")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Attenuation of everolimus-induced cytotoxicity by a protective autophagic pathway involving ERK activation in renal cell carcinoma cells
Authors Zeng YZ, Tian XF , Wang Q, He WY, Fan J, Gou X
Received 21 December 2017
Accepted for publication 9 March 2018
Published 19 April 2018 Volume 2018:12 Pages 911—920
DOI https://doi.org/10.2147/DDDT.S160557
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Yizhou Zeng, Xiaofang Tian, Quan Wang, Weiyang He, Jing Fan, Xin Gou
Department of Urinary Surgery, The First Affiliated Hospital of Chongqing Medical University, Yuzhong District, Chongqing, China
Aim: The mammalian target of rapamycin (mTOR) pathway is a critical target for cancer treatment and the mTOR inhibitor everolimus (RAD001) has been approved for treatment of renal cell carcinoma (RCC). However, the limited efficacy of RAD001 has led to the development of drug resistance. Autophagy is closely related to cell survival and death, which may be activated under RAD001 stimulation. The aim of the present study was to identify the underlying mechanisms of RAD001 resistance in RCC cells through cytoprotective autophagy involving activation of the extracellular signal-regulated kinase (ERK) pathway.
Methods and results: RAD001 strongly induced autophagy of RCC cells in a dose- and time-dependent manner, as confirmed by Western blot analysis. Importantly, suppression of autophagy by the pharmacological inhibitor chloroquine effectively enhanced RAD001-induced apoptotic cytotoxicity, as demonstrated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and Western blot analysis, indicating a cytoprotective role for RAD001-induced autophagy. In addition, as was shown by the MTT assay, flow cytometry, and Western blot analysis, RAD001 robustly activated ERK, but not c-Jun N-terminal kinase and p38. Activation of ERK was inhibited by the pharmacological inhibitor selumetinib (AZD6244), which effectively promoted RAD001-induced cell death. Moreover, employing AZD6244 markedly attenuated RAD001-induced autophagy and enhanced RAD001-induced apoptosis, which play a central role in RAD001-induced cell death. Furthermore, RAD001-induced autophagy is regulated by ERK-mediated phosphorylation of Beclin-1 and B-cell lymphoma 2, as confirmed by Western blot analysis.
Conclusion: These results suggest that RAD001-induced autophagy involves activation of the ERK, which may impair cytotoxicity of RAD001 in RCC cells. Thus, inhibition of the activation of ERK pathway-mediated autophagy may be useful to overcome chemoresistance to RAD001.
Keywords: apoptosis, autophagy, everolimus, ERK, renal cancer, selumetinib
Introduction
Renal cell carcinoma (RCC) is the most common form of kidney cancer, with ~338,000 new diagnoses and 144,000 deaths occurring annually worldwide.1 Surgical resection is generally performed to treat this disease; however, nephrectomy is not a feasible option for about 30% of patients with metastatic disease.2 Therefore, to further improve the quality of life and survival of patients, systemic treatment may be a more effective choice.3
Everolimus (RAD001), a mammalian target of rapamycin (mTOR) inhibitor, has been demonstrated to exert cytotoxicity against human cancers of the breast, stomach, and prostate,4–6 and is currently used as a sequential or second-line therapy for RCC refractory to sunitinib or sorafenib. However, the efficacy of RAD001 is thought to be limited by feedback loops and cross talk with other pathways, resulting in drug resistance. Karam et al7 established and characterized a panel of mouse models of RCC derived from patients undergoing radical nephrectomy. Using these models, resistance to the mTOR inhibitor RAD001 was identified. François et al8 stated that RAD001 rarely induces regression of pancreatic neuroendocrine tumors. In addition, as compared with RAD001 alone, co-treatment seems to be potentially more effective in controlling cell signaling.9,10 Motzer et al11 declared that a combination therapy of RAD001 and lenvatinib showed a favorably synergistic effect in patients with advanced or metastatic RCC, which was the first successful combination therapy approved by the US Food and Drug Administration.12 Thus, based on these findings, it is necessary to explore the underlying mechanism of the drug resistance of RAD001.
Autophagy is a highly conserved intracellular catabolic process that degrades and recycles cellular components for cell survival under certain conditions, and is closely related to cell survival or death. For cancer cells resistant to chemotherapy, autophagy presents either protective or injurious effects. For example, RAD001 is able to induce autophagy in human renal cancer cells, which then promotes tumor cell survival, resulting in a limited anticancer effect.13 The mTOR complex is now regarded as an autophagy switch to promote proliferation and inhibit autophagy, although the potential mechanisms involved in the cell signal pathways for this process are still not fully understood. Interestingly, Butler et al14 found that inhibition of the phosphatidylinositol 3 kinase (PI3K)/protein kinase B (AKT)/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/extracellular signal-regulated kinase (ERK) signaling in prostate cancer. Besides, activation of the PI3K/AKT/mTOR and Ras/MEK/ERK cell signaling pathways is also critical for autophagy. Particularly, cross talk between these two pathways has been well illustrated in previous studies.15–17 Moreover, several studies have shown that inhibition of the ERK pathway enhanced the antitumor activity of RAD001 in pediatric gliomas,18 neuroblastoma,19 and acute myelogenous leukemia.20 Thus, the aim of this study was to identify the underlying mechanisms and biochemical pathways involved in RAD001 resistance in RCC.
Materials and methods
Materials
The small molecular inhibitors RAD001 and AZD6244 were obtained from MedChem Express (Monmouth Junction, NJ, USA). Antibodies against B-cell lymphoma 2 (Bcl-2), phospho-Bcl-2, Beclin-1, ERK, phospho-ERK, p38, phospho-p38, c-Jun N-terminal kinase (JNK), and phospho-JNK were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-cleaved poly ADP ribose polymerase (PARP) and p62 were obtained from BD Biosciences (San Jose, CA, USA). Anti-LC3 and chloroquine (CQ) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Anti-β-actin antibody was purchased from Zoonbio Biotechnology Co., Ltd (Nanjing, China). All secondary antibodies were obtained from Abgent (San Diego, CA, USA). Roswell Park Memorial Institute (RPMI)-1640 medium and trypsin were purchased from HyClone (Logan, UT, USA). Fetal bovine serum was purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell kit was purchased from Beyotime Institute of Biotechnology (Shanghai, China).
Cell culture
The human RCC cell lines 786-O and A498 were purchased from American Type Culture Collection (Manassas, VA, USA) and grown in RPMI-1640 medium supplemented with 10% fetal bovine serum, 1 mmol/L glutamine, 105 U/L penicillin, and 100 g/L streptomycin at 37°C, under a humidified atmosphere containing 5% CO2.
MTT assay
Cell viability was assessed with the MTT assay. Briefly, 786-O and A498 cells were seeded into 96-well plates at 5,000 cells/well and cultivated for 24 hours. After adherence, the 786-O and A498 cells were treated with various concentrations of RAD001 for 24 hours. MTT (5 mg/mL, 20 μL/well) was added to each well and the plate was incubated at 37°C for 4 hours. Dimethyl sulfoxide was then added (150 μL/well) to each well to dissolve any crystals and the plates were agitated for 15 minutes. Absorbance at 570 nm was measured using an Infinite F200/M200 type multifunction microplate reader (Tecan Trading AG, Männedorf, Switzerland). Cell viability was calculated by OD values of treated groups/OD values of control group) ×100%. Each experiment was repeated three times.
Apoptosis analysis by flow cytometry
After treatment with RAD001 and AZD6244, the cells were trypsinized, washed with phosphate-buffered saline, and suspended in 195 μL of Annexin V-fluorescein isothiocyanate (FITC) binding buffer containing 5 μL of Annexin V-FITC and 10 μL of propidium iodide (Beyotime Institute of Biotechnology). After incubation for 10–20 minutes at room temperature in the dark, the cells were subjected to flow cytometry analysis using a FACSCanto 6-color flow cytometer (BD Biosciences).
Western blot analysis
To extract total protein lysates, the cells were lysed in cold radioimmunoprecipitation assay lysis buffer (Beyotime Institute of Biotechnology) containing phenylmethanesulfonyl fluoride (Beyotime Institute of Biotechnology), and then centrifuged at 12,000 rpm for 15 minutes at 4°C to remove debris. Protein concentrations were measured with the bicinchoninic acid protein assay kit (Beyotime Institute of Biotechnology). The protein extracts were heat denatured in sodium dodecyl sulfate polyacrylamide gel electrophoresis Sample Loading Buffer (Beyotime Institute of Biotechnology). Equal amounts of protein from each cell lysate (40 μg/lane) were separated in 12% polyacrylamide gels and transferred on to a membrane. After blocking with 5% nonfat dried milk for 2 hours, the membrane was incubated with the primary antibodies overnight at 4°C. Afterward, the proteins were visualized by enhanced chemiluminescence using horseradish peroxidase-conjugated immunoglobulin G secondary antibodies.
Statistical analysis
Data are expressed as mean ± SD. Statistical analyses were performed with GraphPad Prism 6 software (GraphPad Software, Inc, San Diego, CA, USA) and IBM SPSS Statistics for Windows, version 20.0 (IBM Corporation, Armonk, NY, USA). Based on the completely random design of this experiment, the results were analyzed by one-way analysis of variance or t-test. A probability value of <0.05 was considered statistically significant.
Results
RAD001-induced autophagy in RCC cells
It has been well demonstrated that the mTOR pathway inhibitor RAD001 can induce autophagy in human RCC cells.13 The aim of the present study was to determine whether RAD001 can induce autophagy in RCC cells. For this purpose, different concentrations of RAD001 were added to cultures of 786-O and A498 cells. The results showed that RAD001-induced autophagy of 786-O and A498 cells in a dose-dependent manner, which was indicated by a typical phenotype of autophagy, including upregulation of LC3-II/I and downregulation of p62 (Figure 1A and B). Next, to further validate this effect, 786-O and A498 cells were treated with RAD001 for 0, 1, 2, 4, or 8 hours. As expected, the increased conversion of LC3-I to LC3-II was in line with a gradual decrease in autophagic protein p62 in both cell lines after RAD001 treatment in a time-dependent manner (Figure 1C and D). Taken together, these findings further emphasize that RAD001 is able to induce autophagy in RCC cells.
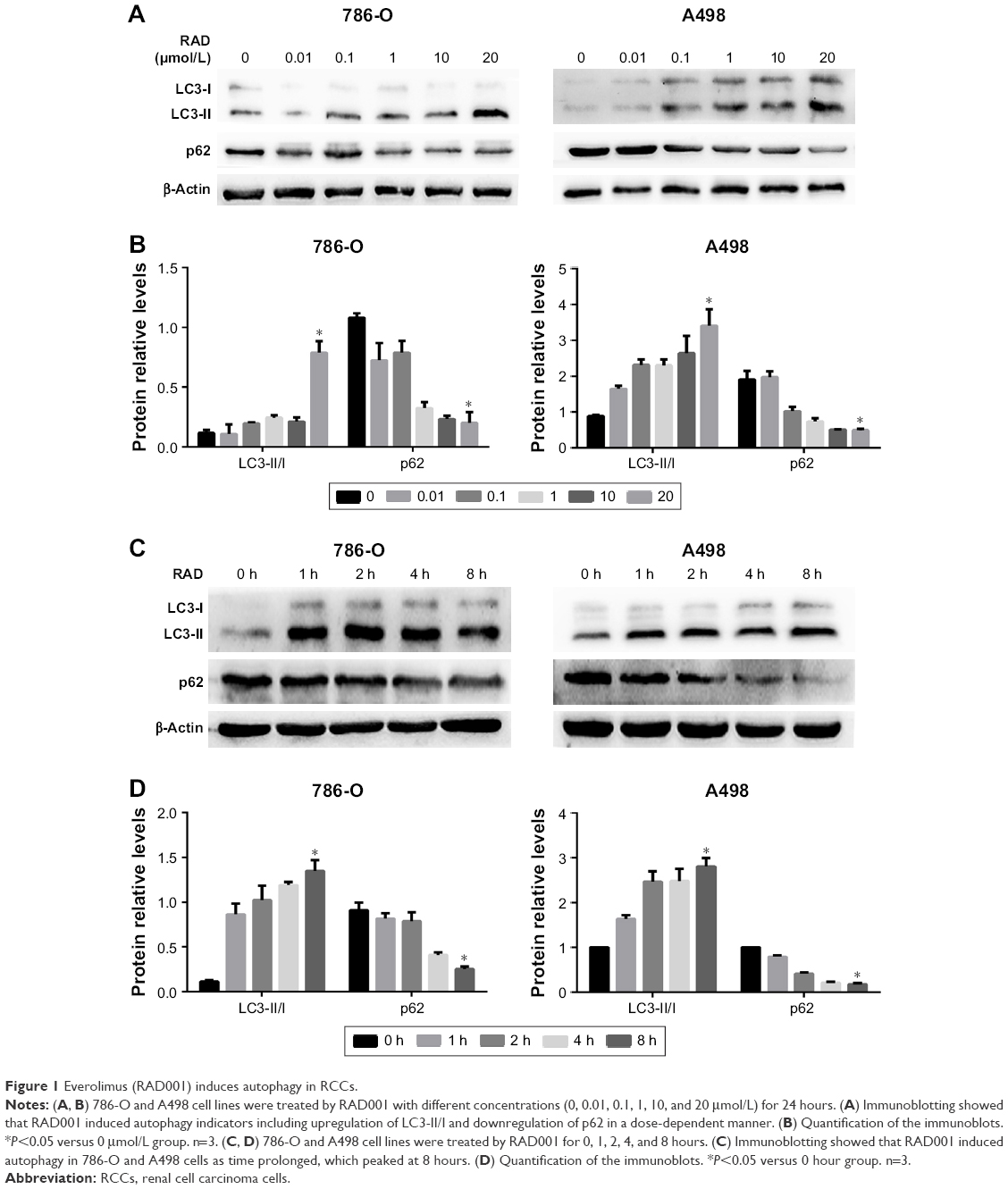 | Figure 1 Everolimus (RAD001) induces autophagy in RCCs. |
Autophagy protected RCC cells against RAD001-induced cell death, which involved activation of the ERK pathway
To further determine whether RAD001 could induce death of RCC cells, the MTT assay was performed with 786-O and A498 cells following exposure to RAD001. Pretreatment of both cell lines with RAD001 (0.01–100 μmol/L) for 24 hours revealed a dose-dependent reduction in cell survival. The half IC50 (half maximal inhibitor concentration) of RAD001 at 24 hours for 786-O and A498 cells was 10 and 1 μmol/L, respectively. Thus, these values were chosen for subsequent experiments (Figure 2A). Based on the observation that RAD001 also induced autophagy, we were particularly interested in investigating whether autophagy was involved in RAD001-induced cytotoxicity. For this purpose, the lysosome inhibitor CQ was used in this study, as it inhibits autophagy via blocking autophagolysosome-mediated degradation of the p62 and LC3-II proteins. Expectedly, the RAD001-induced downregulation of p62 expression was effectively inhibited by CQ, although a higher LC3-II level was observed in 786-O and A498 cells after exposure to co-treatment with both RAD001and CQ, suggesting that RAD001 induces a continuous autophagic flux in both cell lines (Figure 2B and C). These results indicate that autophagy was indeed induced by RAD001. More importantly, in line with these findings, the results of the MTT assay showed that the effect of RAD001-induced cell cytotoxicity was significantly enhanced by inhibition of autophagy in 786-O and A498 cells, suggesting that autophagy may play a protective role in RAD001-induced cytotoxicity of RCC (Figure 2D). Because cell cytotoxicity is closely associated with apoptosis, to further determine whether the protective effects of autophagy for RAD001-induced cell death were due to the activation of apoptotic signaling, Western blot analysis was conducted to measure expression levels of cleaved PARP protein. Our results demonstrated that the apoptosis biomarker cleaved-PARP was significantly upregulated by RAD001 and this effect was enhanced by inhibition of autophagy with CQ (Figure 2E and F). Altogether, these results suggest that autophagy protects RCC cells against RAD001-induced cytotoxicity in part by suppressing the apoptosis biomarker-cleaved PARP.
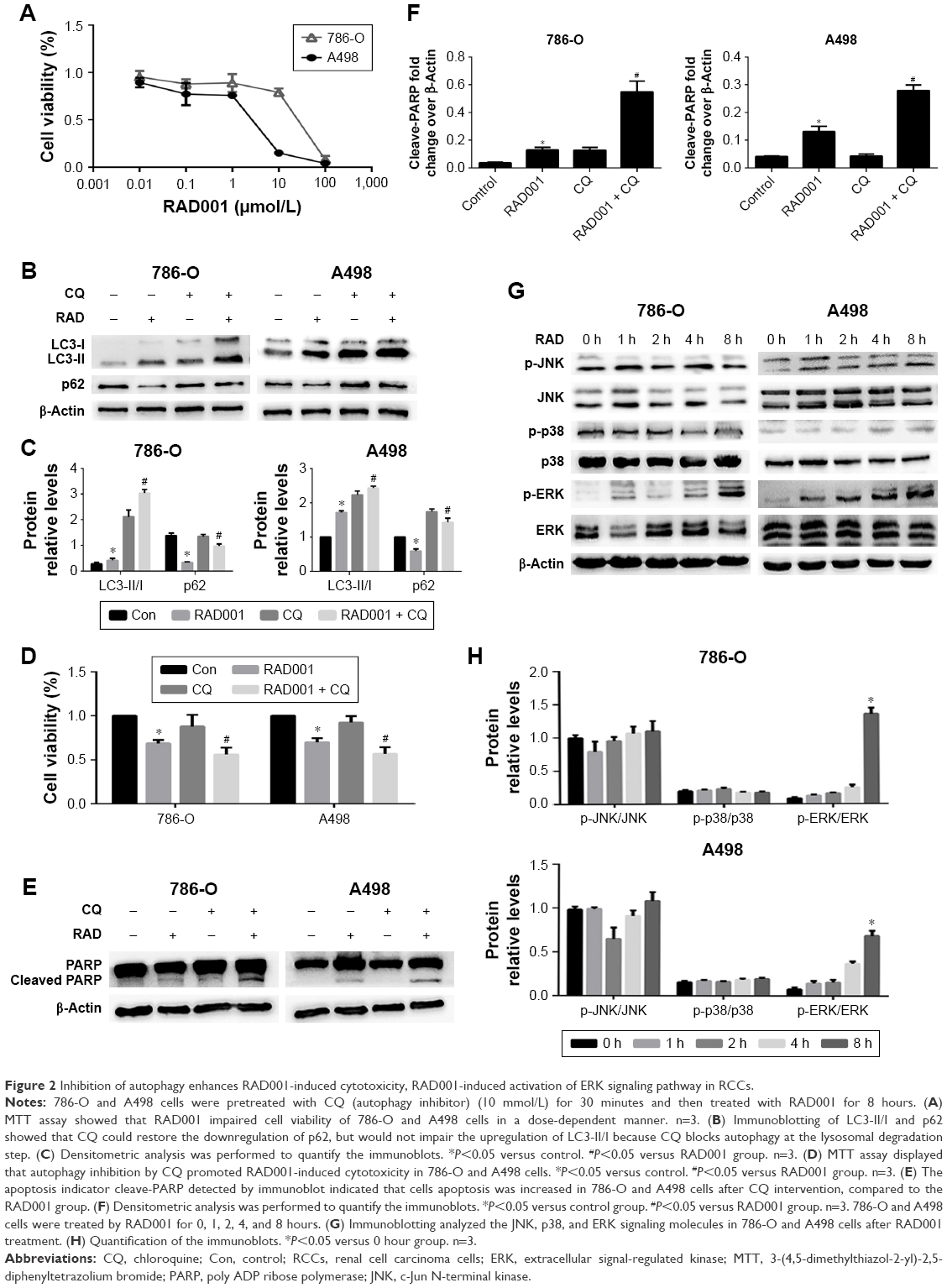 | Figure 2 Inhibition of autophagy enhances RAD001-induced cytotoxicity, RAD001-induced activation of ERK signaling pathway in RCCs. |
We next sought to explore the molecular mechanism by which RAD001 induces death of RCC cells. As it has been recognized that mitogen-activated protein kinases play a critical regulative role in cancer cell death and survival, the focus of the present study was the effect of RAD001-induced cell death on the activity of the JNK, ERK, and p38 cell signaling pathways. Briefly, 786-O and A498 cells were treated with RAD001 for the indicated times, and the activities of the involved signaling pathways were assessed by Western blot analysis. The results showed that following RAD001 pretreatment, p-ERK expression was significantly upregulated in both cell lines. However, the expression levels of p-JNK and p-p38 were largely unchanged following RAD001 exposure (Figure 2G and H).
Inhibition of ERK activation suppressed RAD001-induced autophagy and enhanced RAD001-induced apoptosis
The pharmacological inhibitor AZD6244 was employed to block ERK signaling to investigate the role of ERK in RAD001-induced cell death. The results showed that AZD6244 markedly inhibited activation of the ERK pathway induced by RAD001 (Figure 3A and B). More importantly, consistent with the effective suppressive effects of AZD6244 on ERK, co-treatment of both RAD001 and AZD6244 induced a greater extent of cell death than pretreatment of RAD001 alone, which indicated that inhibition of ERK effectively synergized with RAD001 to impair cell viability (Figure 3C). Overall, these results suggest that RAD001-induced cytotoxicity involves ERK signaling.
 | Figure 3 Selumetinib (AZD6244) significantly enhances RAD001-induced apoptosis of RCCs. |
A series of studies, including those by our group, have demonstrated that ERK signaling is closely related to autophagy in various cancers. To further confirm whether this signaling is involved in RAD001-induced autophagy, the specific inhibitor AZD6244 for ERK was used to interfere with autophagy induction. Indeed, employing AZD6244 markedly attenuated the autophagy effect induced by RAD001 in both 786-O and A498 cells, as indicated by the decreased expression of LC3-II/I and increased expression of p62 (Figure 3D and E). It is well established that cell death after exposure to cytotoxic chemotherapeutics is partially mediated by apoptosis. Moreover, as suggested by the above observation that autophagy protected RCC cells against RAD001-induced cytotoxicity in part by suppressing the apoptotic biomarker-cleaved PARP, we were interested in investigating whether ERK signaling was involved in the apoptosis process that caused RAD001-induced cytotoxicity. To test this hypothesis, we evaluated cell apoptosis by flow cytometry and the protein expression of cleaved PARP by Western blot analysis. Immunoblotting showed that AZD6244 exaggerated RAD001-induced cleavage of PARP (Figure 3F and G), indicating that the RAD001-induced apoptosis was enhanced by AZD6244. Furthermore, the flow cytometry results showed that pretreatment with RAD001 resulted in a higher rate of apoptosis in both cell lines, compared with no RAD001 exposure, suggesting that RAD001 did indeed induce cell apoptosis. Moreover, when these cell lines were treated with both AZD6244 and RAD001, this RAD001-induced apoptosis effect was further enhanced in both cell lines (Figure 3H and I). In summary, these data suggest that inhibition of ERK activation not only suppressed RAD001-induced autophagy, but also enhanced RAD001-induced apoptosis, which plays a central role in RAD001-induced cell death.
ERK mediated autophagy by upregulating expression of Beclin-1 and p-Bcl-2
It is reported that ERK activation mediates phosphorylation of Bcl-2, resulting in dissociation of the Bcl-2/Beclin-1 complex. This process releases Beclin-1 and leads to autophagy induction. Next, we further explored the mechanism whereby RAD001 induced ERK activation and led to the induction of autophagy. Beclin-1 is critical for the initiation of autophagy.21 The downstream factors of the ERK pathway, including Beclin-1 and p-Bcl-2, were upregulated as early as 1 hour after RAD001 treatment in both cell lines (Figure 4A and B). Moreover, inhibition of ERK activation with AZD6244 significantly decreased the expression levels of Beclin-1 and p-Bcl-2 (Figure 4C and D). These results suggest that RAD001-induced ERK-activated autophagy is at least in part due to upregulating the expression levels of Beclin-1 and p-Bcl-2.
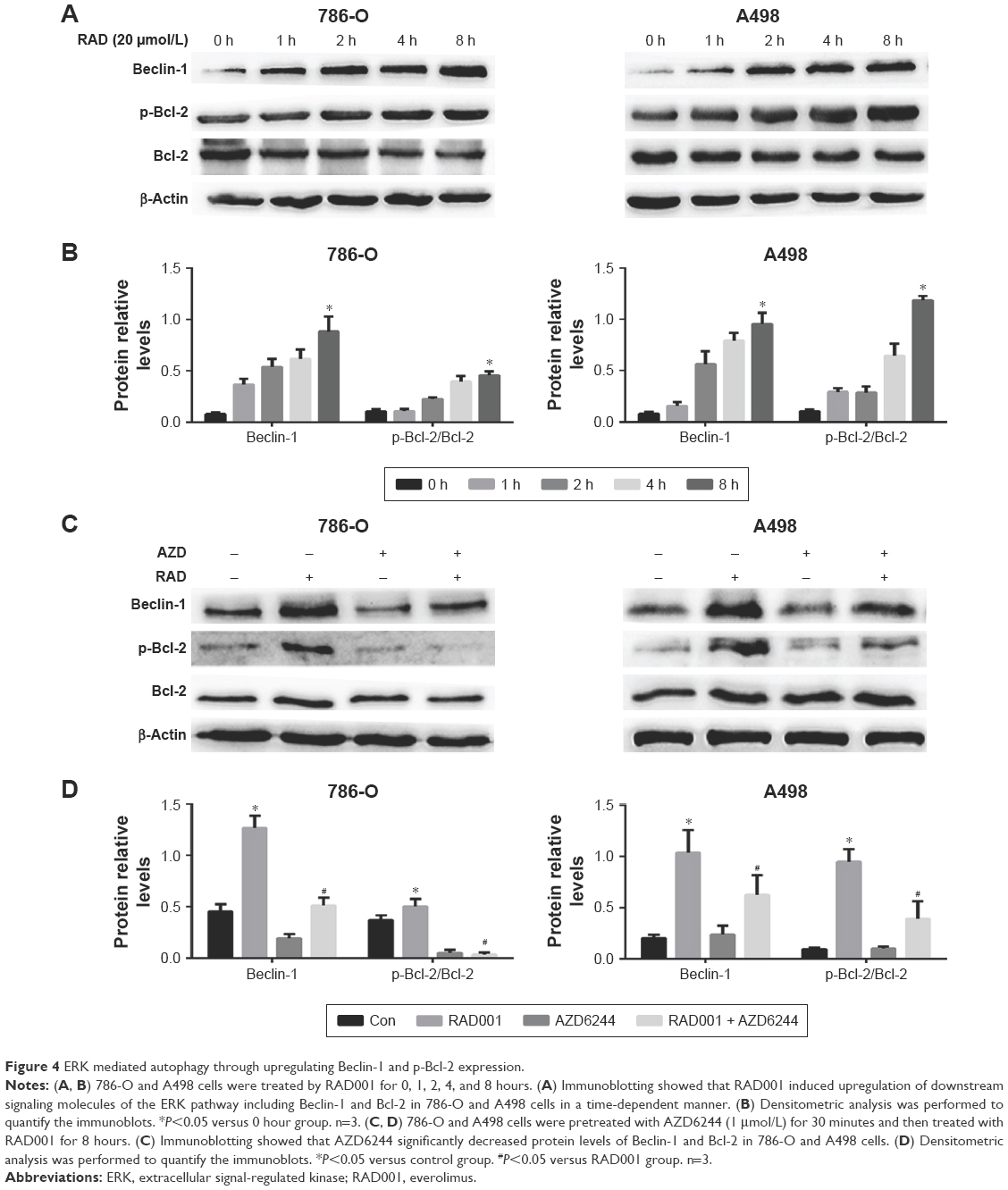 | Figure 4 ERK mediated autophagy through upregulating Beclin-1 and p-Bcl-2 expression. |
Discussion
The results of the present study showed that RAD001-induced autophagy may play an important role in RAD001-induced cytotoxicity of RCC cells, although the underlying mechanism remains unknown. Furthermore, cytoprotective autophagy involving the ERK pathway was induced by RAD001, thereby presenting a novel combination strategy to target ERK signaling. AZD6244 is a highly selective ERK inhibitor. Interestingly, AZD6244 significantly inhibited RAD001-induced autophagy and enhanced RAD001-induced apoptosis of 786-O and A498 cells. These findings suggested that RCC cell resistance to RAD001 may be closely related to cytoprotective autophagy linked to the activation of the ERK pathway.
Evidence has confirmed that RAD001 can substantially induce autophagy, as well as promote the apoptosis of breast cancer cells22 and hepatocellular carcinoma cells.23,24 Similarly, the results of the present experiment showed that RAD001 treatment of RCC cells resulted in the upregulation of LC3-II/I and downregulation of p62, which are typical features of autophagy induction, suggesting that autophagy by 786-O and A498 cells was induced by RAD001 treatment. So, high concentrations of RAD001 were used in this experiment due to the grade of the RCC cell lines 786-O and A498. In addition, it is reported that RAD001 did not induce autophagy in RCC cells at low concentrations such as 0.1 μM.25 Our data showed that the combination of RAD001 with CQ restored downregulation of p62, but further promoted the upregulation of LC3-II/I, which is due to the ability of CQ to effectively block autophagy at the lysosomal degradation step.26 However, currently available autophagy blockers are highly unspecific and lack potency, and are limited by the severe toxicity of these compounds.27–30 Collectively, RAD001-induced LC3 upregulation was due to the excessive production and accumulation of LC3-II. In addition, autophagy inhibition enhanced RAD001-induced apoptosis of RCC cells. However, due to the clinical limitations of the autophagy inhibitor CQ in RCC cells, it is necessary to further explore the effectiveness of this novel combination therapy with RAD001.
Emerging evidence shows that the JNK, p38, and ERK signaling pathways are involved in cell death and autophagy.31–33 Previous studies have indicated that ERK activation mediates phosphorylation of Bcl-2, leading to dissociation of the Bcl-2/Beclin-1 complex, which releases Beclin-1 and triggers autophagy induction.34,35 In the present study, ERK, but not the p38 and JNK pathways, was activated in RCC cells after RAD001 treatment, as well as the upregulation of downstream factors such as Beclin-1 and p-Bcl-2. Targeting the ERK pathway can potentially improve the anticancer efficacy of RAD001. AZD6244 is a highly selective ERK inhibitor and several studies have demonstrated that it enhances the antitumor activity of RAD001 in pediatric gliomas,18 neuroblastoma,19 and acute myelogenous leukemia.20 However, only one study showed that treatment with the combination of sorafenib and AZD6244 enhanced the antitumor activity of sorafenib in RCC.36 Thus, we suggest a novel combined therapeutic approach with RAD001 and AZD6244 for the treatment of RCC. As expected, our results showed that the combination of RAD001 and AZD6244 significantly enhanced apoptosis and impaired the viability of 786-O and A498 cells. Furthermore, RAD001-induced cytoprotective autophagy was significantly impaired by AZD6244 and the blunted dissociation of the Bcl-2/Beclin-1 complexes led to the downregulation of Beclin-1 and p-Bcl-2. Collectively, AZD6244 synergizes with RAD001 to kill RCC cells via inhibition of cytoprotective autophagy, which was linked to ERK/Bcl-2/Beclin-1 signaling.
In conclusion, the underlying mechanism of RAD001 resistance in RCC cells may be related to cytoprotective autophagy involving the ERK pathway, which plays a critical role in RAD001-induced cell death. However, there were still some limitations to this study. As this was only a preclinical study, further assessment on patient-derived RCC is required. The findings described here are only part of the complex mechanisms involved in RAD001 resistance, and other mechanisms remain to be characterized. Notably, these findings have important preclinical implications and the combination of RAD001 with AZD6244 may be a promising tumor-targeted strategy for the treatment of RCC.
Acknowledgments
This study was supported by funding from the Natural Science Foundation of China (grant no 81372758) and the Chongqing Health Bureau (2013-1-006). We sincerely thank Lixue Chen, Wenhui Yan, Xiaojuan Deng, and Guangchen Qin of the Central Laboratory of the First Affiliated Hospital of Chongqing Medical University for their assistance with these experiments.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. | ||
Fisher R, Gore M, Larkin J. Current and future systemic treatments for renal cell carcinoma. Semin Cancer Biol. 2013;23:38–45. | ||
Bedke J, Gauler T, Grunwald V, et al. Systemic therapy in metastatic renal cell carcinoma. World J Urol. 2017;35:179–188. | ||
Hurvitz SA, Kalous O, Conklin D, et al. In vitro activity of the mTOR inhibitor everolimus, in a large panel of breast cancer cell lines and analysis for predictors of response. Breast Cancer Res Treat. 2015;149:669–680. | ||
Lee SJ, Lee J, Lee J, et al. Phase II trial of capecitabine and everolimus (RAD001) combination in refractory gastric cancer patients. Invest New Drugs. 2013;31:1580–1586. | ||
Wedel S, Hudak L, Seibel JM, et al. Inhibitory effects of the HDAC inhibitor valproic acid on prostate cancer growth are enhanced by simultaneous application of the mTOR inhibitor RAD001. Life Sci. 2011;88:418–424. | ||
Karam JA, Zhang XY, Tamboli P, et al. Development and characterization of clinically relevant tumor models from patients with renal cell carcinoma. Eur Urol. 2011;59:619–628. | ||
François RA, Maeng K, Nawab A, et al. Targeting focal adhesion kinase and resistance to mTOR inhibition in pancreatic neuroendocrine tumors. J Natl Cancer Inst. 2015;107:djv123. | ||
Huang JJ, Li ZM, Huang Y, et al. Schedule-dependent inhibition of T-cell lymphoma cells by cotreatment with the mTOR inhibitor everolimus and anticancer drugs. Invest New Drugs. 2012;30:223–235. | ||
Calderwood SK. Tumor heterogeneity, clonal evolution, and therapy resistance: an opportunity for multitargeting therapy. Discov Med. 2013;15:188–194. | ||
Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473–1482. | ||
Colwell J. FDA approves drug combo for kidney cancer. Cancer Discov. 2016;6:687–688. | ||
Grimaldi A, Santini D, Zappavigna S, et al. Antagonistic effects of chloroquine on autophagy occurrence potentiate the anticancer effects of everolimus on renal cancer cells. Cancer Biol Ther. 2015;16:567–579. | ||
Butler DE, Marlein C, Walker HF, et al. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget. 2017;8:56698–56713. | ||
Sheppard KE, Cullinane C, Hannan KM, et al. Synergistic inhibition of ovarian cancer cell growth by combining selective PI3K/mTOR and RAS/ERK pathway inhibitors. Eur J Cancer. 2013;49:3936–3944. | ||
Roberts PJ, Usary JE, Darr DB, et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res. 2012;18:5290–5303. | ||
Satonaka H, Ishida K, Takai M, et al. (-)-Epigallocatechin-3-gallate down-regulates doxorubicin-induced overexpression of P-glycoprotein through the coordinate inhibition of PI3K/Akt and MEK/ERK signaling pathways. Anticancer Res. 2017;37:6071–6077. | ||
Olow A, Mueller S, Yang X, et al. BRAF status in personalizing treatment approaches for pediatric gliomas. Clin Cancer Res. 2016;22:5312–5321. | ||
Kiessling MK, Curioni-Fontecedro A, Samaras P, et al. Targeting the mTOR complex by everolimus in NRAS mutant neuroblastoma. PLoS One. 2016;11:e0147682. | ||
Nishioka C, Ikezoe T, Yang J, Yokoyama A. Inhibition of MEK/ERK signaling induces apoptosis of acute myelogenous leukemia cells via inhibition of eukaryotic initiation factor 4E-binding protein 1 and down-regulation of Mcl-1. Apoptosis. 2010;15:795–804. | ||
Sahni S, Merlot AM, Krishan S, et al. Gene of the month: BECN1. J Clin Pathol. 2014;67:656–660. | ||
Lui A, New J, Ogony J, et al. Everolimus downregulates estrogen receptor and induces autophagy in aromatase inhibitor-resistant breast cancer cells. BMC Cancer. 2016;16:487. | ||
Zhang X, Yang H, Yue S, et al. The mTOR inhibition in concurrence with ERK1/2 activation is involved in excessive autophagy induced by glycyrrhizin in hepatocellular carcinoma. Cancer Med. 2017;6:1941–1951. | ||
Altmeyer A, Josset E, Denis JM, et al. The mTOR inhibitor RAD001 augments radiation induced growth inhibition in a hepatocellular carcinoma cell line by increasing autophagy. Int J Oncol. 2012;41:1381–1386. | ||
Zou Y, Wang J, Leng X, et al. The selective ERK inhibitor selumetinib enhances the antitumor activity of everolimus against renal cell carcinoma in vitro and in vivo. Oncotarget. 2017;8:20825–20833. | ||
Wang A, Zhang H, Liang Z, et al. Antagonistic effects of chloroquine on autophagy occurrence potentiate the anticancer effects of everolimus on renal cancer cells. Cancer Biol Ther. 2015;16:567–579. | ||
Yang Y, Hu L, Zheng H, et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin. 2013;34:625–635. | ||
Shi TT, Yu XX, Yan LJ, et al. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol. 2017;79:287–294. | ||
Rosenfeld MR, Ye X, Supko JG, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10:1359–1368. | ||
Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol. 2010;176:1181–1192. | ||
Liu T, Wu L, Wang D, et al. Role of reactive oxygen species-mediated MAPK and NF-κB activation in polygonatum cyrtonema lectin-induced apoptosis and autophagy in human lung adenocarcinoma A549 cells. J Biochem. 2016;160:315–324. | ||
Hung AC, Tsai CH, Hou MF, et al. The synthetic β-nitrostyrene derivative CYT-Rx20 induces breast cancer cell death and autophagy via ROS-mediated MEK/ERK pathway. Cancer Lett. 2016;371:251–261. | ||
Huang XL, Zhang H, Yang XY, et al. Activation of a C-Jun N-terminal kinase-mediated autophagy pathway attenuates the anticancer activity of gemcitabine in human bladder cancer cells. Anticancer Drugs. 2017;28:596–602. | ||
Dai JP, Zhao XF, Zeng J, et al. Drug screening for autophagy inhibitors based on the dissociation of Beclin1-Bcl2 complex using BiFC technique and mechanism of eugenol on anti-influenza A virus activity. PLoS One. 2013;8:e61026. | ||
He W, Wang Q, Srinivasan B, et al. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene. 2014;33:3004–3013. | ||
Yuen JS, Sim MY, Sim HG, et al. Combination of the ERK inhibitor AZD6244 and low-dose sorafenib in a xenograft model of human renal cell carcinoma. Int J Oncol. 2012;41:712–720. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.