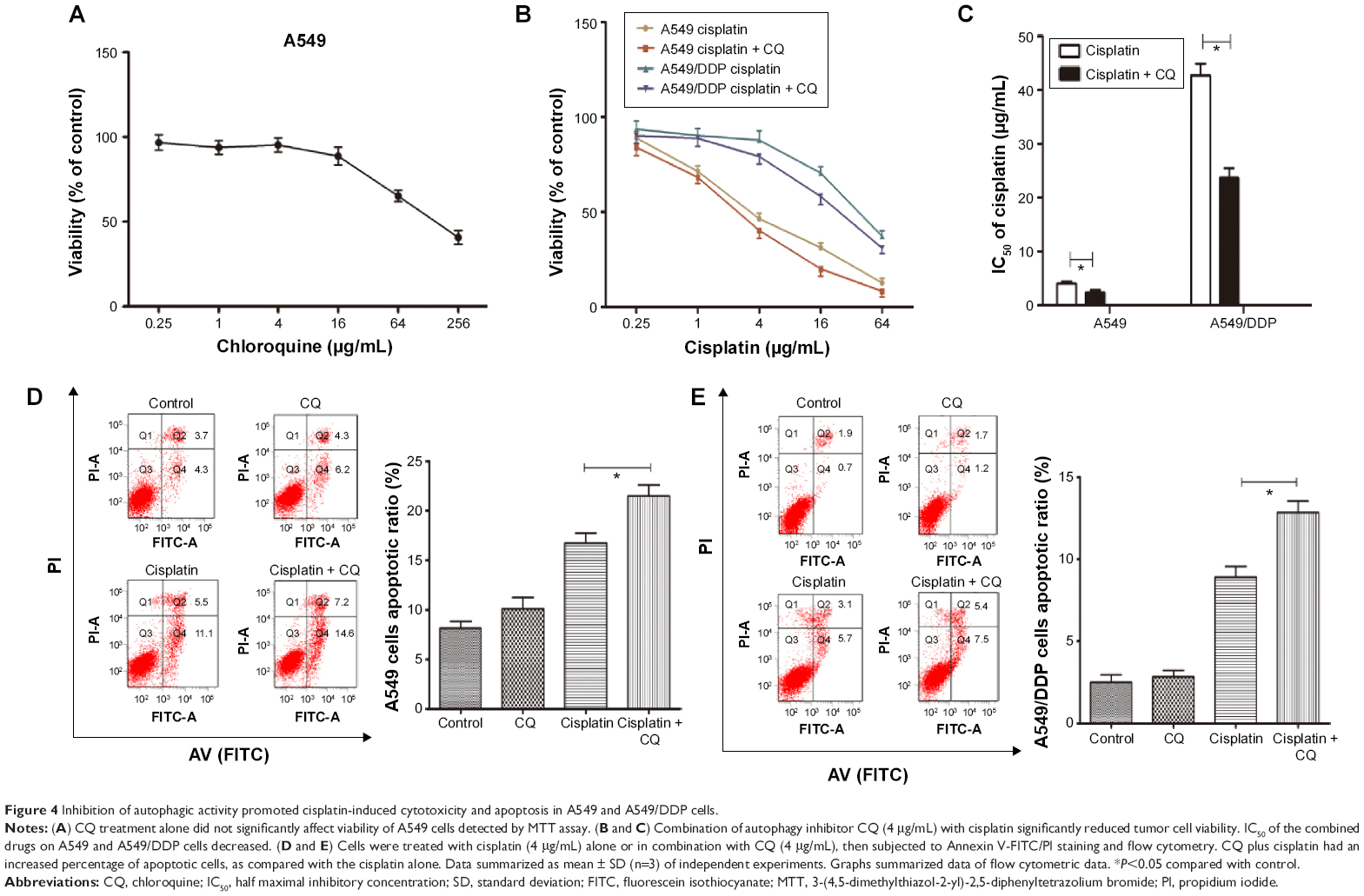 | Figure 4 Inhibition of autophagic activity promoted cisplatin-induced cytotoxicity and apoptosis in A549 and A549/DDP cells. |
AMPK/mTOR signaling pathway involved in autophagy activation in lung ADC cells treated by cisplatin
We next explored the mechanism of autophagy induced by cisplatin treatment in A549 and cisplatin-refractory A549/DDP cells. mTOR is an evolutionarily conserved serine/threonine kinase that regulates autophagy.24 AMPK is one of the main stress-sensing enzymes and can actively regulate metabolism and cell proliferation. Notably, AMPK is also a key regulator of autophagy.25 Phosphorylation of AMPK leads to inhibition of mTOR, which activates autophagy.26 We, thus, analyzed and compared levels of p-AMPK, AMPK, p-mTOR, and mTOR after treatment with cisplatin. Figure 5A and B show that cisplatin increased AMPK phosphorylation in A549 cells, which was accompanied by decreasing mTOR phosphorylation. Cisplatin treatment had no distinct effect on total mTOR and AMPK levels. Comparing this to cisplatin-alone treatment, co-treatment of A549 cells with CQ and cisplatin decreased AMPK phosphorylation and increased mTOR phosphorylation (Figure 5C). In addition, the AMPK inhibitor Compound C plus cisplatin downregulated p-AMPK and upregulated p-mTOR as well as depressed LC3B cleavage (Figure 5D). These data imply that the AMPK/mTOR signaling pathway is a vital regulator involved in cisplatin-induced autophagic responses in A549 and A549/DDP cells.
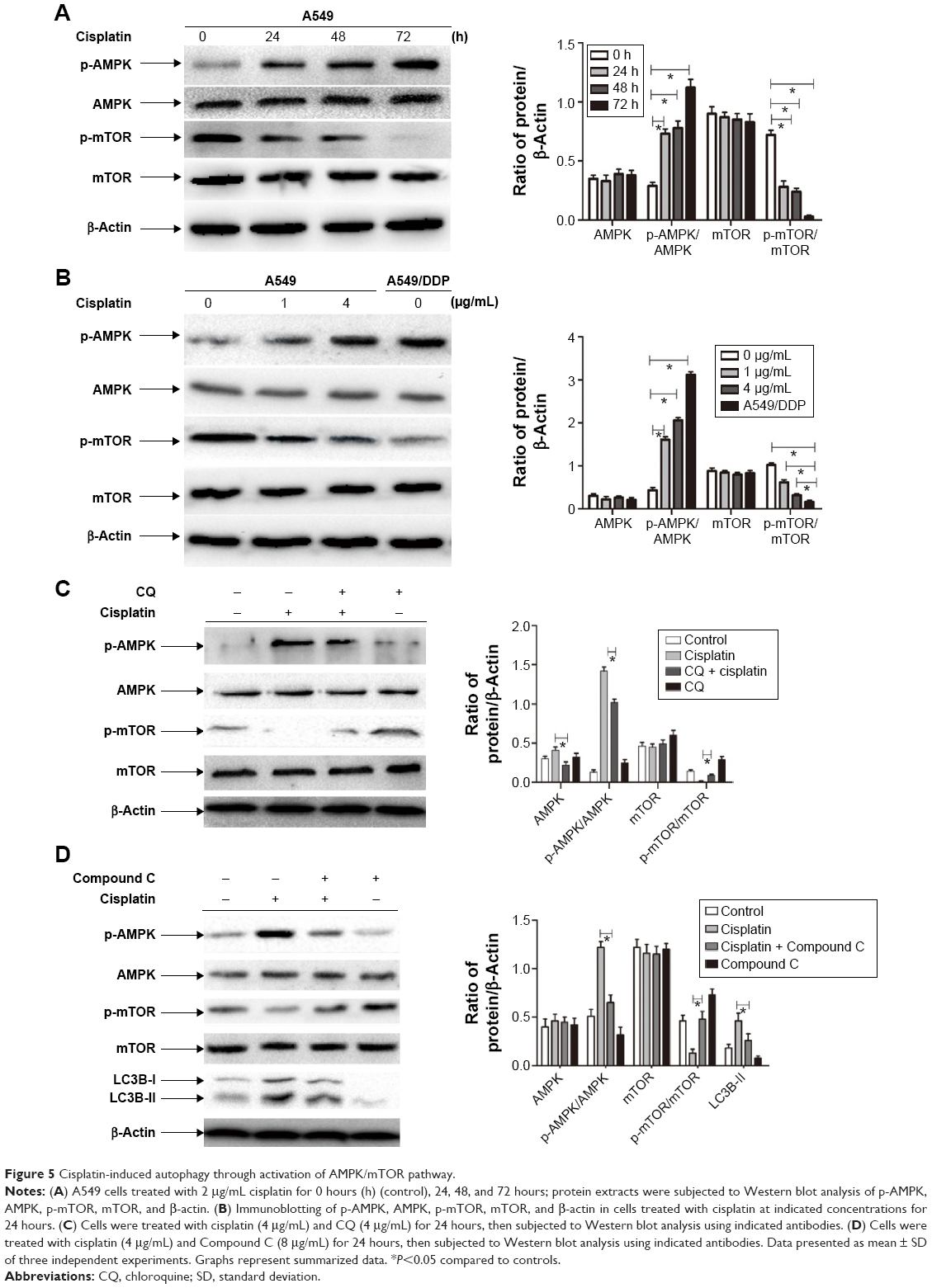 | Figure 5 Cisplatin-induced autophagy through activation of AMPK/mTOR pathway. |
Discussion
Although autophagy as a potential anticancer target for malignant tumors has been demonstrated,27 accumulating evidence has shown the role of autophagy in tumor-suppression or promotion for anticancer treatment to be conflicting. Some autophagy inducers, as single agents or in combination with other anticancer drugs, have been proposed to be a potential therapeutic strategy. This includes the mTORC1 inhibitors temsirolimus,28 rapamycin,29,30 and imatinib,31 whereas there is growing evidence showing that autophagy served a prosurvival function against anticancer drugs and may be related to chemotherapy resistance. Therefore, inhibitors of autophagy could also be used as a novel therapeutic strategy to improve chemotherapy efficacy.32–34 In this study, our findings further confirmed that the extent of autophagy in lung ADC cells, by detecting Beclin-1 and LC3B, was increased, consistent with the response to cisplatin treatment. The preliminary conclusion is that cisplatin induces an autophagic response.
In the autophagosome, LC3B-II is present both in the exterior and interior of the vesicle, which will be, in part, degraded after its fusion with lysosomes. Thus, increase in LC3B-II levels may be due to augmentation of autophagosome induction or suppressed autophagosome maturation. Inhibition of autophagosome-lysosome fusion using lysosomal protease inhibitors could monitor LC3B turnover and be an indicator of the total autophagic flux.20,35 Our current data showed LC3B-II accumulation after treatment of A549 cells with cisplatin in presence of the lysosomotropic reagent, CQ. In agreement with these observations, the indirect immunofluorescence staining assay confirmed that CQ increased distribution of LC3B localization in A549 cells during cisplatin treatment. MDC-labeled vacuoles assay provided independent evidence that cisplatin treatment triggers autophagy.
Here, we deployed an autophagy inhibitor to determine its role in the antineoplastic properties of cisplatin and found that its combination with CQ significantly enhanced the anti-proliferative effect on lung cancer cells compared to cisplatin alone in A549 cells and A549/DDP cells. CQ induced a prominent sensitization effect of cisplatin on A549/DDP cells. Autophagy and apoptosis are highly interconnected but this relationship has not been well elucidated. Thus, the pivotal molecular mechanism behind the regulation of autophagy and apoptosis induced by cisplatin should be further explored. Our present study allows us to conclude that autophagy appears to have a prosurvival role in protecting lung ADC cells from a cisplatin-mediated anti-proliferation effect and causing resistance to cisplatin. Autophagy could regulate degradation, recycle the damaged cytoplasmic contents, and prevent cisplatin-induced DNA damage.
There is little knowledge to date on the relationship between autophagy and cisplatin treatment and on the mechanisms of cisplatin-induced autophagic response in lung cancer. mTOR serves as a central regulator by negatively regulating autophagy. AMPK inhibits mTOR and initiates autophagy.36,37 Thus, the AMPK/mTOR signaling pathway may be involved in regulating autophagy in response to various anticancer agents.38,39 In this study, we elucidated that autophagy triggered by cisplatin was mediated by activation of the AMPK/mTOR pathway in lung ADC. Treatment of A549 cells with cisplatin augmented levels of phosphorylated AMPK, downregulated phosphorylated mTOR protein, and upregulated LC3B and Beclin-1 expression. In agreement with these facts, the AMPK inhibitor Compound C plus cisplatin suppressed phosphorylation of AMPK and increased phosphorylation of mTOR as well as depressed LC3B cleavage, as compared with the cisplatin-alone treatment.
mTOR comprises two functionally distinct complexes, termed mTORC1 and mTORC2.40 Although studies have suggested AMPK regulates cell growth and autophagy via suppression of mTORC1 pathway,41,42 in the current study, there is less evidence showing that mTORC1 or mTORC2 is involved in autophagy induced by cisplatin treatment. Therefore, more research is needed to further explore the association between autophagy induced by cisplatin and mTORC1 and mTORC2 in lung ADC cells.
In summary, as shown in Figure 6, our study demonstrates that autophagy contributes to development of cisplatin resistance in human lung ADC, thus playing a prosurvival role in protecting cells from cisplatin-mediated cytotoxicity. To our knowledge, this is the first report of the effect of cisplatin on induction of autophagic response via activation of the AMPK/mTOR signaling pathway in lung ADC. Therefore, use of autophagy inhibitors is a potentially novel strategy to potentiate the efficacy of cisplatin in patients with cisplatin-otherwise-resistant lung cancer.
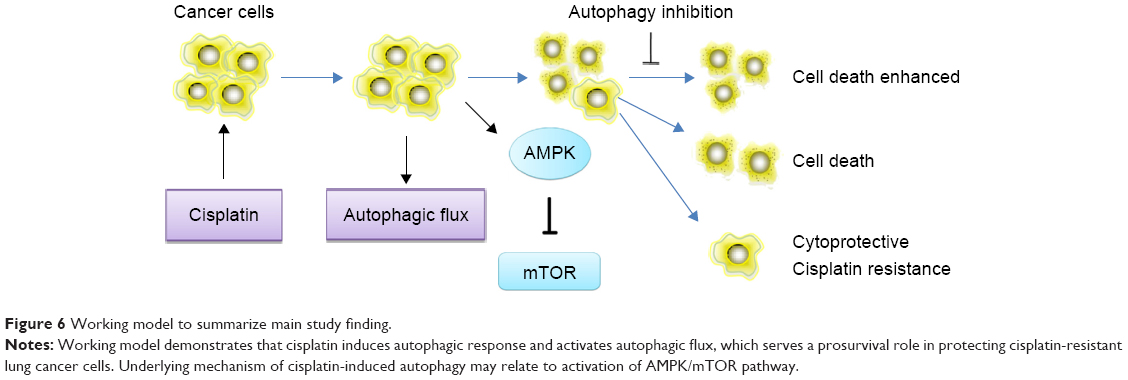 | Figure 6 Working model to summarize main study finding. |
Acknowledgments
The present study was supported by grants from the National Natural Science Foundation of China (number 81101777), and the social development research project of Shaanxi Provincial Department of Science and Technology (number 2013K12-08-3).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Vansteenkiste J, De Ruysscher D, Eberhardt WE, et al. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24 Suppl 6:vi89–vi98. | ||
Wang MC, Liang X, Liu ZY, et al. In vitro synergistic antitumor efficacy of sequentially combined chemotherapy/icotinib in nonsmall cell lung cancer cell lines. Oncol Rep. 2015;33(1):239–249. | ||
Peters S, Adjei AA, Gridelli C, et al. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23 Suppl 7:vii56–vii64. | ||
Gkiozos I, Charpidou A, Syrigos K. Developments in the treatment of non-small cell lung cancer. Anticancer Res. 2007;27(4C):2823–2827. | ||
Gironés R, López P, Chulvi R, Cañabate M, Dolores T. Ten years of lung cancer in a single center: gender, histology, stage and survival. Journal of Cancer Metastasis and Treatment. 2015;1(3):201–207. | ||
Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–1883. | ||
Lee HY, Mohammed KA, Goldberg EP, Kaye F, Nasreen N. Cisplatin loaded albumin mesospheres for lung cancer treatment. Am J Cancer Res. 2015;5(2):603–615. | ||
Galluzzi L, Vitale I, Michels J, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. | ||
Brozovic A, Osmak M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett. 2007;251(1):1–16. | ||
Chen N, Karantza V. Autophagy as a therapeutic target in cancer. Cancer Biol Ther. 2011;11(2):157–168. | ||
Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–822. | ||
Wu WK, Coffelt SB, Cho CH, et al. The autophagic paradox in cancer therapy. Oncogene. 2012;31(8):939–953. | ||
Sui X, Chen R, Wang Z, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. | ||
Sirichanchuen B, Pengsuparp T, Chanvorachote P. Long-term cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Mol Cell Biochem. 2012;364(1–2):11–18. | ||
Ren JH, He WS, Nong L, et al. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother Radiopharm. 2010;25(1):75–80. | ||
Cho KH, Park JH, Kwon KB, et al. Autophagy induction by low-dose cisplatin: the role of p53 in autophagy. Oncol Rep. 2014;31(1):248–254. | ||
Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. | ||
Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. | ||
Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–545. | ||
Lee SW, Kim HK, Lee NH, et al. The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett. 2015;360(2):195–204. | ||
Niemann A, Takatsuki A, Elsasser HP. The lysosomotropic agent monodansylcadaverine also acts as a solvent polarity probe. J Histochem Cytochem. 2000;48(2):251–258. | ||
Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114(Pt 20):3619–3629. | ||
Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32. | ||
Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131. | ||
Liu B, Cheng Y, Liu Q, Bao JK, Yang JM. Autophagic pathways as new targets for cancer drug development. Acta Pharmacol Sin. 2010;31(9):1154–1164. | ||
Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–730. | ||
Shi WY, Xiao D, Wang L, et al. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012;3:e275. | ||
Takeuchi H, Kondo Y, Fujiwara K, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65(8):3336–3346. | ||
Cirstea D, Hideshima T, Rodig S, et al. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther. 2010;9(4):963–975. | ||
Can G, Ekiz HA, Baran Y. Imatinib induces autophagy through BECLIN-1 and ATG5 genes in chronic myeloid leukemia cells. Hematology. 2011;16(2):95–99. | ||
Yu L, Gu C, Zhong D, et al. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett. 2014;355(1):34–45. | ||
Lefort S, Joffre C, Kieffer Y, et al. Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers. Autophagy. 2014;10(12):2122–2142. | ||
Jiang L, Huang S, Zhang D, et al. Inhibition of autophagy augments chemotherapy in human salivary adenoid cystic carcinoma. J Oral Pathol Med. 2014;43(4):265–272. | ||
Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. | ||
Yang ZJ, Chee CE, Huang S, Sinicrope F. Autophagy modulation for cancer therapy. Cancer Biol Ther. 2011;11(2):169–176. | ||
Leone RD, Amaravadi RK. Autophagy: a targetable linchpin of cancer cell metabolism. Trends Endocrinol Metab. 2013;24(4):209–217. | ||
Din FV, Valanciute A, Houde VP, et al. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012;142(7):1504–1515.e1503. | ||
Aryal P, Kim K, Park PH, Ham S, Cho J, Song K. Baicalein induces autophagic cell death through AMPK/ULK1 activation and downregulation of mTORC1 complex components in human cancer cells. FEBS J. 2014;281(20):4644–4658. | ||
Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014;36:121–129. | ||
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–1023. | ||
Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.