")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Arsenic sulfide amplifies JQ1 toxicity via mitochondrial pathway in gastric and colon cancer cells
Authors Tan Z, Zhang X, Kang T, Zhang L, Chen S
Received 20 July 2018
Accepted for publication 24 September 2018
Published 14 November 2018 Volume 2018:12 Pages 3913—3927
DOI https://doi.org/10.2147/DDDT.S180976
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Zhen Tan,* Xiuli Zhang,* Ting Kang, Lian Zhang, Siyu Chen
Department of Oncology, Xin Hua Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
*These authors contributed equally to this work
Purpose: Gastric and colon cancers have been the leading causes of cancer mortality in the world with limited therapy. Small molecules binding to bromodomains of bromodomain-containing protein 4 (BRD4) exert strong antitumor activities against hematological malignancies, while generally have limited efficacy in advanced solid tumors. Here, we found that the bromodomain and extra-terminal (BET)-bromodomain inhibitor JQ1, when combined with arsenic sulfide (As4S4, abbreviated as AS), synergistically decreased the expression of nuclear factor of activated T-cells (NFATs) as well as the downstream oncogene c-Myc and largely induced cell apoptosis via mitochondrial pathway in gastric and colon cancer cell lines.
Methods: The synergistic cytotoxicity of AS and JQ1 in gastric and colon cancer cells was determined by MTT assay and verified by FACS assay. Western blot analysis and quantitative real-time PCR (qPCR) assay were used to detect the expression of NFATs and downstream apoptotic proteins. The mitochondrial transmembrane potential was determined by FACS assay, and the metastasis of cancer cells was detected by the wound-healing assay.
Results: AS and JQ1 synergistically induced cell apoptosis in gastric and colon cancer cells by downregulating NFATs and upregulating apoptotic proteins. Combination of AS and JQ1 was associated with the decreased mitochondrial transmembrane potential, the cytochrome c release, and the subsequent caspase-3 activation.
Conclusion: Thus, our data indicate that AS can effectively enhance the cytotoxicity of BET inhibitors in gastric and colon cancer cells through mitochondrial-mediated apoptosis induction.
Keywords: arsenic sulfide, JQ1, NFAT, apoptosis, mitochondria
Introduction
Gastric cancer (GC) and colon cancer have been the leading causes of cancer mortality in the United States.1 GC is a malignant tumor derived from gastric epithelial cells and is associated with poor prognosis2 and also the third major cause of global cancer-related death.1,3 Due to the propensity for metastasis and resistance to chemotherapy,4 patients with advanced colorectal cancer (CRC) still suffer from dismal prognosis. Recently, despite the large advances in therapeutic approaches, including surgical methods, radiotherapy, chemotherapy, and novel molecular-targeted therapy for GC and colon cancer, the prognosis remains unsatisfactory, and the effective treatments are still lagging for these diseases.3,5 Thus, the novel therapeutic strategies for these lethal diseases are in need of continued investigation to ameliorate the prognosis of GC and colon cancer patients.
A class of epigenetic readers that contain bromodomains can recognize acetylated lysines of histone and nonhistone proteins via their bromodomains.6 BRD4, one of the bromodomain and extraterminal (BET) family proteins, functions well in transcription, cell cycle control, inflammatory cytokine production, and cancer procession.7,8 Small molecules binding to bromodomains of BRD4 exert strong antitumor activities, suppressing the proliferation and transformation potential of various cancer cells. JQ1, a BRD4 inhibitor, targets the domain of BET proteins that interacts with histones, thereby competitively inhibiting the binding of BET proteins to chromatin and decreasing the expression of RNA species dependent on this mechanism of transcription.9 JQ1 shows potent chemotherapeutic activity against various hematological malignancies, including acute myeloid leukemia,10 multiple myeloma,11 and acute lymphoblastic leukemia (ALL).12 In these malignancies, JQ1 shows its antiproliferative activity mainly through the inhibition of c-Myc expression, a well-known oncogene whose constitutive overexpression is responsible for several types of cancers.13 JQ1 also has the activity in eliminating solid tumors, including lung adenocarcinoma (LAC), neuroblastoma,14 and medulloblastoma,15 emerging as a promising therapeutic strategy for cancer treatment. Nevertheless, in advanced solid tumors, BET inhibitors generally have limited efficacy and do not achieve ideal cancer remission in tumor-bearing mice in most preclinical tumor models.16,17 These findings prove that solid cancers might be intrinsic resistant to monotherapy of BET inhibitors, which pose a severe challenge for the potential clinical utility of this kind of novel drugs in the near future.16,17
Arsenic sulfide (As4S4, abbreviated as AS), the active ingredient of realgar, has been widely used in traditional Chinese medicine for several centuries. Previous studies have shown the modest cytotoxicity in hematological malignant diseases and solid tumors such as GC and colon cancer.18–20 AS potently inhibited the expression of a panel of oncogenes, such as Bcl-2 family proteins, nuclear factor of activated T-cell (NFAT) family proteins, and c-Myc.20,21 AS has also exerted synergistic effect with various anticancer agents.22 Here, we investigated how AS overcame JQ1 resistance and exert synergistic cytotoxicity in both GC and colon cancer cells. Our data suggested that AS and JQ1 exerted anticancer activity synergistically through mitochondrial pathway in GC and colon cancer cells, with the increased inhibition of Bcl-2, NFATs, and c-Myc.
Materials and methods
Cell culture and reagents
AGS, MGC803, HCT116, and SW480 cells were obtained from the Chinese Academy of Sciences Committee Type Culture Collection Cell Bank (Shanghai, China). AGS cells were grown in DMEM/F12 media (Hyclone, Logan, UT, USA) containing 10% FBS; MGC803 cells were grown in 1640 media (Hyclone) containing 10% FBS; HCT116 and SW480 cells were grown in DMEM media (Hyclone) containing 10% FBS. Highly purified AS was supplied by the Shanghai Institute of Hematology (Shanghai, China) and was prepared as previously described.23 The stock solution of AS (133.36 μM) was diluted in complete culture medium to obtain working solutions. MTT was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Antibodies for NFATc1 and NFATc2 were purchased from Sigma-Aldrich Co. and Santa Cruz Biotechnology Inc. (Dallas, TX, USA), respectively. Antibodies for NFATc3 and NFATc4 were purchased from Abcam (Cambridge, MA, USA). Antibodies for Brd4, p-gsk3β, gsk3β, Bax, Bcl-2, p53, and c-Myc were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies for cytochrome C, COX IV, caspase-3, and cleaved caspase-3 were purchased from ABclonal (Wuhan, China), and anti-β-actin was obtained from Proteintech Group (Wuhan, China).
DAPI-stained images
AGS and HCT116 cells were seeded at a density of 1.25×104 cells/mL in six-well plates for 24 hours. Cells were exposed to AS or JQ1 alone or in combination in different concentrations when they were in the logarithmic growth phase and appropriate density. After corresponding treatments for 24 hours, cells were then fixed with 4% paraformaldehyde (PFA) for 15 minutes, washed with dulbecco’s phosphate-buffered saline (DPBS) (×3), and covered with DAPI staining solution (Beyotime, Wuhan, China) for 5 minutes in the dark. The cells were again washed with DPBS (×3) to remove the DAPI staining solution and observed under fluorescence microscopy.
Cell viability evaluation by MTT assay
Cell viability after AS and JQ1 treatment was evaluated by the MTT assay. AGS and HCT16 cells were seeded in 96-well plates at a density of 5×103 cells/0.1 mL per well. Twenty milliliters of MTT stock solution were added to each well after drug treatment. The supernatant was discarded, and 100 mL DMSO was added to dissolve the precipitate after incubation in the dark at 37°C for an additional 4 hours. The absorbance at 562 nm was used to determine cell viability using a microplate reader (Bio-TEK, Winoski, VT, USA). Cells incubated without any treatment were used as a control. Culture medium without cells was used as a blank control. Each sample was assayed in quadruples. The percentage of cell viability was calculated as follows:
|
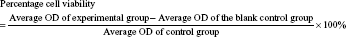
Western blotting
Cells were exposed to AS or JQ1 alone or in combination in different concentrations for 24 hours. Protein was extracted by lysing the cells in RIPA buffer with phenylmethanesulfonyl fluoride (Beyotime, Wuhan, China). Protein concentrations were determined using a microplate reader (Bio-TEK) with the enhanced BCA Protein Assay kit (Beyotime). Samples were diluted in a sample diluting buffer, heated at 95°C for 5 minutes, and loaded onto the gel and electrophoresed. Equal amounts of protein in each lane were separated by 8%–10% SDS-PAGE and transferred to a 0.45 μm polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). After blocking the membrane in 5% nonfat milk, the membrane was incubated with primary antibodies at 4°C overnight. Unbound antibodies were washed with PBS containing 0.1% Tween-20, followed by incubating with labeled secondary antibody at room temperature for 1 hour, washing again, and adding Immobilon Western Chemilum HRP Substrate (Millipore), and image was acquired using GelDoc XR System (Bio-Rad Laboratories Inc., Hercules, CA, USA). Band intensity was quantified using ImageJ software (NIH, Bethesda, MD, USA).
The extraction and isolation of mitochondrial and cytoplasmic protein
AGS cells were collected by a desired protocol after incubation with AS or JQ1 alone or in combination. The extraction and isolation of mitochondrial and cytoplasmic fractions were performed according to the ApoAlert® Cell Fractionation Kit (Clontech, Shiga, Japan). The AGS cells were first centrifuged at 600× g for 5 minutes at 4°C to remove supernatant and then resuspended in 1 mL of ice-cold wash buffer. Next, cells were centrifuged at 600× g for another 5 minutes at 4°C and were then resuspended in 0.8 mL of ice-cold Fractionation Buffer Mix (2 μL Protease Inhibitor Cocktail+1 μL DTT+1 mL 1× Fraction Buffer) after displacing supernatant. After the incubation on ice for 10 minutes, cells were then homogenized 50 passes in an ice-cold tissue grinder. The homogenate was then transferred to a 1.5 mL microcentrifuge tube, followed by centrifugation at 700× g for 10 minutes at 4°C. After centrifugation, the supernatant was transferred to a new, 1.5 mL tube and was centrifuged at 10,000× g for 25 minutes at 4°C. Finally, the pellet was resuspended in 0.1 mL Fractionation Buffer Mix as the mitochondrial fraction, and the supernatant was collected as the cytosolic fraction.
Quantitative real-time PCR (qPCR) analysis
Total RNA was extracted using RNeasy Mini Kit (Qiagen NV, Venlo, the Netherlands) according to the manufacturer’s protocol. First-strand cDNA synthesis and qPCR were performed as previously described.20 Genes were amplified using the primers as follows: NFATc1: 5′-ggagatggaagcgaaaactg-3′ (forward) and 5′-gcgggaaggtaggtgaaac-3′ (reverse); NFATc3: 5′-cacaccactttgcttaccacat-3′ (forward) and 5′-ccgttctgggtcatttatctgt-3′ (reverse); NFATc4: 5′-cttcccttcccagagtgatg-3′ and 5′-accttcctccagcgtgatac-3′ (reverse); GAPDH: 5′-ggcacagtcaaggctgagaatg-3′ (forward) and 5′-atggtggtgaagacgccagta-3′ (reverse). The primers were synthesized and purchased from Sangon Biotech (Shanghai, China). All qPCR reactions were run at the conditions: 3 minutes at 94°C followed by 40 seconds at 94°C, 40 seconds at 60°C, and 25 seconds at 72°C for 40 cycles. The expression data were normalized using the house-keeping gene GAPDH.
Annexin V/propidium iodide (PI) assays for apoptosis
AGS cells were seeded into six-well plates at a density of 1×105 cells per well and then maintained in the aforementioned medium, which was supplemented with AS or JQ1 alone or in combination. After drug treatment for 24 hours, the cell apoptosis was detected by flow cytometry (FCM) with an annexin V–fluorescein isothiocyanate (FITC)/PI apoptosis detection kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions.
Briefly, AGS cells were washed once in a PBS and once in a 1× binding buffer. Then, the AGS cells were resuspended in a 1× binding buffer, and 5 μL of annexin V was added to each sample. After incubation for 10 minutes at room temperature, the cells were washed with a 1× binding buffer. The apoptotic cells were then determined using a flow cytometer (FACSCalibur; BD Biosciences) after adding 5 μL of PI staining solution. Both early and late apoptotic cells were included in cell death detection.
FCM analysis of mitochondrial potential
The mitochondrial membrane potential (MMP) of AGS was detected using an MMP assay kit (JC-1; Beyotime). In accordance with the manufacturer’s instructions, AGS cells were seeded in six-well culture plates, pretreated with AS or JQ1 alone or in combination for 24 hours. After washing twice with D-Hank’s solution, the cells were then collected and digested with 0.5 mL TrypLE for 1 minute and centrifuged at 1,000 rpm at 4°C for 5 minutes. Five microliters of JC-1 dye (200 μM) were added to each sample and incubated at 37°C for 30 minutes and then measured using FCM (FACSCalibur; BD Biosciences).
Wound-healing assay
The wound-healing assay was performed as previously described.24 AGS cells were plated in six-well plates at a density of 5×105 cells/well and serum starved for 12 hours. Then, wounds were scratched with pipette tips, and the suspended cells were washed away with PBS. The cells were cultured with the corresponding concentration of AS or JQ1 or in combination in media containing 1% serum for 24 or 48 hours at 37°C and 5% CO.2 Wounds were photographed with a DMI3000 B inverted microscope (Leica Microsystems, Wetzlar, Germany) at 0, 6, 12, and 24 hours, and the migration rates of each group were calculated in accordance with the following formula:
|
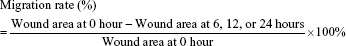
Statistical analyses
Statistical analyses were performed using SPSS software version 19.0 (IBM Corporation, Armonk, NY, USA). All data were presented as mean (SD). Data analysis was performed using one-way ANOVA, followed by either the least significant difference procedure (when the variance was equal) or the Games–Howell procedure (when the variance was unequal). Tukey’s post hoc test was used for multiple group comparisons, and Student’s t-test was used for single comparisons. A two-sided P<0.05 was considered as statistically significant.
Results
AS and JQ1 exerted synergistic cytotoxicity against GC and colon cancer cells
Previous studies showed that AS was able to exert anticancer activity in GC and colon cancer cells;19,20,22 here, we investigated whether AS could synergistically enhance the JQ1-mediated cytotoxicity in GC and colon cancer cells. First, we examined the inhibitory effect of AS or JQ1 alone on the cell viability in a panel of four GC and colon cancer cell lines that include GC cell lines, ie, AGS and MGC803, and colon cancer cell lines, ie, HCT116 and SW480. We found that treatment with AS at 1 μM for 24 hours partially inhibited cell viability in GC cells AGS and MGC803, while in colon cells HCT116 and SW480, treatment with 5 μM AS caused mild cell loss. This might attribute to the different drug sensitivity to AS between GC cells and colon cancer cells. We then treated the GC lines and colon cancer cell lines with different concentrations of AS in the following experiments. JQ1 at 1 μM was able to induce cell damage in all these four cell lines (Figure 1A). When cotreated with AS plus JQ1 for 24 hours, all the GC and colon cancer cells showed a significant decrease in cell viability compared with the single AS treatment. We then further assessed the apoptotic changes in nuclear morphology through DAPI staining. We found that in both GC cell line AGS and colon cancer cell line HCT116, the combination of AS and JQ1 resulted in the enhanced cytotoxicity compared with AS or JQ1 alone. In combination, signs of nuclear apoptosis including nuclear chromatin condensation and disassembled and fragmented nuclei were more obvious (Figure 1B). These results showed that AS profoundly enhanced the JQ1-induced cytotoxicity in GC and colon cancer cells.
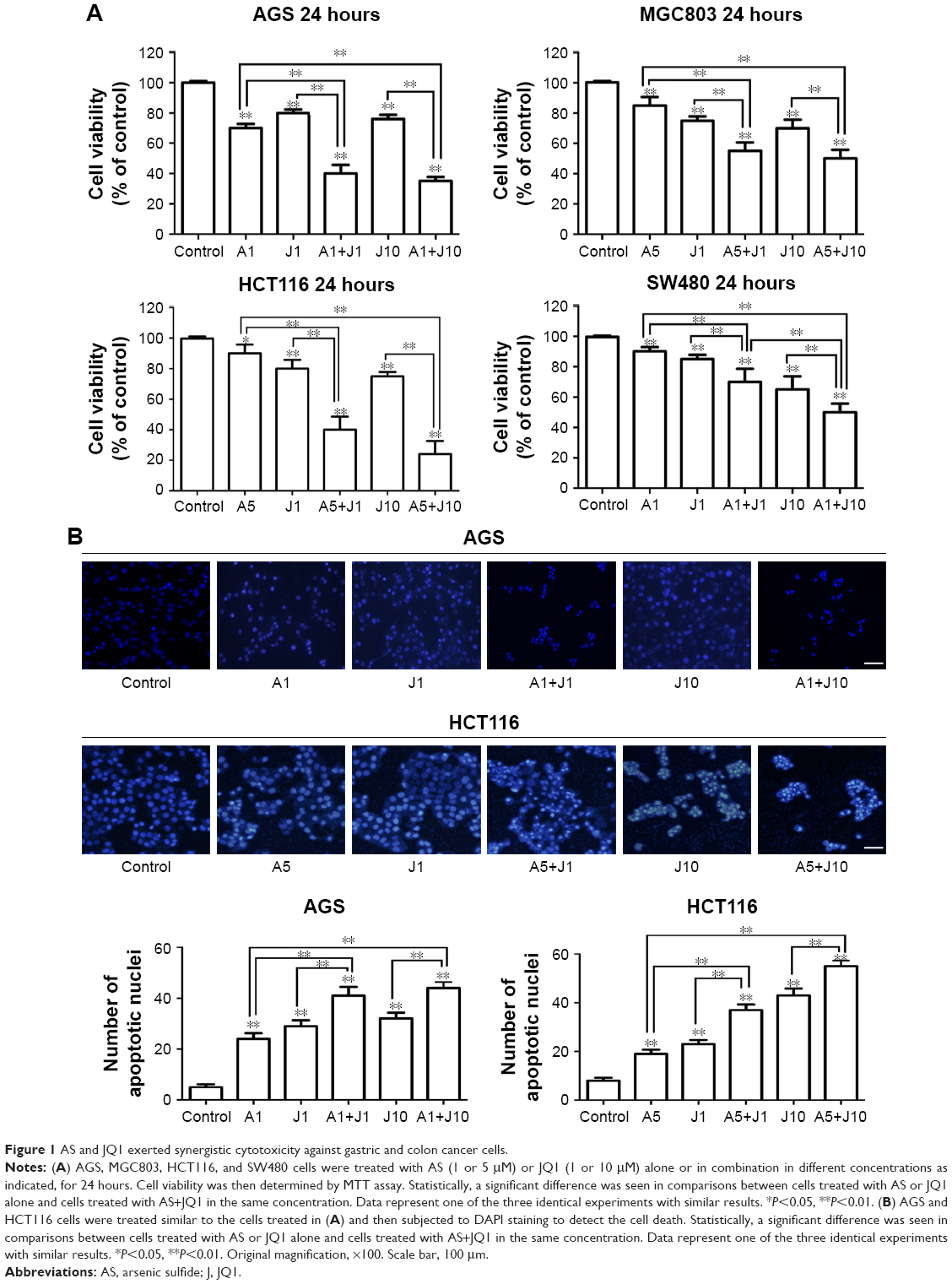 | Figure 1 AS and JQ1 exerted synergistic cytotoxicity against gastric and colon cancer cells. |
AS and JQ1 synergistically inhibited NFAT proteins in GC and colon cancer cells in both protein and RNA levels
Involved in cell cycle regulation, cell differentiation, and cell survival, NFAT proteins have also been proved to regulate tumor cell invasion and metastasis. Previous studies showed that AS exerted anticancer activity via NFAT pathway.20,21 Here, we investigated whether AS combined with JQ1 showed further inhibition of NFAT proteins in GC and colon cancer cells. We first analyzed the inhibitory effect of AS or JQ1 single treatment or the combined effect on NFAT proteins (NFATc1, NFATc3, and NFATc4) with the Western blotting analysis in four GC and colon cancer cells mentioned above. The results showed that AS combined with JQ1 profoundly reduced the protein levels of NFATc1, NFATc3, and NFATc4 in all these cell lines (Figure 2). However, the NFATc2 protein level did not show synchronous change after treatment with AS and JQ1 (data not shown). This might attribute to the intact-p53 gene in HCT116 cells and AGS cells, while p53 is mutated in MGC803 cells and SW480 cells. Then, we further confirmed this enhanced inhibitory effect in real-time PCR (Figure 3), which showed consistent results with the Western blotting analysis. When treated with both AS and JQ1, the mRNA levels of NFATc1, NFATc3, and NFATc4 decreased significantly compared with a single AS or JQ1 treatment in these four cell lines. These results suggested that AS and JQ1 synergistically inhibited NFAT proteins in GC and colon cancer cell lines in both mRNA and protein levels.
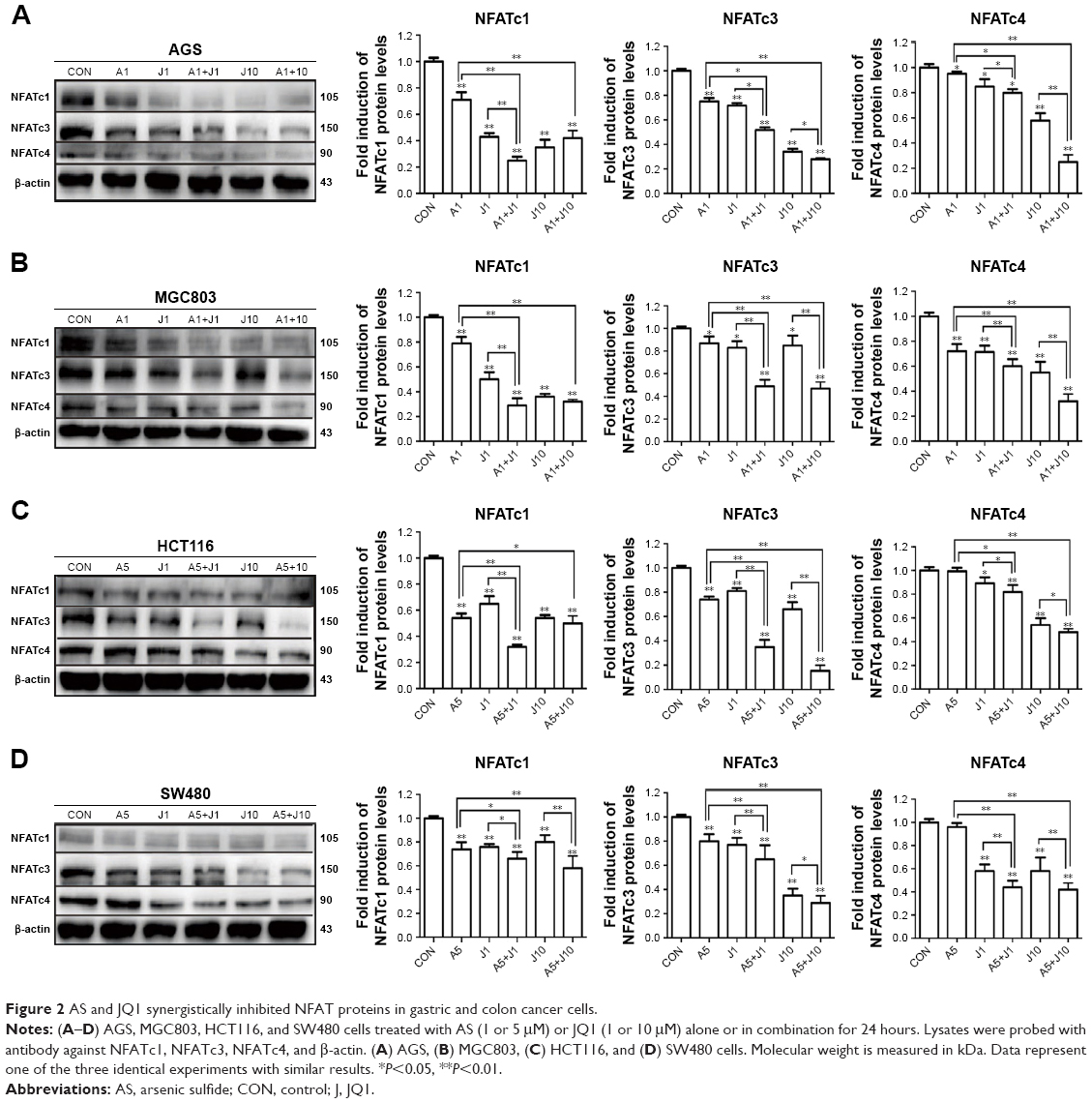 | Figure 2 AS and JQ1 synergistically inhibited NFAT proteins in gastric and colon cancer cells. |
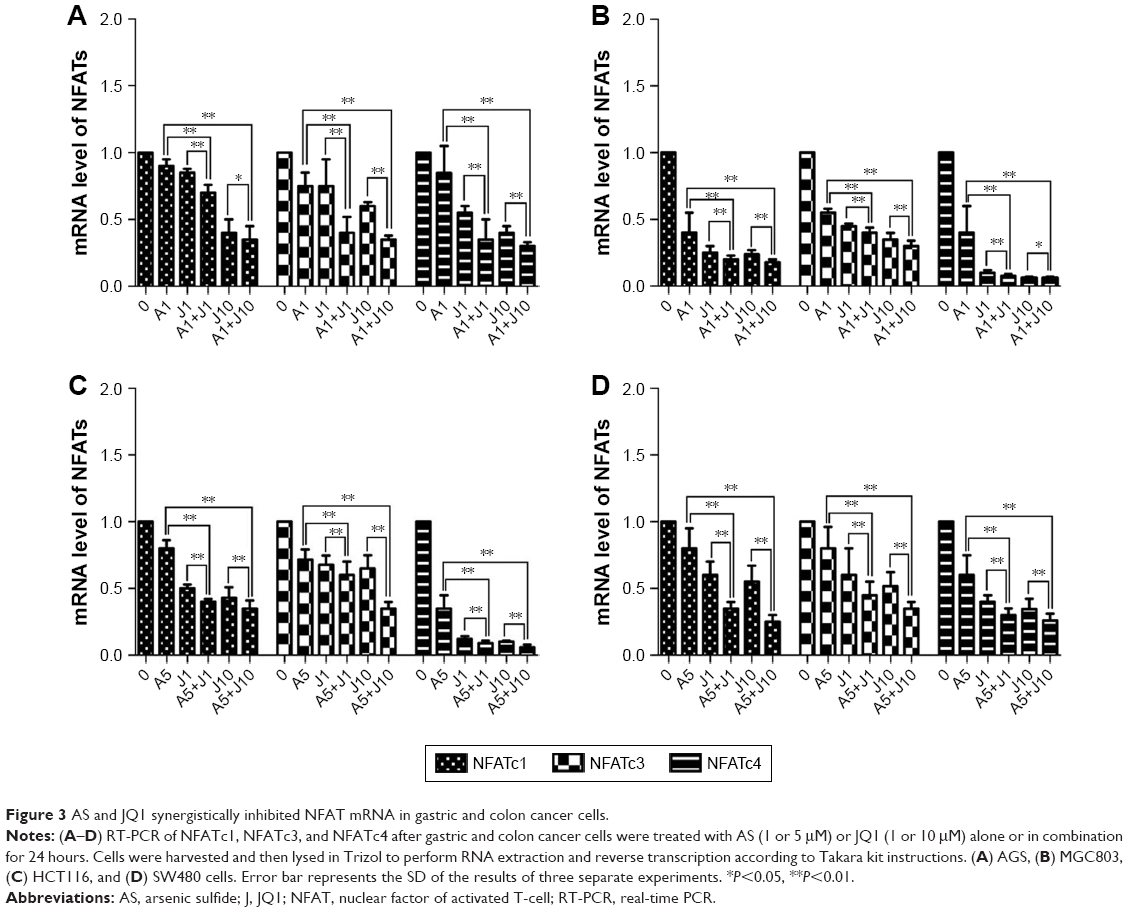 | Figure 3 AS and JQ1 synergistically inhibited NFAT mRNA in gastric and colon cancer cells. |
Combination treatment with AS and JQ1 synchronously modulated apoptosis-related proteins in GC and colon cancer cells
Apart from NFAT proteins, AS also modulates several apoptosis-related proteins.20 Hence, we investigated whether co-treatment with AS and JQ1 had a synergistic modulation in these apoptosis-related proteins via Western blotting analysis. Brd4, which is targeted by JQ1, seems not modulated by JQ1 in a dose-dependent manner, and this might contribute to the drug resistance of JQ1 in solid tumor therapy. BET domain proteins facilitate the transcription of target genes, including c-Myc.11 The previous study showed that JQ1 was able to reduce the expression of c-Myc in multiple myeloma models.11 Next, we investigated whether co-treatment with AS and JQ1 was able to synergistically decrease the expression of oncogene c-Myc. As shown in Figure 4, the c-Myc expression in AS and JQ1 co-treatment groups was remarkably lower than the AS- or JQ1-treated groups. AS and JQ1 also synergistically upregulated the pro-apoptotic protein Bax and downregulated the anti-apoptotic protein Bcl-2 in all these GC and colon cancer cells. Caspases are the central components in the execution of apoptosis. In general, they are involved in apoptosis and are divided into initiator and executioner caspases. Here, we also found that the combination of AS and JQ1 synergistically activated the caspase-3, the key executioner caspase in apoptosis. By using Western blot analysis, we found that the procaspase-3 was significantly decreased in cells co-treated with AS and JQ1, accompanied by an increase in their levels in cleaved caspase-3 (Figure 4).
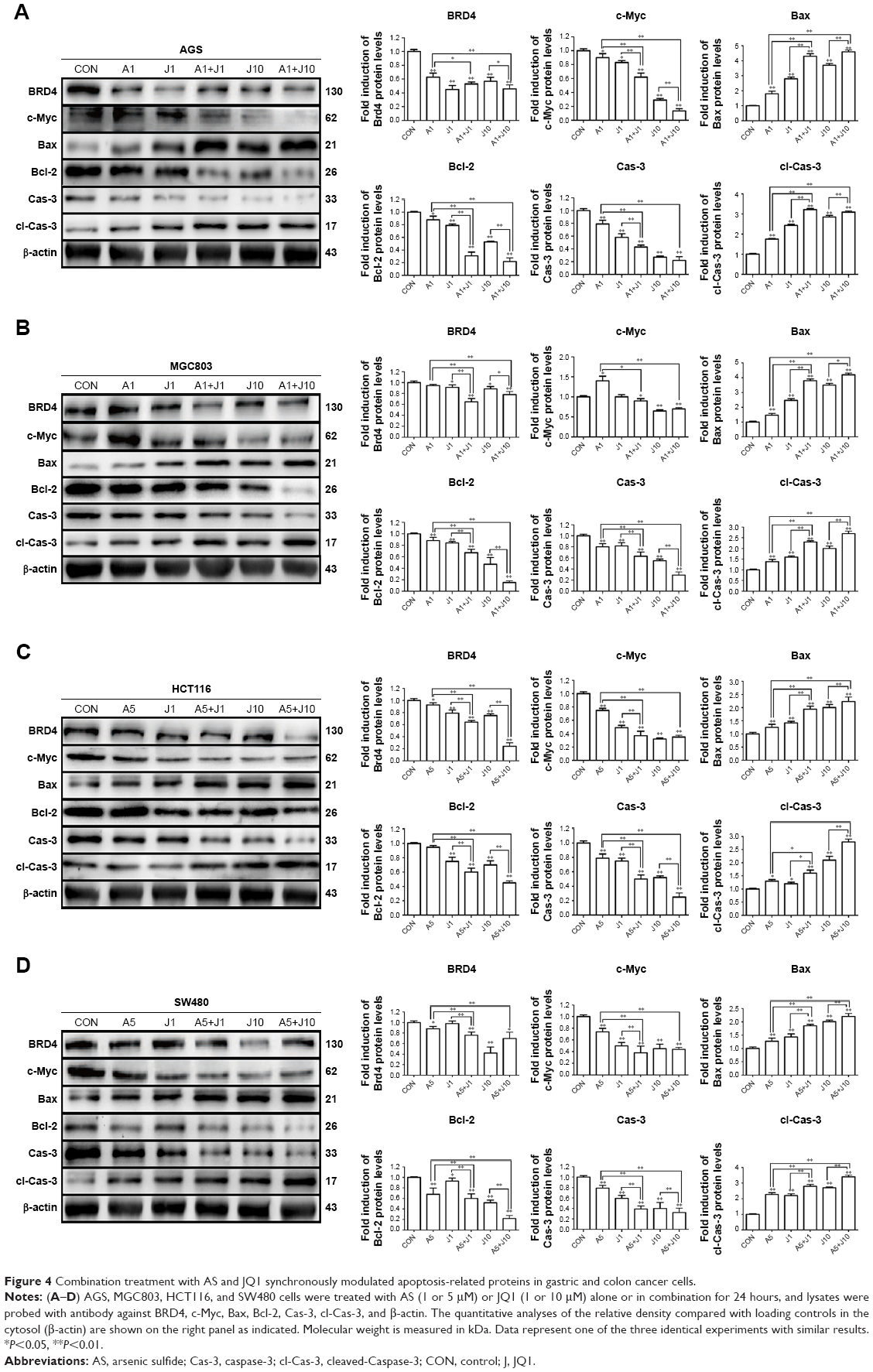 | Figure 4 Combination treatment with AS and JQ1 synchronously modulated apoptosis-related proteins in gastric and colon cancer cells. |
Combination treatment with AS and JQ1 induced mitochondrial-mediated apoptosis in GC and colon cancer cells
To determine whether AS and JQ1 synergistically induce cell apoptosis, AGS cells were treated with control solvent, 1 μmol/L AS, 1 μmol/L JQ1, 10 μmol/L JQ1, or the combination AS and JQ1 for 24 hours, followed by staining with annexin V–FITC and PI. Cells positively stained with annexin V alone (early apoptosis) or annexin V plus PI (late apoptosis) were counted as apoptotic cells. Treatment with 1 μmol/L AS or JQ1 alone slightly increased the percentage of apoptotic cells, while combination treatment with AS and JQ1 synergistically increased the apoptosis to 10.6% with 1 μmol/L JQ1 and 25.6% with 10 μmol/L JQ1, respectively (Figure 5A).
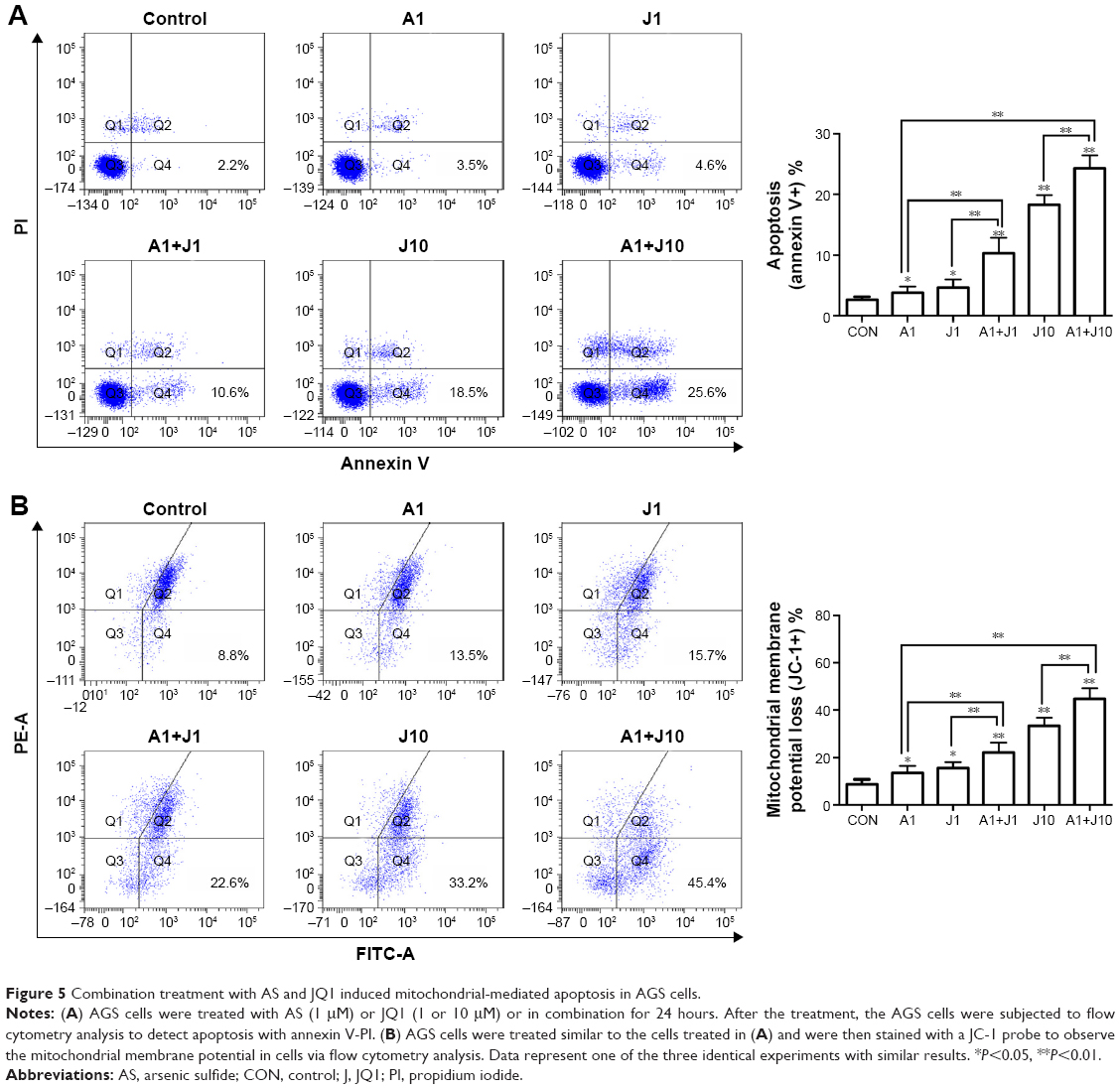 | Figure 5 Combination treatment with AS and JQ1 induced mitochondrial-mediated apoptosis in AGS cells. |
The mitochondrion is a therapeutic target of various anticancer drugs. In mammal cells, arsenic genotoxicity is mainly mediated by mitochondrial damage, including mitochondrial oxidative dysfunction. Arsenic alters the mitochondrial function by decreasing cytochrome c oxidase function and oxygen consumption.25,26 Apoptotic stimuli disrupt the electron transport in the mitochondria, leading to the MMP collapse. Therefore, we further examined the effect of the combination of AS and JQ1 on MMP in AGS cells (Figure 5B). A microprobe JC-1 assay kit was used to assess the change in MMP after exposing the cells to AS, JQ1, or the combination. JC-1 probe aggregated in normal mitochondria, and the color changed from red to green when membrane potential collapsed. After incubation with corresponding treatments for 24 hours, the MMP of AGS cells was analyzed with FCM. As shown in Figure 5B, treatment with 1 μmol/L AS or JQ1 alone mildly damaged the MMP, causing 13.5% and 15.7% MMP loss, respectively, whereas combination treatment of 1 μmol/L AS and JQ1 caused 22.6% MMP loss. The rate of MMP loss increased to 33.2% when treated with 10 μmol/L JQ1 alone. Consistently, 1 μmol/L AS plus 10 μmol/L JQ1 exerted additional toxicity on MMP and caused another 12.2% loss compared with treatment with 10 μmol/L JQ1 alone. These results revealed that co-treatment with AS and JQ1 synergistically impaired MMP in GC cells.
Mitochondrial depolarization elevates mitochondrial outer membrane permeabilization, facilitating the leakage of cytochrome c from dysfunctional mitochondria into the cytoplasm, which activates the caspase cascade and mitochondria-mediated apoptosis.27 To further confirm whether mitochondrial pathway was involved in the apoptosis induced by the combination treatment, we then performed subcellular fractionations in these four GC and colon cancer cells. We observed that the protein level of cytochrome c was markedly increased in the cytosolic fraction of cells co-treated with AS and JQ1, accompanied by a reduction of their levels in membrane fraction, as determined by Western blot analysis (Figure 6). Treatment with 1 μmol/L AS or JQ1 alone mildly induced cytochrome c release from mitochondria to the cytoplasm. While the treatment with a combination of 1 μmol/L AS and JQ1 caused a modest increase in cytochrome c level in the cytosolic fraction, accompanied by a marked reduction in the membrane fraction. Treatment with 10 μmol/L JQ1 alone was able to induce significant cytochrome c leakage from the mitochondria to the cytoplasm. However, treatment with 1 μmol/L AS plus 10 μmol/L JQ1 caused additional cytochrome c release in all these GC and colon cancer cells. All these data indicated that the combination of AS and JQ1 caused damage of mitochondrial membrane integrity, prompting the cytochrome c release into the cytoplasm and leading to the mitochondria-mediated apoptosis.
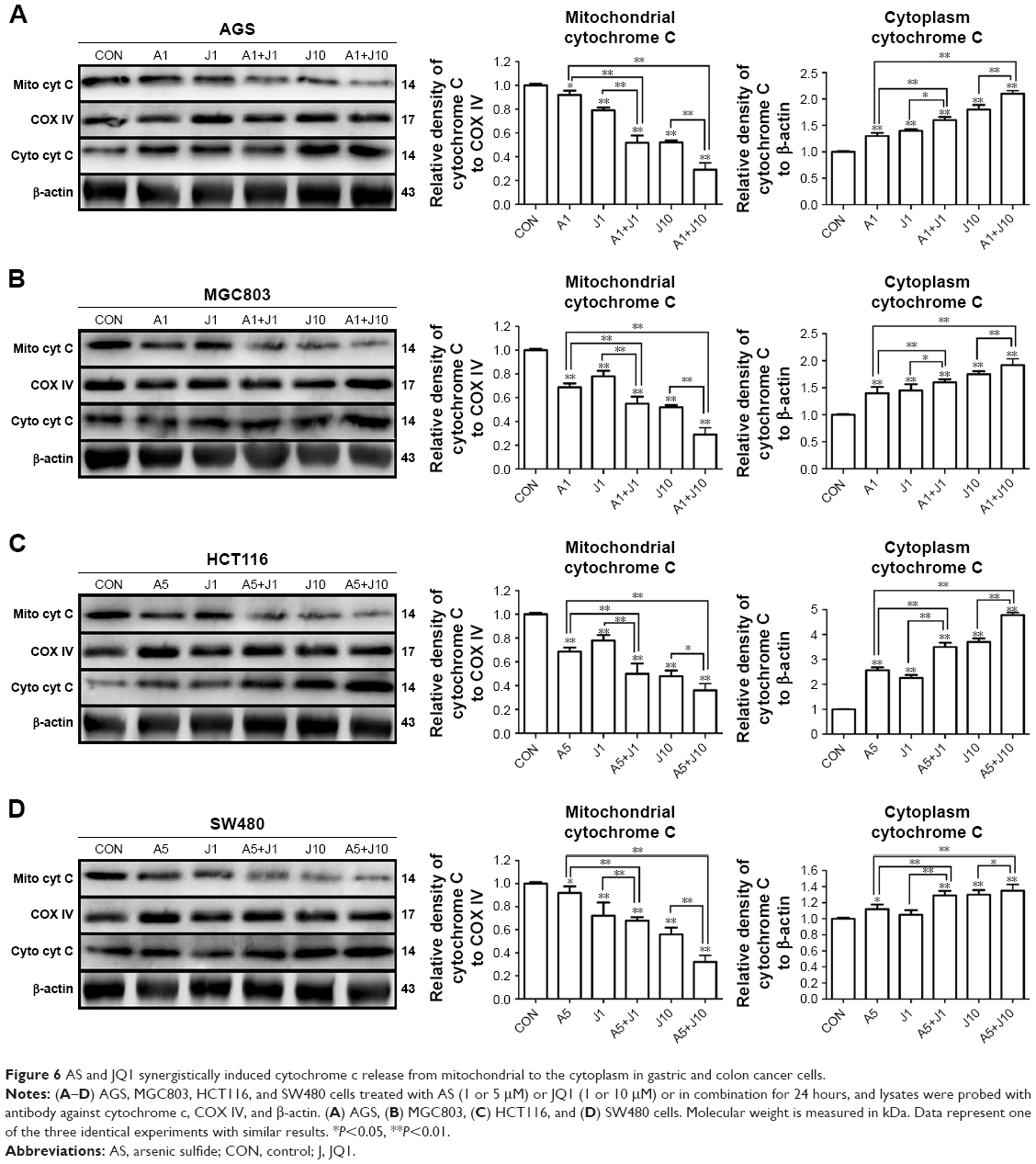 | Figure 6 AS and JQ1 synergistically induced cytochrome c release from mitochondrial to the cytoplasm in gastric and colon cancer cells. |
AS and JQ1 synergistically suppressed the motility of GC cells
Metastasis is the main characteristic of tumors and is responsible for the poor prognosis of advanced cancer. A wound healing assay was performed to verify whether co-treatment with AS and JQ1 synergistically attenuated the metastatic capability of GC cells. The AGS cells were treated with the single agent or the combination for 24 hours. As shown in Figure 7A, the wound-healing ability in AGS cells decreased slightly when treated with 1 μmol/L AS or JQ1, compared with the control group. 1 μmol/L combination treatment with AS and JQ1 or 10 μmol/L JQ1 alone modestly decreased the wound-healing ability. However, when co-treated with 1 μmol/L AS and 10 μmol/L JQ1, the AGS wound-healing ability reached another trough. The migration rates of AGS cells after corresponding treatment at 6 hours were 18.34%, 12.45%, 10.23%, 7.55%, 9.43%, and 5.31%, respectively. The rates at 12 hours were 28.48%, 20.58%, 24.65%, 15.49%, 12.75%, and 8.34%, respectively. Meanwhile, the rates at 24 hours were 39.33%, 29.63%, 31.67%, 25.47%, 16.27%, and 10.18%, respectively (Figure 7A).
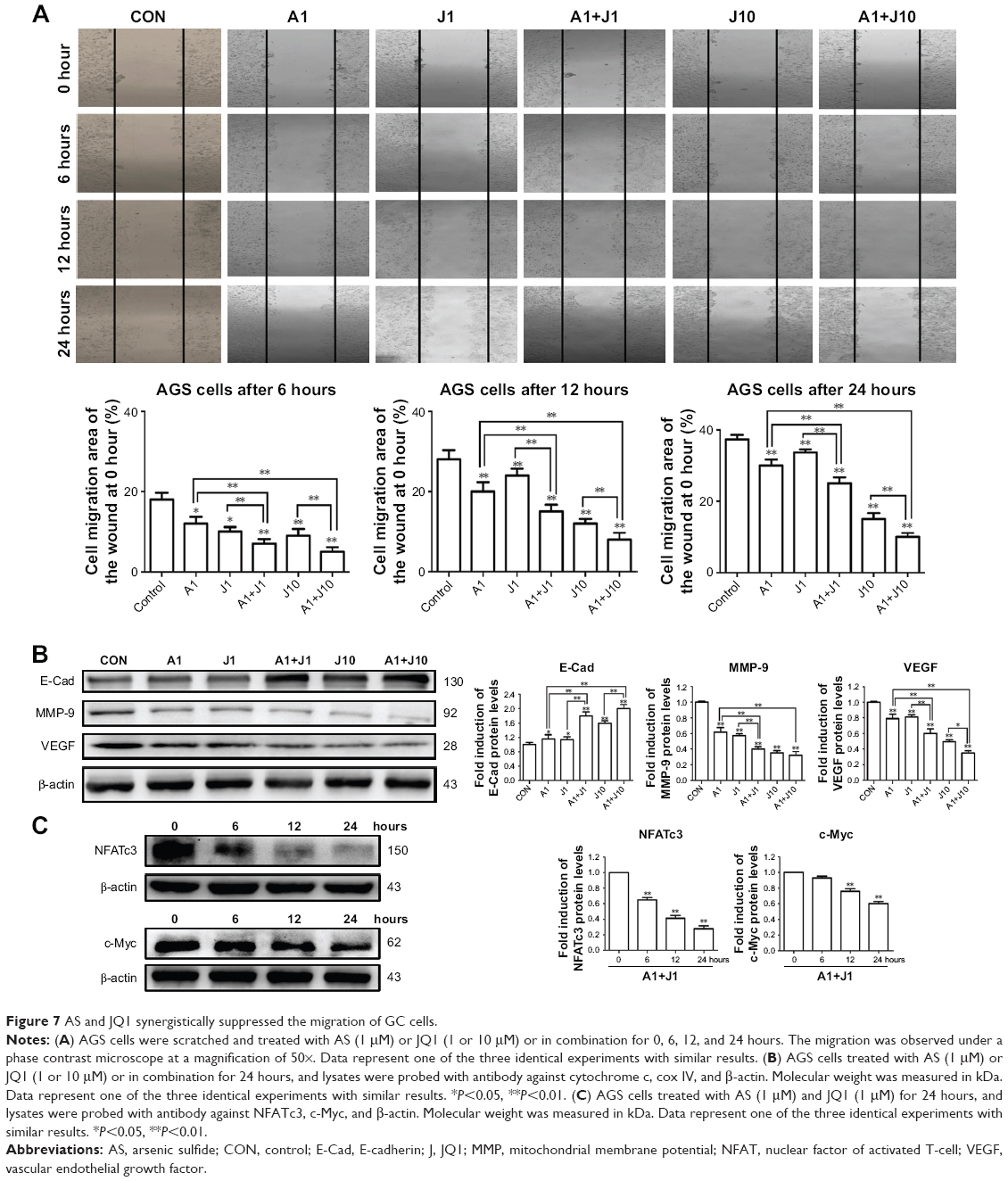 | Figure 7 AS and JQ1 synergistically suppressed the migration of GC cells. |
Various proteins are responsible for tumor invasion and metastasis, including MMP-9, E-cadherin, and VEGF. MMP-9 is highly expressed in tumor cells, and its expression is associated with invasiveness, tumor growth, and angiogenesis. E-catenin is one of the critical members of the complex adhesive connection of E-cadherin/catenin and plays a critical role in epithelial-mesenchymal transition and the metastatic process. VEGF, an effective factor that can induce angiogenesis, is closely associated with tumor metastasis and prognosis. To explore whether the decreased cell migration and invasion induced by co-treatment were associated with the activity of MMP-9, E-cadherin, and VEGF, we detected these proteins in AGS cells treated with AS, JQ1, or the combination for 24 hours via Western blot analysis. In consistency, with the wound-healing assay, the levels of metastasis-related proteins, including MMP-9, E-cadherin, and VEGF, were significantly decreased by co-treatment of AS and JQ1 (Figure 7B).
To further confirm that NFATc3 and c-Myc are key molecules involved in the apoptotic signaling pathway, we also examined their expression at earlier time points by performing Western blot analysis. The protein level of NFATc3 was significantly inhibited by AS and JQ1 at 6 hours, while the significant inhibition of c-Myc was observed at 12 hours. This suggests that NFATc3 and c-Myc are involved in the apoptotic signaling pathways induced by AS and JQ1 (Figure 7C).
Discussion
The current study evaluated the effects of BET inhibitor JQ1 combined with AS on the cellular viability of GC and colon cancer cells. The GC and colon cancer are the leading causes of cancer death in the world with unsatisfactory prognosis and few satisfying therapies. Previous studies have illustrated that the pharmacologic inhibition of BET proteins induces cell growth arrest and cell apoptosis in various disease models. Evidence of safety and efficacy of the BET inhibitors in preclinical testing is now promoting the rapid development of these novel specific compounds for further clinical evaluation. As a first-in-class BET inhibitor, JQ1 interferes with the ability of BRD4 to identify the acetylated chromatin marks that facilitate gene transcriptional activation through competitively binding to the acetyl-lysine recognition area of BRD4. Clinical studies have also shown favorable responses of JQ1 in hematological malignancies, while the bioactivity of these novel agents is generally limited in solid cancers. Cancer remission can be hardly induced by BET inhibitors in most preclinical tumor models, and few obvious clinical responses were induced within solid tumors. Hence, overcoming drug resistance of BET inhibitors through combinational strategies in diverse types of cancers has been the focus of recent studies. It has been reported that JQ1 synergized ABT-263 against CRC cells.28 Oridonin synergistically enhances JQ1-triggered apoptosis in hepatocellular cancer cells.29 The histone deacetylase inhibitor panobinostat and JQ1 synergistically exert anticancer effects in aggressive neuroblastoma.30 Here, we reported another combinational strategy to improve the cytotoxicity of BET inhibitors in cancer therapy by co-treatment with AS and JQ1. In this strategy, AS showed the ability to override the apoptosis resistance to BET inhibitors in GC and colon cancer cells. AS has excellent cytotoxic activity in acute promyelocytic leukemia and exerts a synergistic inhibitory effect on cell growth and c-Myc expression in colorectal cells with cyclosporine A.20 It has been proved that both JQ1 and AS showed antitumor activity in several cancer types via suppressing c-Myc transcription. This supplys the rationale to evaluate that combination treatment of AS and JQ1 may exert synergistic cell killing effect in GC and colon cancer.
The ineffectiveness of apoptosis induction contributes to JQ1 resistance in solid tumor cells.16 Here, we demonstrated that, by inhibiting the activity of the BET protein BRD4 and the resulting inhibition of c-Myc, co-treatment with AS and JQ1 attenuated the expression of several prosurvival genes, including NFATs and Bcl-2, in GC and colon cancer cells. This resulted in the JQ1-mediated growth inhibition and apoptosis of GC and colon cancer cells. Our findings also showed that co-treatment with AS and JQ1 induced mitochondrial dysfunction of GC and colon cancer cells. Treatment with AS and JQ1 is highly effective in inducing MMP collapse and cytochrome c release, leading to the activation of the apoptosis cascade. We also demonstrated here that co-treatment with AS and JQ1 exerted synergistic in vitro lethality against GC and colon cancer cells. In addition to their abilities to induce apoptosis, our findings also highlight that co-treatment with AS and JQ1 synergistically decreased the invasion and metastasis ability of GC and colon cancer cells.
As reported for AML and diffuse large B-cell lymphoma cells, inhibition of BRD4 by JQ1 also attenuates the levels of c-Myc. BRD4 preferentially localizes to “super-enhancer” regions upstream of a variety of oncogenes, including c-Myc, to regulate their expression.31 c-Myc is a master regulator of cell metabolism and is central to the pathogenesis of many human cancers, by the coordinated upregulation of a transcriptional program influencing metabolic adaptation, cell division, and survival.13,32 c-Myc also promotes transformation and maintenance of stem cells in genetically engineered mouse models of glioblastoma.33 The angiogenesis and invasion of tumor cells also involve c-Myc activation.34,35 Furthermore, conditional transgenic models featuring tunable transcriptional suppression have shown that even transient inactivation of c-Myc results in sustained regression of tumors.36
Consistent with this, our findings demonstrated that AS and JQ1 synergistically repressed the c-Myc expression while exerted in vitro lethality against GC and colon cancer cells. c-Myc was previously known to be activated by NFATc1,37,38 while NFATc3 overexpression also markedly increased c-Myc,20 suggesting that c-Myc was under the regulation of NFATs. The NFAT involves in the cancer initiation and progression, including cell proliferation, invasion, and metastasis.21 NFATc1 promotes cell proliferation in hepatocellular carcinoma cells,39 NFATc2 increases the invasiveness in breast cancer cells,40 and NFATc3 and NFATc4 contribute to the progression of colon cancer.20 Here, we found that AS combined with JQ1 profoundly reduced the protein levels of NFATc1, NFATc3, and NFATc4 in most GC and colon cancer cells, which was in consistency with the downregulated c-Myc protein level and the impaired cell viability.
JQ1 resistance in solid tumor cells leads to the ineffectiveness of apoptosis induction, while mitochondrial protection impairs BET bromodomain inhibitor-mediated cell death. This provides a rationale for the combination therapeutic strategies through mitochondrial-mediated apoptosis in cancer therapy. Mitochondrion, containing key regulators of caspases, is the important mediator of the intrinsic apoptosis pathway.41 Once mitochondria membrane permeability was altered with the decline of MMP, the pro-apoptotic molecules will release into the cytoplasm and trigger the downstream apoptosis proceeding. AS, the main component of realgar, showed modest cytotoxic activity against solid tumors via inducing cell cycle arrest and apoptosis. When combined with cisplatin, irinotecan, or celecoxib, AS showed additional cytotoxicity against tumor cells.22 To verify whether the collapse of MMP occurred among apoptosis induced by the combination treatment of AS and JQ1, the FCM was performed with JC-1 staining. In the current study, we found that co-treatment of AS and JQ1 decreased the MMP profoundly, showing further inhibition of the MMP than treatment with either AS or JQ1 alone. Accompanied by MMP depolarization, an apoptotic insult leads to matrix condensation and leakage of cytochrome c into the intermembrane space, facilitating cytochrome c release and the cell death. Hence, we then detected the expression of cytochrome c and caspase-3 with Western blot analysis. The results proved that the combination treatment of AS and JQ1 synergistically induced cytochrome c release and activated downstream caspase-3. Released cytochrome c then targeted the cytosolic protein Apaf-1 in the presence of dATP, facilitating the formation of protein complexes known as “apoptosome”. The apoptosome further activated pro-caspase-9, which in turn leads to the activation of caspase-3 and DNA fragmentation. AS and JQ1 also synergistically increased the level of Bax while inhibiting the protein level of Bcl2. As both Bcl-2 and Bax are key molecules in regulating mitochondria-mediated apoptosis, their imbalance contributes to the dysfunction of mitochondria and the release of pro-apoptotic factors. The Bcl-2 protein may be degraded by activated caspase-3 and thus weakens its anti-apoptotic property. Once the procaspase-3 is activated after cleavage, the occurrence of apoptosis is irreversible.
We previously reported that AS in combination with JQ1 showed enhanced cytotoxicity on GC and colon cancer cells through inducing cell apoptosis with the underlying mechanism unknown.22 Here, in this article, we further elucidated that the combination of AS and JQ1 showed synergistic cytotoxicity on GC and colon cancer cells via mitochondria-mediated apoptosis pathway (Figure 8). In addition, AS and JQ1 co-treatment effectively suppressed the NFATs and c-Myc proteins, which are responsible for the decreased cell viability and impaired invasiveness and metastasis.21,34,35 c-Myc was previously known to be activated by NFATc137,38 and NFATc3.21 Once the NFATs were decreased, the expression of the downstream c-Myc was also suppressed, which leads to the increase in apoptotic proteins, including Bcl-2, Bax, and caspase-3 and cell apoptosis. The combination of AS and JQ1 has a synergistic inhibitory effect on tumor cells while reducing the dose of each single drug, which may be instructive for clinical treatment strategy. In conclusion, the data of the present study indicated that AS and JQ1 combinational therapy might be a prognostic strategy for the cancer cure and also warrants further investigation in multiple myeloma and acute myeloid leukemia.
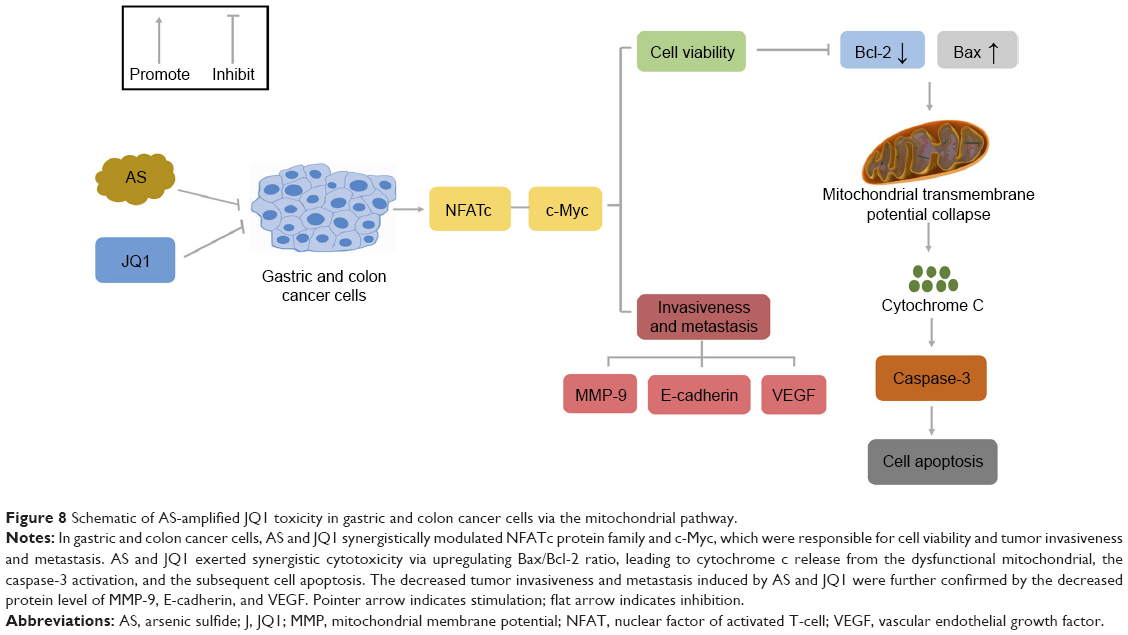 | Figure 8 Schematic of AS-amplified JQ1 toxicity in gastric and colon cancer cells via the mitochondrial pathway. |
Conclusion
AS effectively enhances the cytotoxicity of JQ1 in GC and colon cancer cells through mitochondrial-mediated apoptosis induction. The combinational strategy of AS and JQ1 may provide potential application of BET bromodomain inhibitor in cancer therapy.
Acknowledgment
This work was supported by the National Science Foundation of China (81274142 and 81874353) and the Science and Technology Commission of Shanghai Municipality (17401931300 and 11ZR1423400).
Author contributions
Zhen Tan and Xiuli Zhang designed and performed most of the experiments. Ting Kang performed some Western blotting experiments. Lian Zhang provided some advice. Siyu Chen conceived the concept and designed and supervised all the experiments. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388(10060):2654–2664. | ||
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Griffin MR, Bergstralh EJ, Coffey RJ, Beart RW Jr, Melton LJ 3rd. Predictors of survival after curative resection of carcinoma of the colon and rectum. Cancer. 1987;60(9):2318–2324. | ||
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399(6735):491–496. | ||
Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282(18):13141–13145. | ||
Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12(7):465–477. | ||
Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108(40):16669–16674. | ||
Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528. | ||
Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. | ||
Ott CJ, Kopp N, Bird L, et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120(14):2843–2852. | ||
Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. | ||
Puissant A, Frumm SM, Alexe G, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3(3):308–323. | ||
Bandopadhayay P, Bergthold G, Nguyen B, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res. 2014;20(4):912–925. | ||
Kurimchak AM, Shelton C, Duncan KE, et al. Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Rep. 2016;16(5):1273–1286. | ||
Mazur PK, Herner A, Mello SS, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21(10):1163–1171. | ||
Chen SJ, Zhou GB, Zhang XW, Mao JH, de Thé H, Chen Z. From an old remedy to a magic bullet: molecular mechanisms underlying the therapeutic effects of arsenic in fighting leukemia. Blood. 2011;117(24):6425–6437. | ||
Zhang L, Kim S, Ding W, et al. Arsenic sulfide inhibits cell migration and invasion of gastric cancer in vitro and in vivo. Drug Des Devel Ther. 2015;9:5579–5590. | ||
Ding W, Tong Y, Zhang X, Pan M, Chen S. Study of arsenic sulfide in solid tumor cells reveals regulation of nuclear factors of activated T-cells by PML and p53. Sci Rep. 2016;6:19793. | ||
Zhang X, Kang T, Zhang L, Tong Y, Ding W, Chen S. NFATc3 mediates the sensitivity of gastric cancer cells to arsenic sulfide. Oncotarget. 2017;8(32):52735–52745. | ||
Zhang L, Tong Y, Zhang X, Pan M, Chen S. Arsenic sulfide combined with JQ1, chemotherapy agents, or celecoxib inhibit gastric and colon cancer cell growth. Drug Des Devel Ther. 2015;9:5851–5862. | ||
Zhang L, Tian W, Kim S, Ding W, Tong Y, Chen S. Arsenic sulfide, the main component of realgar, a traditional Chinese medicine, induces apoptosis of gastric cancer cells in vitro and in vivo. Drug Des Devel Ther. 2015;9:79–92. | ||
Pan X, Han H, Wang L, et al. Nitidine Chloride inhibits breast cancer cells migration and invasion by suppressing c-Src/FAK associated signaling pathway. Cancer Lett. 2011;313(2):181–191. | ||
Partridge MA, Huang SX, Hernandez-Rosa E, Davidson MM, Hei TK. Arsenic induced mitochondrial DNA damage and altered mitochondrial oxidative function: implications for genotoxic mechanisms in mammalian cells. Cancer Res. 2007;67(11):5239–5247. | ||
Liu SX, Davidson MM, Tang X, et al. Mitochondrial damage mediates genotoxicity of arsenic in mammalian cells. Cancer Res. 2005;65(8):3236–3242. | ||
Liu T, Chen XM, Sun JY, et al. Palmitic Acid-Induced Podocyte Apoptosis via the Reactive Oxygen Species-Dependent Mitochondrial Pathway. Kidney Blood Press Res. 2018;43(1):206–219. | ||
Wu Z, Hu Z, Han X, et al. The BET-Bromodomain Inhibitor JQ1 synergized ABT-263 against colorectal cancer cells through suppressing c-Myc-induced miR-1271-5p expression. Biomed Pharmacother. 2017;95:1574–1579. | ||
Zhang HP, Li GQ, Guo WZ, et al. Oridonin synergistically enhances JQ1-triggered apoptosis in hepatocellular cancer cells through mitochondrial pathway. Oncotarget. 2017;8(63):106833–106843. | ||
Shahbazi J, Liu PY, Atmadibrata B, et al. The Bromodomain Inhibitor JQ1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin Cancer Res. 2016;22(10):2534–2544. | ||
Lovén J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. | ||
Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. 2013;3(8):a014217. | ||
Zheng H, Ying H, Yan H, et al. Pten and p53 converge on c-Myc to control differentiation, self-renewal, and transformation of normal and neoplastic stem cells in glioblastoma. Cold Spring Harb Symp Quant Biol. 2008;73:427–437. | ||
Ciribilli Y, Borlak J. Oncogenomics of c-Myc transgenic mice reveal novel regulators of extracellular signaling, angiogenesis and invasion with clinical significance for human lung adenocarcinoma. Oncotarget. 2017;8(60):101808–101831. | ||
Li H, Jin Y, Hu Y, et al. The PLGF/c-MYC/miR-19a axis promotes metastasis and stemness in gallbladder cancer. Cancer Sci. 2018;109(5):1532–1544. | ||
Jain M, Arvanitis C, Chu K, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297(5578):102–104. | ||
Buchholz M, Schatz A, Wagner M, et al. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006;25(15):3714–3724. | ||
Köenig A, Linhart T, Schlengemann K, et al. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterology. 2010;138(3):1189–1199. | ||
Wang S, Kang X, Cao S, Cheng H, Wang D, Geng J. Calcineurin/NFATc1 pathway contributes to cell proliferation in hepatocellular carcinoma. Dig Dis Sci. 2012;57(12):3184–3188. | ||
Foldynová-Trantírková S, Sekyrová P, Tmejová K, et al. Breast cancer-specific mutations in CK1epsilon inhibit Wnt/beta-catenin and activate the Wnt/Rac1/JNK and NFAT pathways to decrease cell adhesion and promote cell migration. Breast Cancer Res. 2010;12(3):R30. | ||
Javadov S, Karmazyn M. Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell Physiol Biochem. 2007;20(1–4):1–22. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.