")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Animal models of ischemic stroke and their application in clinical research
Authors Fluri F, Schuhmann M, Kleinschnitz C
Received 18 March 2015
Accepted for publication 9 April 2015
Published 2 July 2015 Volume 2015:9 Pages 3445—3454
DOI https://doi.org/10.2147/DDDT.S56071
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Shu-Feng Zhou
Felix Fluri, Michael K Schuhmann, Christoph Kleinschnitz
Department of Neurology, University Clinic Wuerzburg, Wuerzburg, Germany
Abstract: This review outlines the most frequently used rodent stroke models and discusses their strengths and shortcomings. Mimicking all aspects of human stroke in one animal model is not feasible because ischemic stroke in humans is a heterogeneous disorder with a complex pathophysiology. The transient or permanent middle cerebral artery occlusion (MCAo) model is one of the models that most closely simulate human ischemic stroke. Furthermore, this model is characterized by reliable and well-reproducible infarcts. Therefore, the MCAo model has been involved in the majority of studies that address pathophysiological processes or neuroprotective agents. Another model uses thromboembolic clots and thus is more convenient for investigating thrombolytic agents and pathophysiological processes after thrombolysis. However, for many reasons, preclinical stroke research has a low translational success rate. One factor might be the choice of stroke model. Whereas the therapeutic responsiveness of permanent focal stroke in humans declines significantly within 3 hours after stroke onset, the therapeutic window in animal models with prompt reperfusion is up to 12 hours, resulting in a much longer action time of the investigated agent. Another major problem of animal stroke models is that studies are mostly conducted in young animals without any comorbidity. These models differ from human stroke, which particularly affects elderly people who have various cerebrovascular risk factors. Choosing the most appropriate stroke model and optimizing the study design of preclinical trials might increase the translational potential of animal stroke models.
Keywords: permanent and transient middle cerebral artery occlusion, photothrombosis, endothelin-1, microsphere/macrosphere, thromboembolic clot model, rat, mouse
Introduction
Stroke is the fifth cause of death in the USA1 and the leading medical cause of acquired adult disability worldwide.2 Approximately 80% of strokes are ischemic in nature and result from thromboembolic occlusion of a major cerebral artery or its branches. Vascular occlusion results in deprivation of oxygen and energy, followed by the formation of reactive oxygen species, release of glutamate, accumulation of intracellular calcium, and induction of inflammatory processes.3 This sequence of events, termed the ischemic cascade, leads to irreversible tissue injury (infarction). The ischemic penumbra – that is, the area of ischemic brain tissue surrounding the infarcted core – is potentially salvageable if an appropriate treatment is administered within a specified therapeutic window.4 Two major approaches have been developed to treat ischemic stroke: neuroprotection and reperfusion. The latter therapeutic strategy uses thrombolytic drugs or mechanical devices to recanalize occluded vessels. The only approved medical treatment for acute ischemic stroke is intravenous thrombolysis with recombinant tissue plasminogen activator (rtPA).5 However, the therapeutic window of rtPA treatment is up to 4.5 hours after stroke, and consequently, rtPA is applicable as treatment in only up to 5% of all patients.6 Thus, there is an urgent need for other, more widely applicable, treatment options.
Over the last four decades, a variety of animal stroke models have been developed, with the aim of identifying the mechanisms that underlie cerebral ischemia and developing new agents for stroke therapy. Animal stroke models are an indispensable tool for several reasons: (1) Human ischemic stroke is extremely diverse in its manifestations, causes, and anatomic localization, whereas an experimental ischemic stroke is highly reproducible, well controllable, and standardized, allowing more precise analysis of stroke pathophysiology and drug effects. (2) Molecular, genetic, biochemical, and physiological investigations often require invasive direct access to brain tissue. (3) Pathophysiological events occurring during the first minute of an ischemic stroke are most often not detectable by imaging techniques used in human stroke and thus can be studied only in an animal model. (4) Perfusion and vasculature (eg, collaterals) are essential in the pathophysiology of stroke and cannot be modeled in in vitro models. However, with the exception of rtPA, which was first investigated in a rabbit stroke model,7 no preclinical-tested neuroprotective agent, either as a single therapy8 or as combination therapy,9 has been translated into effective stroke therapies. The poor translational power of preclinical stroke models has led to the establishment of the Stroke Therapy Academic Industry Roundtable (STAIR), which issues recommendations to improve the quality of preclinical stroke research.10 However, even strict adherence to these recommendations has not resulted in major progress in clinical translation. The success of preclinical stroke research in developing new therapeutics might rely – at least in part – on the selection of the appropriate animal stroke model to use. This review deals with the most common rodent stroke models and also discusses the advantages and limitations of each model, as well as factors influencing the outcome of the model (Table 1).
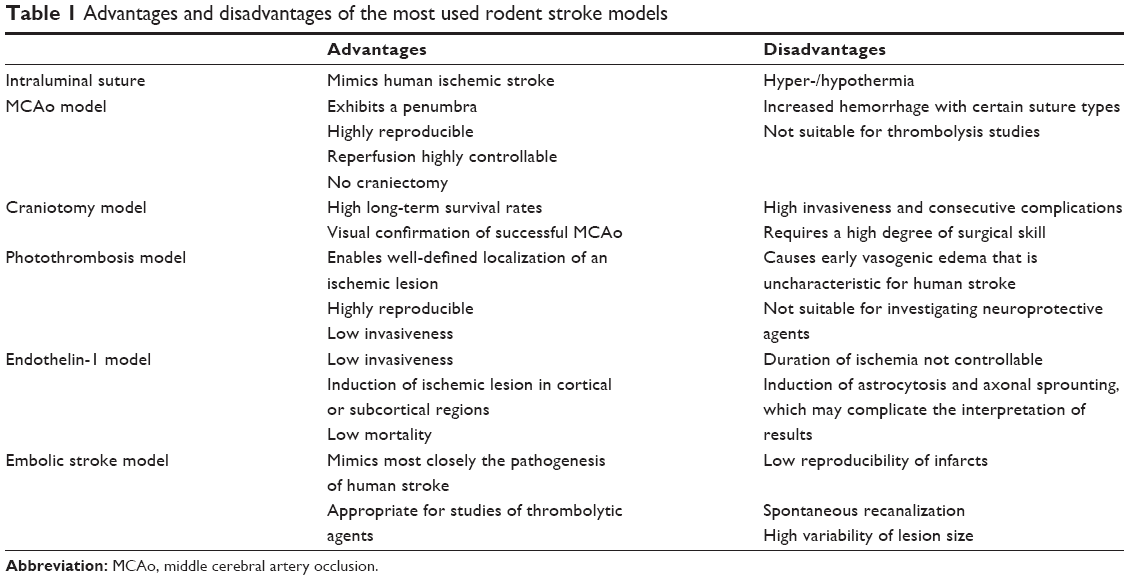 | Table 1 Advantages and disadvantages of the most used rodent stroke models |
Selection of animals for ischemic stroke
The majority of stroke experiments are carried out in small animals (eg, mice, rats, rabbits). The use of small animals presents clear advantages – lower cost and greater acceptability from an ethical perspective – compared to larger animals. The rat is one of the most commonly used animals in stroke studies for many reasons: the cerebral vasculature and physiology of the rat is similar to that of humans;11 its moderate body size allows easy monitoring of physiologic parameters; its small brain size is well suited to fixation procedures (eg, in vivo freeze trapping for biochemical analysis);12 there is a relative homogeneity within strains;13 and most of all, it is easy to conduct reproducible studies. As the mouse is the most appropriate animal in which genetic modifications can be created, it is widely used in transgenic technology studies that assess the molecular pathophysiology of stroke.14,15 According to the STAIR recommendations, a positive result achieved from a drug study in a species should always be verified in another species and should be replicable in a further stroke model (ie, permanent and then transient occlusion model).10
Intraluminal suture MCAo model
The middle cerebral artery (MCA) and its branches are the cerebral vessels that are most often affected in human ischemic stroke, accounting for approximately 70% of infarcts.16 Thus, techniques that occlude this artery are closest to human ischemic stroke.17–19 Among the occlusive MCA stroke models, intra-arterial suture occlusion of the MCA (MCAo) is the most common method in rodents, which has been used in >40% of approximately 2,600 experiments on neuroprotection after ischemic stroke.20
Technically, the MCAo model is less invasive and does not require craniectomy and thus avoids damage to cranial structures. This technique involves temporarily occluding the common carotid artery (CCA), introducing a suture directly into the internal carotid artery (ICA), and advancing the suture until it interrupts the blood supply to the MCA (Figure 1). In a modified manner, the suture is inserted into the transected external carotid artery (ECA), using the ECA trunk as a path to advance a suture through the ICA. Laser Doppler flowmetry can be a useful tool for ensuring complete MCAo.21 This method enables permanent MCAo or transient ischemia with reperfusion. The ECA approach is a better choice for transient MCAo because it maintains the anatomic integrity that is required for reperfusion.22
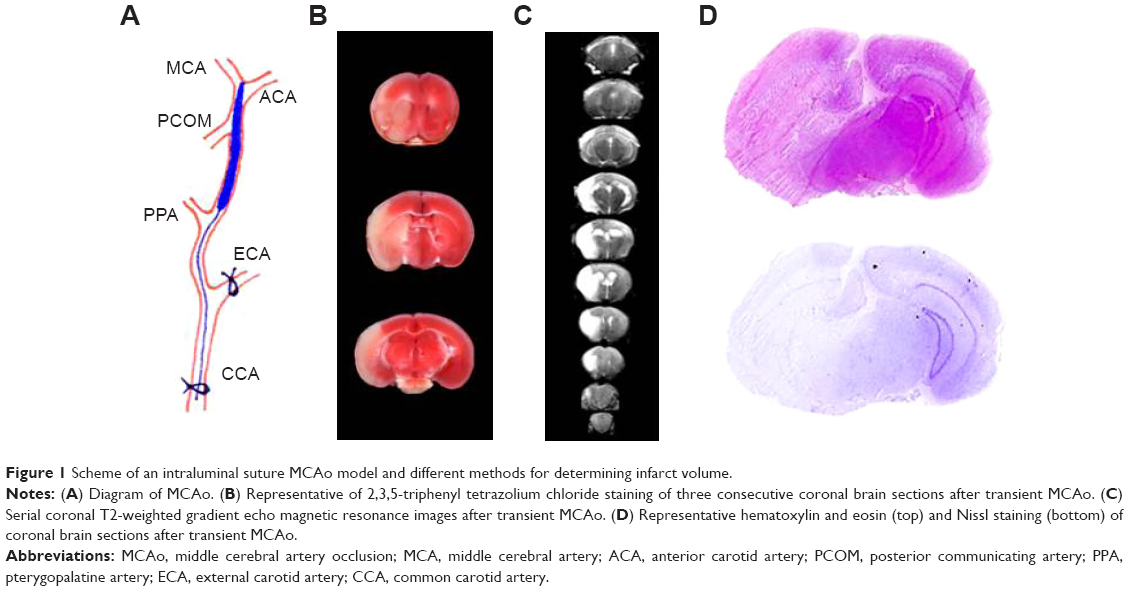 | Figure 1 Scheme of an intraluminal suture MCAo model and different methods for determining infarct volume. |
The most common durations of MCAo using a suture are 60 minutes, 90 minutes, and 120 minutes, or permanent MCAo in rats; the success rate of inducing an infarction is 88%–100%22 and that of subarachnoid hemorrhage (SAH) is 12%.21 In mouse models, the success rates of SAH and after transient MCAo of 60 minutes are similar to those of rats.22,23 Depending on the duration of MCAo, this method leads to territorial infarction in rats, involving the striatum and the overlying frontoparietal and temporal cortices, as well as some portion of the occipital cortex, but also induces infarction in the thalamus and hypothalamus.24 Infarction after MCAo follows an almost stereotypical progression from early ischemia in the striatum to delayed infarction in the cortex overlying the striatum. The MCA branches supplying the striatum are end arteries, which – in contrast to the cortical branches – do not form collaterals with the adjacent vascular territories.25 Interestingly, cerebral blood flow (CBF) in cortical branches returns to baseline values within 120 minutes of reperfusion, whereas CBF values in the striatum remain depressed.26 These observations might support findings that striatal infarction in rats is resistant to most neuroprotective agents.27 The underlying mechanism of reduced CBF in the striatum compared to the cortex is not yet understood in detail; an obstructive effect of adhesive leukocytes in the cerebral microvasculature has been suggested to contribute to this finding.28
In mice, infarction involves a substantial proportion of the hemisphere (including most of the cortex, striatum, thalamus, hippocampus, and subventricular zone) and is similar to that of rats undergoing MCAo.29,30 The extent and distribution of ischemic injury is highly sensitive to increasing occlusion duration in mice; the difference between 15 minutes and 30 minutes of MCAo results in a fivefold increase in infarct volume (9±2 mm3 vs 56±6 mm3) and also includes regions outside the MCAo territory.30 No ischemic lesions are observed in mice subjected to MCAo below or equal to 10 minutes, whereas an MCAo of 15 minutes leads to a detectable infarction.31
Reproducibility of infarction is affected by different factors, such as suture diameter (3-0 or 4-0 sutures for rats; 6-0 sutures for mice). It has been shown that the size of the suture correlates well with the volume of infarct.32 Insertion length of the suture also influences infarct size and may vary from 18 mm to 22 mm in rats, depending on the age of the animal, rat strain, and suture tip.33 When the suture is positioned 15–16 mm from the CCA bifurcation, only the hypothalamic and anterior choroidal arteries are occluded, leading to small subcortical infarcts that might be a potential model of lacunar stroke.34 Sutures coated with silicone35 or poly-L-lysine36 adhere better to the adjacent vascular endothelium than uncoated sutures and thus cause larger infarcts and reduce inter-animal variability compared to uncoated sutures.36 Furthermore, there is a strong correlation between silicone-coating length and infarct size – that is, a silicone-coating length of 2.0–3.3 mm is required to completely occlude the MCA in rats; a coating length >3.3 mm results in additional occlusion of the anterior choroidal artery, posterior cerebral artery, and hypothalamic artery.37
A large body of literature suggests that infarct size is affected by different rat and mouse strains,13,38–43 whereas the extension of the penumbra (as measured by magnetic resonance perfusion/diffusion-weighted imaging mismatch) is independent of rodent strains.44 MCAo in spontaneously hypertensive rats (SHRs) and stroke-prone SHRs results in relatively large infarcts,38,45 with low variation in size. In contrast, Sprague–Dawley rats exhibit small infarct volumes40 with considerable variability,38 which is even dependent on the choice of vendor.46 It is of note that the strain most commonly used in preclinical stroke studies is Sprague–Dawley;47 approximately 60% of all neuroprotection data come from this strain.20 A recently published meta-analysis found that the Wistar strain yielded the lowest variability of infarct size.13 Mouse strains also exhibit profound differences in infarct volume after MCAo. C57Bl/6 mice are reported to develop larger infarct volumes than Sv129 mice when subjected to permanent MCAo.29,48 Strain differences in arterial collaterals and excitotoxic cell death might partially contribute to these interstrain differences.49 In the brain, pial collateral number and diameter vary by 35-fold and fourfold, respectively, among 21 inbred mouse strains; additionally, collaterals are highly heritable.50 This genetic-dependent variation in collaterals has been identified as a major factor underlying differences in ischemic tissue injury.51 However, certain mouse strains share similar collateral vessel anatomy but exhibit significantly different infarct volumes after permanent (distal) MCAo. This finding might be associated with two novel gene loci, Civq4 and Civq7, which have been suggested to be involved in collateral-independent infarct evolution.52 Furthermore, the age and weight of animals also play a crucial role as arterial dimensions/diameters vary with age. The optimal suture diameter for rats weighing 275–320 g is approximately 0.38 mm for silicone rubber-coated monofilaments.53 In mice, a 15 g increase in body weight can result in a doubling of the required thread diameter, from 100 m to 200 m.54
The MCAo model has several advantages: First, this model mimics human ischemic stroke, which often originates from an MCAo, and exhibits a penumbra that is similar to that of human stroke. Furthermore, the MCAo model is characterized by large infarct volumes and high reproducibility. The reperfusion and thus the duration of ischemia is precisely controllable. Additionally, the procedure is relatively easy to perform and not time-consuming. The MCAo model is considered to be suitable for reproducing ischemic stroke and subsequent neuronal cell death, cerebral inflammation, and blood–brain barrier (BBB) damage, as well as producing good results in behavior tests.20 However, this technique may lead to inadequate MCAo, depending on the type of suture, or may be followed by vessel rupture and subsequent SAH. These shortcomings can be solved by using a silicone-coated suture and laser Doppler-guided placement of the suture; the latter might reduce the incidence of subarachnoid bleeding.21 In MCAo of 60-minute duration, hypothalamic damage is always seen,55 whereas it rarely occurs in human stroke. Hypothalamic ischemia results in a hyperthermic response in rats that persists for at least 1 day after MCAo55 and thus may affect further analysis. Hypothalamic damage is also observed in mice after MCAo, but the surface/volume ratio of the mouse leads to temperature loss in the postoperative period.56 There are also concerns with regard to the different pathophysiologies of the permanent and transient MCAo model.57 In the transient MCAo model, primary core damage may recover, and a secondary delayed injury evolves after a free interval of up to 12 hours. This is a long therapeutic window that is not seen in human stroke. In contrast, the permanent MCAo is characterized by primary core damage that expands in peripheral brain regions and achieves its maximum at approximately 3 hours after MCAo.57 Thus, due to the two different pathophysiologies, unequal results may be expected from the permanent and transient MCAo model, which has been suggested to contribute to the failure of neuroprotective agents in clinical trials.
Craniectomy model
This method includes direct surgical MCAo requiring a craniectomy and section of the dura mater to expose the MCA. There are two main distal occlusion models of the MCA. The first technique involves separating the parotid gland and temporalis muscle, transecting the zygomatic arch, and removing the skull overlying the MCA.19 The MCA is occluded by electrocoagulation and additional transection, resulting in permanent occlusion,19 or alternatively by a microaneurysm clip, hooks (used to lift the MCA from the brain surface until blood flow is interrupted), ligatures, or photochemical MCAo in mice.58–61 The second technique – the so-called three-vessel occlusion (3VO) model – involves the additional occlusion of both CCAs, which reduces the collateral blood flow and thus consolidates the ischemic damage.62 The infarct size varies depending on whether the MCA and CCAs are permanently or transiently occluded.63 Cerebral ischemia induced by this method compromises most of the frontal, parietal, temporal, and rostral occipital cortices, the underlying white matter, and a marginal part of the striatum.59 This technique avoids the thalamic, hypothalamic, hippocampal, and midbrain damage seen in the suture MCAo model. Compared to the suture MCAo model, this procedure induces smaller infarcts.
The main advantages of this model are good reproducibility in infarct size and neurologic deficits, low mortality, and visual confirmation of successful MCAo. Additionally, this technique allows subsequent reperfusion of ischemic tissue. The main disadvantage is the craniectomy technique, which may lead to injury of the underlying cortex or rupture of a vessel by drilling or electrocoagulation. Furthermore, this procedure affects intracranial pressure and BBB function, and requires significant surgical skills to be performed. In order to overcome this limitation, Sugimori et al suggested inducing distal MCAo using a photochemical approach through the intact skull of the mouse.61
Photothrombosis model
The photothrombotic stroke model is based on intravascular photo-oxidation, which leads to well-defined ischemic lesions in the cortex64 and modifications in the striatum.65 A photoactive dye (eg, Rose bengal, erythrosin B) is injected intraperitoneally (mice)66 or intravenously (rats),64 and the intact skull is irradiated within minutes by a light beam at a specific wavelength.64 This procedure generates oxygen radicals that lead to endothelial damage, platelet activation, and aggregation in both pial and intraparenchymal vessels within the irradiated area. This model is further characterized by rapid progression of ischemic cell death.67 The area of irradiation can be determined using stereotactical coordinates, which enable the cerebral ischemia to be induced in a desired region. Further advantages of the photothrombotic model are the minimal surgical intervention required, the high reproducibility of the lesion, and the low mortality. Disadvantages of this model are due to its end-arterial occlusive nature. The rapidly evolving ischemic damage and the endothelial injury at the same time are associated with early cytotoxic (intracellular) and simultaneous vasogenic (extracellular) edema formation;68 in addition, only a little or no ischemic penumbra and local collateral flow/reperfusion occur in this model. These pathomechanisms are different from those seen in human stroke, in which the cytotoxic edema is a hallmark of acute cerebral ischemia.69 However, modifications of photothrombosis-induced stroke have been developed, using a circular laser beam and modified irradiation parameters.70 The ring interior zone is characterized by a hypoperfusion and thus might mimic ischemic penumbra or “tissue at risk”,71 although there are some concerns regarding this concept.27 Recently, photothrombotic stroke models were modified to induce an infarct in the striatum65 or in the internal capsule of rats72 using a stereotactically implanted optic fiber. The aforementioned photothrombotic model is an example of white matter infarction and resulted in damage to the internal capsule that produced prolonged motor deficits.72 Recently, induction and imaging of photothrombotic stroke in conscious and freely moving rats has been demonstrated;73 this technique allowed real-time CBF monitoring without the influence of anesthetics. Photothrombotic ischemia has also been established in freely moving mice in order to investigate the link between the development of motor cortex ischemia and motor deficits.74
Although the photothrombotic ischemic stroke model has some limitations, this procedure allows ongoing processes in the perilesional area and the contralateral cortex to be elucidated and helps to gain a better understanding of neuronal repair after stroke. Harrison et al have used the photothrombotic stroke model to study the functional organization of the sensorimotor cortex at multiple time points before and after stroke.75 They were able to demonstrate the feasibility of longitudinal sensorimotor mapping and characterized the spontaneous cortical reorganization that occurs in the absence of any intervention in mice. Such a detailed analysis of pathogenetic processes would be more difficult with stroke models of large infarction with variable extension.
Endothelin-1 model
Another model of focal stroke is based on the application of endothelin-1 (ET-1). ET-1 is a potent and long-acting vasoconstrictive peptide.76 It can be applied directly onto the exposed MCA,77 as an intracerebral (stereotactic) injection,78 or onto the cortical surface,79 which leads to a dose-dependent ischemic lesion with marginal ischemic edema.78,79 The first two modes of delivery produce an ischemic lesion comparable to that induced by permanent MCAo,80 whereas a cortical injection provides a semicircular infarct that involves all cortical layers.79 The vasoconstrictive property of ET-1 was also used to establish a model of white matter ischemia in the internal capsule81 and anterior cerebral artery occlusion.82 The latter resulted in ischemic infarction of the medial prefrontal cortex and the anteromedial basal forebrain, and thus led to executive impairments.82 After ET-1 administration, a rapid CBF reduction (70%–90%) is seen, followed by a reperfusion that occurs over several hours.83 It should be noted that ET-1 is approximately four times more potent in conscious rats than in anesthetized rats.84 Advantages of the ET-1 model are the less invasive technique, the low mortality, as well as the possibility of inducing direct focal ischemia in deep and superficial brain regions. However, this model is limited by the fact that ET-1 receptors and ET-1-converting enzyme are also expressed by neurons and astrocytes.85 ET-1 has been shown to induce astrocytosis and facilitates axonal sprouting,86 which may interfere with the interpretation of neural repair experiments.27
Embolic stroke model
Embolic stroke models can be divided into two categories: microsphere-/macrosphere-induced stroke models and thromboembolic clot models. The microsphere model involves spheres with a diameter of 20–50 μm of different materials such as dextran, supraparamagnetic iron oxide, TiO2, and ceramic.87 The procedure involves inserting microspheres into the MCA or ICA via the ECA using a microcatheter, which are flushed passively into the cerebral circulation by the blood flow. The microsphere-induced stroke model results in multifocal and heterogeneous infarcts.88 The lesion size increases slowly up to 24 hours after injection.89 The size and dose of the injected microspheres can be used to regulate the extent and severity of the resulting lesions.90 The brain has good tolerance levels for microembolization in animals: showers of microemboli can be created in the rat cerebral vasculature, with development of only a few small areas of injury.91 The macrosphere model uses spheres with a diameter of 100–400 μm, which are instilled into the ICA. The intra-arterial embolization of macrospheres provides reproducible occlusion of the MCA main stem, resulting in focal ischemic lesions that are comparable to the suture MCAo model.88 In contrast to the MCAo model, this technique preserves the blood supply to the hypothalamus and thus avoids hypothalamic infarctions and subsequent hyperthermia.
The thromboembolic clot model is based on the application of spontaneously formed clots or thrombin-induced clots from autologous blood.92,93 Another method of inducing clots is to inject thrombin directly into the intracranial segment of the ICA94 or into the MCA.95,96 This model mimics more closely the mechanism of vascular occlusion that is seen in a large proportion of human strokes and therefore allows the study of thrombolytic agents alone97 or combined with neuroprotective drugs,98 as well as thrombolytic processes such as the development of lesion size.99 The volume and localization of infarcts in the thromboembolic clot model depend on the size and elasticity of the clot.100 However, infarct volume in this model is smaller and more variable compared to an ischemic lesion due to suture MCAo.101 Within 1 hour after clot injection, reopened microvessels in the cortex may be found. By 3 hours, cortical vessels are observed; striatal vessels clear by 24 hours,102 probably due to endogenous thrombolysis,93 or thrombus extravasation.103 Thus, the composition and stability of the thromboembolic clots are important in avoiding spontaneous clot dissolution.93,100 Disintegration of clots and recanalization of occluded vessels depend on the concentration of thrombin and erythrocytes in the clots.97 Interestingly, the fibrinolytic systems in rats are tenfold less sensitive to rtPA than the human system,104 and thus seem to require a tenfold higher dose (10 mg/kg) of rtPA to achieve successful thrombolysis. However, recently published data provide evidence that a dose of 0.9 mg/kg rtPA is also appropriate for preclinical stroke studies in rodents.105 Furthermore, a dose of 10 mg/kg shows a loss of effectiveness beyond 4 hours after stroke, and increases infarct volume as well as hemorrhagic transformation.106
Intravascular installation of clots often leads to multifocal infarcts, with significant variability in lesion volume and localization; furthermore, a high degree of autolysis results in early and uncontrollable reperfusion.93,102 Therefore, this model is not suitable for validating neuroprotective effects. Additionally, the rate of cerebral hemorrhage and mortality is high.96 Previous analyses of clots induced by the thrombin injection model in mice have revealed that they are mainly composed of polymerized fibrin containing a low number of cells and platelets,95 and thus differ from human clots after thromboembolic stroke: approximately 75% of these clots demonstrate platelet/fibrin accumulation, neutrophil/monocyte deposition, and a significant accumulation of erythrocytes.107 It remains to be seen whether further improvements of the clot model might provide the opportunity to study more precisely the effect of reperfusion strategies in animal models.
Conclusion
In the last three decades, significant progress has been made in understanding the pathophysiology that underlies ischemic stroke. Several studies have shown that the ischemic cascade has many intervening and interlinking steps that have provided a number of potential drug targets, as discussed in Moskowitz et al.108 A large number of agents have revealed neuroprotective efficacy in preclinical studies but have failed to translate to efficacious therapies, irrespective of whether neuroprotective drugs were administered as a single agent8 or in combination.9 This is probably due to shortcomings in the design and conduct of both clinical and preclinical studies.109 This raises the question as to what extent the choice of animal stroke model might contribute to this failure. In the permanent MCAo model, the infarcted core expands into more peripheral regions supplied by the MCA and reaches its maximal size within 3 hours. This time is the therapeutic window, during which infarct expansion can be alleviated.57 In the transient MCAo model, recirculation results in an immediate reoxygenation of the ischemic tissue and prompts recovery of energy metabolism in the infarcted core. However, after an interval of 6–12 hours, secondary delayed cell death develops in the region of primary energy failure,57 which can be alleviated by numerous interventions during the interval between primary and secondary tissue damage. In contrast, the therapeutic responsiveness of permanent focal stroke in humans declines significantly within 3 hours after stroke onset. Thus, a stroke model with prompt reperfusion (ie, a long therapeutic window) might be critical when attempting to translate these results achieved through this method to clinical studies. Although thrombolytic trials have always emphasized the importance of timing, there has rarely been the same emphasis in clinical trials of neuroprotection. This discrepancy is well illustrated by analysis of the animal and human study data for tirilazad, in which the median time to successful treatment in animal experiments was 10 minutes, whereas in unsuccessful human trials, it was approximately 5 hours.110 Another major problem of animal stroke models is that studies are mostly conducted in young animals without any comorbidity. These models differ from human stroke, which particularly affects elderly people who have a multiplicity of cerebrovascular risk factors. However, optimizing the study design of preclinical trials might increase the translational potential of animal stroke models.
Acknowledgments
This work was supported by the Sonderforschungsbereich 688 and the Interdisziplinäres Zentrum für klinische Forschung Würzburg.
Disclosure
The authors report no disclosures relevant to the paper.
References
Mozaffarian D, Benjamin EJ, Go AS, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2015 update a report from the American Heart Association. Circulation. 2015;131(4):e29–e322. | ||
Murray CJ, Vos T, Lozano R, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2197–2223. | ||
Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79(4):1431–1568. | ||
Fisher M. The ischemic penumbra: identification, evolution and treatment concepts. Cerebrovasc Dis. 2004;17(suppl 1):1–6. | ||
Adams HP Jr, del Zoppo G, Alberts MJ, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Circulation. 2007;115(20):e478–e534. | ||
Fonarow GC, Smith EE, Saver JL, et al. Timeliness of tissue-type plasminogen activator therapy in acute ischemic stroke: patient characteristics, hospital factors, and outcomes associated with door-to-needle times within 60 minutes. Circulation. 2011;123(7):750–758. | ||
Zivin JA, Fisher M, DeGirolami U, Hemenway CC, Stashak JA. Tissue plasminogen activator reduces neurological damage after cerebral embolism. Science. 1985;230(4731):1289–1292. | ||
O’Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59(3):467–477. | ||
O’Collins VE, Macleod MR, Cox SF, et al. Preclinical drug evaluation for combination therapy in acute stroke using systematic review, meta-analysis, and subsequent experimental testing. J Cereb Blood Flow Metab. 2011;31(3):962–975. | ||
Fisher M, Feuerstein G, Howells DW, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40(6):2244–2250. | ||
Yamori Y, Horie R, Handa H, Sato M, Fukase M. Pathogenetic similarity of strokes in stroke-prone spontaneously hypertensive rats and humans. Stroke. 1976;7(1):46–53. | ||
Pontén U, Ratcheson RA, Salford LG, Siesjö BK. Optimal freezing conditions for cerebral metabolites in rats. J Neurochem. 1973;21(5):1127–1138. | ||
Ström JO, Ingberg E, Theodorsson A, Theodorsson E. Method parameters’ impact on mortality and variability in rat stroke experiments: a meta-analysis. BMC Neurosci. 2013;14(1):41. | ||
Kraft P, Göb E, Schuhmann MK, et al. FTY720 ameliorates acute ischemic stroke in mice by reducing thrombo-inflammation but not by direct neuroprotection. Stroke. 2013;44(11):3202–3210. | ||
Göb E, Reymann S, Langhauser F, et al. Blocking of plasma kallikrein ameliorates stroke by reducing thromboinflammation. Ann Neurol. 2015;77(5):784–803. | ||
Bogousslavsky J, Melle GV, Regli F. The Lausanne Stroke Registry: analysis of 1,000 consecutive patients with first stroke. Stroke. 1988; 19(9):1083–1092. | ||
Koizumi J, Yoshida Y, Nakazawa T, Ooneda G. Experimental studies of ischemic brain edema. I. A new experimental model of cerebral embolism in which recirculation can introduced into the ischemic area. Jpn J Stroke. 1986;8(1):1–8. | ||
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989; 20(1):84–91. | ||
Tamura A, Graham DI, McCulloch J, Teasdale GM. Focal cerebral ischaemia in the rat: 1. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1981;1(1):53–60. | ||
Howells DW, Porritt MJ, Rewell SS, et al. Different strokes for different folks: the rich diversity of animal models of focal cerebral ischemia. J Cereb Blood Flow Metab. 2010;30(8):1412–1431. | ||
Schmid-Elsaesser R, Zausinger S, Hungerhuber E, Baethmann A, Reulen H-JA. Critical reevaluation of the intraluminal thread model of focal cerebral ischemia evidence of inadvertent premature reperfusion and subarachnoid hemorrhage in rats by laser-doppler flowmetry. Stroke. 1998;29(10):2162–2170. | ||
Liu S, Zhen G, Meloni BP, Campbell K, Winn HR. Rodent stroke model guidelines for preclinical stroke trials (1st edition). J Exp Stroke Transl Med. 2009;2(2):2–27. | ||
Tsuchiya D, Hong S, Kayama T, Panter SS, Weinstein PR. Effect of suture size and carotid clip application upon blood flow and infarct volume after permanent and temporary middle cerebral artery occlusion in mice. Brain Res. 2003;970(1–2):131–139. | ||
Garcia JH, Liu K-F, Ho K-L. Neuronal necrosis after middle cerebral artery occlusion in Wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke. 1995;26(4):636–643. | ||
Zülch K. Cerebrovascular pathology and pathogenesis as a basis of neuroradiological diagnosis. In: Diethelm L, Heuck F, Olsson O, Strnad F, Vieten H, Zuppinger A, editors. Handbuch Der Medizinischen Radiologie. Vol. 14, 1A. Berlin: Springer; 1981:1–192. | ||
Takagi K, Zhao W, Busto R, Ginsberg MD. Local hemodynamic changes during transient middle cerebral artery occlusion and recirculation in the rat: a [14C]iodoantipyrine autoradiographic study. Brain Res. 1995;691(1–2):160–168. | ||
Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. Neuro Rx. 2005;2(3):396–409. | ||
del Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22(10):1276–1283. | ||
Maeda K, Hata R, Hossmann K-A. Regional metabolic disturbances and cerebrovascular anatomy after permanent middle cerebral artery occlusion in C57Black/6 and SV129 mice. Neurobiol Dis. 1999;6(2): 101–108. | ||
McColl BW, Carswell HV, McCulloch J, Horsburgh K. Extension of cerebral hypoperfusion and ischaemic pathology beyond MCA territory after intraluminal filament occlusion in C57Bl/6J mice. Brain Res. 2004;997(1):15–23. | ||
Pedrono E, Durukan A, Strbian D, et al. An optimized mouse model for transient ischemic attack. J Neuropathol Exp Neurol. 2010; 69(2):188–195. | ||
Türeyen K, Vemuganti R, Sailor KA, Dempsey RJ. Ideal suture diameter is critical for consistent middle cerebral artery occlusion in mice. Neurosurgery. 2005;56:196–200. | ||
Zarow GJ, Karibe H, States BA, Graham SH, Weinstein PR. Endovascular suture occlusion of the middle cerebral artery in rats: effect of suture insertion distance on cerebral blood flow, infarct distribution and infarct volume. Neurol Res. 1997;19(4):409–416. | ||
He Z, Yamawaki T, Yang S, Day AL, Simpkins JW, Naritomi H. Experimental model of small deep infarcts involving the hypothalamus in rats changes in body temperature and postural reflex. Stroke. 1999; 30(12):2743–2751. | ||
Bouley J, Fisher M, Henninger N. Comparison between coated vs uncoated suture middle cerebral artery occlusion in the rat as assessed by perfusion/diffusion weighted imaging. Neurosci Lett. 2007; 412(3):185–190. | ||
Belayev L, Busto R, Zhao W, Ginsberg MD. Quantitative evaluation of blood–brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996;739(1):88–96. | ||
Guan Y, Wang Y, Yuan F, et al. Effect of suture properties on stability of middle cerebral artery occlusion evaluated by synchrotron radiation angiography. Stroke. 2012;43(3):888–891. | ||
Duverger D, MacKenzie ET. The quantification of cerebral infarction following focal ischemia in the rat: influence of strain, arterial pressure, blood glucose concentration, and age. J Cereb Blood Flow Metab. 1988;8(4):449–461. | ||
Bardutzky J, Shen Q, Henninger N, Bouley J, Duong TQ, Fisher M. Differences in ischemic lesion evolution in different rat strains using diffusion and perfusion imaging. Stroke. 2005;36(9):2000–2005. | ||
Walberer M, Stolz E, Müller C, et al. Experimental stroke: ischaemic lesion volume and oedema formation differ among rat strains (a comparison between Wistar and Sprague–Dawley rats using MRI). Lab Anim. 2006;40(1):1–8. | ||
Barone FC, Knudsen DJ, Nelson AH, Feuerstein GZ, Willette RN. Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. J Cereb Blood Flow Metab. 1993;13(4):683–692. | ||
Majid A, He YY, Gidday JM, et al. Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke. 2000;31(11):2707–2714. | ||
Cheng M-H, Lin L-L, Liu J-Y, Liu A-J. The outcomes of stroke induced by middle cerebral artery occlusion in different strains of mice. CNS Neurosci Ther. 2012;18(9):794–795. | ||
Reid E, Graham D, Lopez-Gonzalez MR, Holmes WM, Macrae IM, McCabe C. Penumbra detection using PWI/DWI mismatch MRI in a rat stroke model with and without comorbidity: comparison of methods. J Cereb Blood Flow Metab. 2012;32(9):1765–1777. | ||
Dogan A, Başkaya MK, Rao VL, Rao AM, Dempsey RJ. Intraluminal suture occlusion of the middle cerebral artery in spontaneously hypertensive rats. Neurol Res. 1998;20(3):265–270. | ||
Oliff HS, Weber E, Miyazaki B, Marek P. Infarct volume varies with rat strain and vendor in focal cerebral ischemia induced by transcranial middle cerebral artery occlusion. Brain Res. 1995;699(2):329–331. | ||
O’Collins VE, Donnan GA, Macleod MR, Howells DW. Animal models of stroke versus clinical stroke: comparison of infarct size, cause, location, study design, and efficacy of experimental therapies. In: Michael Conn P, editor. Animal Models for the Study of Human Disease. Waltham: Academic Press; 2013:531–568. | ||
Connolly ES Jr, Winfree CJ, Stern DM, Solomon RA, Pinsky DJ. Procedural and strain-related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery. 1996; 38(3):523–531. [discussion 532]. | ||
Yang G, Kitagawa K, Matsushita K, et al. C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res. 1997;752(1–2):209–218. | ||
Zhang H, Prabhakar P, Sealock R, Faber JE. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J Cereb Blood Flow Metab. 2010;30(5):923–934. | ||
Sealock R, Zhang H, Lucitti JL, Moore SM, Faber JE. Congenic fine-mapping identifies a major causal locus for variation in the native collateral circulation and ischemic injury in brain and lower extremity. Circ Res. 2014;114(4):660–671. | ||
Chu P-L, Keum S, Marchuk DA. A novel genetic locus modulates infarct volume independently of the extent of collateral circulation. Physiol Genomics. 2013;45(17):751–763. | ||
Spratt NJ, Fernandez J, Chen M, et al. Modification of the method of thread manufacture improves stroke induction rate and reduces mortality after thread-occlusion of the middle cerebral artery in young or aged rats. J Neurosci Methods. 2006;155(2):285–290. | ||
Hata R, Mies G, Wiessner C, et al. A reproducible model of middle cerebral artery occlusion in mice: hemodynamic, biochemical, and magnetic resonance imaging. J Cereb Blood Flow Metab. 1998;18(4): 367–375. | ||
Li F, Omae T, Fisher M. Spontaneous hyperthermia and its mechanism in the intraluminal suture middle cerebral artery occlusion model of rats. Stroke. 1999;30(11):2464–2471. | ||
Barber PA, Hoyte L, Colbourne F, Buchan AM. Temperature-regulated model of focal ischemia in the mouse a study with histopathological and behavioral outcomes. Stroke. 2004;35(7):1720–1725. | ||
Hossmann K-A. The two pathophysiologies of focal brain ischemia: implications for translational stroke research. J Cereb Blood Flow Metab. 2012;32(7):1310–1316. | ||
Shigeno T, Teasdale GM, McCulloch J, Graham DI. Recirculation model following MCA occlusion in rats. J Neurosurg. 1985;63(2):272–277. | ||
Buchan AM, Xue D, Slivka A. A new model of temporary focal neocortical ischemia in the rat. Stroke. 1992;23(2):273–279. | ||
Popa-Wagner A, Schröder E, Schmoll H, Walker LC, Kessler C. Upregulation of MAP1B and MAP2 in the rat brain after middle cerebral artery occlusion: effect of age. J Cereb Blood Flow Metab. 1999; 19(4):425–434. | ||
Sugimori H, Yao H, Ooboshi H, Ibayashi S, Iida M. Krypton laser-induced photothrombotic distal middle cerebral artery occlusion without craniectomy in mice. Brain Res Brain Res Protoc. 2004;13(3):189–196. | ||
McAuley MA. Rodent models of focal ischemia. Cerebrovasc Brain Metab Rev. 1995;7(2):153–180. | ||
Yanamoto H, Nagata I, Niitsu Y, Xue J-H, Zhang Z, Kikuchi H. Evaluation of MCAO stroke models in normotensive rats: standardized neocortical infarction by the 3VO technique. Exp Neurol. 2003;182(2): 261–274. | ||
Watson BD, Dietrich WD, Busto R, Wachtel MS, Ginsberg MD. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol. 1985;17(5):497–504. | ||
Kuroiwa T, Xi G, Hua Y, Nagaraja TN, Fenstermacher JD, Keep RF. Development of a rat model of photothrombotic ischemia and infarction within the caudoputamen. Stroke. 2009;40(1):248–253. | ||
Kleinschnitz C, Braeuninger S, Pham M, et al. Blocking of platelets or intrinsic coagulation pathway–driven thrombosis does not prevent cerebral infarctions induced by photothrombosis. Stroke. 2008;39(4):1262–1268. | ||
Dietrich WD, Ginsberg MD, Busto R, Watson BD. Photochemically induced cortical infarction in the rat. 1. Time course of hemodynamic consequences. J Cereb Blood Flow Metab. 1986;6(2):184–194. | ||
Lee VM, Burdett NG, Carpenter A, et al. Evolution of photochemically induced focal cerebral ischemia in the rat magnetic resonance imaging and histology. Stroke. 1996;27(11):2110–2119. | ||
Provenzale JM, Jahan R, Naidich TP, Fox AJ. Assessment of the patient with hyperacute stroke: imaging and therapy. Radiology. 2003; 229(2):347–359. | ||
Hilger T, Blunk JA, Hoehn M, Mies G, Wester P. Characterization of a novel chronic photothrombotic ring stroke model in rats by magnetic resonance imaging, biochemical imaging, and histology. J Cereb Blood Flow Metab. 2004;24(7):789–797. | ||
Hu X, Wester P, Brännstörm T, Watson BD, Gu W. Progressive and reproducible focal cortical ischemia with or without late spontaneous reperfusion generated by a ring-shaped, laser-driven photothrombotic lesion in rats. Brain Res Brain Res Protoc. 2001;7(1):76–85. | ||
Kim HS, Kim D, Kim RG, et al. A rat model of photothrombotic capsular infarct with a marked motor deficit: a behavioral, histologic, and microPET study. J Cereb Blood Flow Metab. 2014;34(4):683–689. | ||
Lu H, Li Y, Yuan L, Li H, Lu X, Tong S. Induction and imaging of photothrombotic stroke in conscious and freely moving rats. J Biomed Opt. 2014;19(9):96013. | ||
Yu C-L, Zhou H, Chai A-P, Yang Y-X, Mao R-R, Xu L. Whole-scale neurobehavioral assessments of photothrombotic ischemia in freely moving mice. J Neurosci Methods. 2015;239:100–107. | ||
Harrison TC, Silasi G, Boyd JD, Murphy TH. Displacement of sensory maps and disorganization of motor cortex after targeted stroke in mice. Stroke. 2013;44(8):2300–2306. | ||
Yanagisawa M, Kurihara H, Kimura S, Goto K, Masaki T. A novel peptide vasoconstrictor, endothelin, is produced by vascular endothelium and modulates smooth muscle Ca2+ channels. J Hypertens Suppl. 1988;6(4):S188–S191. | ||
Robinson MJ, Macrae IM, Todd M, Reid JL, McCulloch J. Reduction of local cerebral blood flow to pathological levels by endothelin-1 applied to the middle cerebral artery in the rat. Neurosci Lett. 1990; 118(2):269–272. | ||
Hughes PM, Anthony DC, Ruddin M, et al. Focal lesions in the rat central nervous system induced by endothelin-1. J Neuropathol Exp Neurol. 2003;62(12):1276–1286. | ||
Fuxe K, Bjelke B, Andbjer B, Grahn H, Rimondini R, Agnati LF. Endothelin-1 induced lesions of the frontoparietal cortex of the rat. A possible model of focal cortical ischemia. Neuroreport. 1997; 8(11):2623–2629. | ||
Sharkey J, Ritchie IM, Kelly PAT. Perivascular microapplication of endothelin-1: a new model of focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab. 1993;13(5):865–871. | ||
Frost SB, Barbay S, Mumert ML, Stowe AM, Nudo RJ. An animal model of capsular infarct: endothelin-1 injections in the rat. Behav Brain Res. 2006;169(2):206–211. | ||
Ward NM, Sharkey J, Marston HM, Brown VJ. Simple and choice reaction-time performance following occlusion of the anterior cerebral arteries in the rat. Exp Brain Res. 1998;123(3):269–281. | ||
Biernaskie J, Corbett D, Peeling J, Wells J, Lei H. A serial MR study of cerebral blood flow changes and lesion development following endothelin-1-induced ischemia in rats. Magn Reson Med. 2001; 46(4):827–830. | ||
Bogaert L, Scheller D, Moonen J, et al. Neurochemical changes and laser Doppler flowmetry in the endothelin-1 rat model for focal cerebral ischemia. Brain Res. 2000;887(2):266–275. | ||
Nakagomi S, Kiryu-Seo S, Kiyama H. Endothelin-converting enzymes and endothelin receptor B messenger RNAs are expressed in different neural cell species and these messenger RNAs are coordinately induced in neurons and astrocytes respectively following nerve injury. Neuroscience. 2000;101(2):441–449. | ||
Uesugi M, Kasuya Y, Hayashi K, Goto K. SB209670, a potent endothelin receptor antagonist, prevents or delays axonal degeneration after spinal cord injury. Brain Res. 1998;786(1–2):235–239. | ||
Hossmann K-A. Cerebral ischemia: models, methods and outcomes. Neuropharmacology. 2008;55(3):257–270. | ||
Gerriets T, Li F, Silva MD, et al. The macrosphere model: evaluation of a new stroke model for permanent middle cerebral artery occlusion in rats. J Neurosci Methods. 2003;122(2):201–211. | ||
Mayzel-Oreg O, Omae T, Kazemi M, et al. Microsphere-induced embolic stroke: an MRI study. Magn Reson Med. 2004;51(6):1232–1238. | ||
Fukuchi K, Kusuoka H, Watanabe Y, Nishimura T. Correlation of sequential MR images of microsphere-induced cerebral ischemia with histologic changes in rats. Invest Radiol. 1999;34(11):698–703. | ||
Rapp JH, Pan XM, Neumann M, Hong M, Hollenbeck K, Liu J. Microemboli composed of cholesterol crystals disrupt the blood–brain barrier and reduce cognition. Stroke. 2008;39(8):2354–2361. | ||
Overgaard K, Sereghy T, Boysen G, Pedersen H, Høyer S, Diemer NH. A rat model of reproducible cerebral infarction using thrombotic blood clot emboli. J Cereb Blood Flow Metab. 1992;12(3):484–490. | ||
Niessen F, Hilger T, Hoehn M, Hossmann K-A. Differences in clot preparation determine outcome of recombinant tissue plasminogen activator treatment in experimental thromboembolic. Stroke. 2003; 34(8):2019–2024. | ||
Zhang Z, Zhang RL, Jiang Q, Raman SB, Cantwell L, Chopp M. A new rat model of thrombotic focal cerebral ischemia. J Cereb Blood Flow Metab. 1997;17(2):123–135. | ||
Orset C, Macrez R, Young AR, et al. Mouse model of in situ thromboembolic stroke and reperfusion. Stroke. 2007;38(10):2771–2778. | ||
Ansar S, Chatzikonstantinou E, Wistuba-Schier A, et al. Characterization of a new model of thromboembolic stroke in C57 black/6J mice. Transl Stroke Res. 2013;5(4):526–533. | ||
Overgaard K. Thrombolytic therapy in experimental embolic stroke. Cerebrovasc Brain Metab Rev. 1994;6(3):257–286. | ||
Zhang L, Zhang ZG, Zhang C, Zhang RL, Chopp M. Intravenous administration of a GPIIb/IIIa receptor antagonist extends the therapeutic window of intra-arterial tenecteplase-tissue plasminogen activator in a rat stroke model. Stroke. 2004;35(12):2890–2895. | ||
Brinker G, Franke C, Hoehn M, Uhlenküken U, Hossmann KA. Thrombolysis of cerebral clot embolism in rat: effect of treatment delay. Neuroreport. 1999;10(16):3269–3272. | ||
Ren M, Lin ZJ, Qian H, et al. Embolic middle cerebral artery occlusion model using thrombin and fibrinogen composed clots in rat. J Neurosci Methods. 2012;211(2):296–304. | ||
Beech JS, Williams SC, Campbell CA, et al. Further characterisation of a thromboembolic model of stroke in the rat. Brain Res. 2001; 895(1–2):18–24. | ||
Wang CX, Todd KG, Yang Y, Gordon T, Shuaib A. Patency of cerebral microvessels after focal embolic stroke in the rat. J Cereb Blood Flow Metab. 2001;21(4):413–421. | ||
Lam CK, Yoo T, Hiner B, Liu Z, Grutzendler J. Embolus extravasation is an alternative mechanism for cerebral microvascular recanalization. Nature. 2010;465(7297):478–482. | ||
Korninger C, Collen D. Studies on the specific fibrinolytic effect of human extrinsic (tissue-type) plasminogen activator in human blood and in various animal species in vitro. Thromb Haemost. 1981; 46(2):561–565. | ||
Haelewyn B, Risso J-J, Abraini JH. Human recombinant tissue-plasminogen activator (alteplase): why not use the “human” dose for stroke studies in rats? J Cereb Blood Flow Metab. 2010;30(5):900–903. | ||
Kano T, Katayama Y, Tejima E, Lo EH. Hemorrhagic transformation after fibrinolytic therapy with tissue plasminogen activator in a rat thromboembolic model of stroke. Brain Res. 2000;854(1–2): 245–248. | ||
Smith WS, Sung G, Starkman S, et al; MERCI Trial Investigators. Safety and efficacy of mechanical embolectomy in acute ischemic stroke results of the MERCI trial. Stroke. 2005;36(7):1432–1438. | ||
Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67(2):181–198. | ||
Sutherland BA, Minnerup J, Balami JS, Arba F, Buchan AM, Kleinschnitz C. Neuroprotection for ischaemic stroke: translation from the bench to the bedside. Int J Stroke. 2012;7(5):407–418. | ||
Sena E, Wheble P, Sandercock P, Macleod M. Systematic review and meta-analysis of the efficacy of tirilazad in experimental. Stroke. 2007;38(2):388–394. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.