")
Back to Journals » Drug Design, Development and Therapy » Volume 14
An LC-MS/MS Bioanalytical Assay for the Determination of Gilteritinib in Rat Plasma and Application to a Drug–Drug Interaction Study
Authors Wang Q, Chen Z , Chen D, Ye X
Received 25 December 2019
Accepted for publication 12 March 2020
Published 26 May 2020 Volume 2020:14 Pages 2061—2067
DOI https://doi.org/10.2147/DDDT.S243760
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Qiong Wang,1 Zhe Chen,1 Dingwen Chen,1 Xia-yan Ye2
1Department of Pharmacy, Wenzhou People’s Hospital, Wenzhou, Zhejiang 325000, People’s Republic of China; 2Department of Brain Surgery, The People’s Hospital of Lishui, Lishui, Zhejiang 323000, People’s Republic of China
Correspondence: Xia-yan Ye
The People’s Hospital of Lishui, No.15 Dazhong street, Liandu District, Lishui, Zhejiang, People’s Republic of China
Email [email protected]
Background: Gilteritinib, a novel, potent FLT3/AXL inhibitor, was recently approved in Japan and USA for the treatment of adult patients who have relapsed or refractory acute myeloid leukemia (AML) with a FLT3 mutation.
Purpose and Methods: In this study, we aimed to develop and validate a sensitive and simple ultra performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) method for the quantification of gilteritinib in plasma and to investigate whether CYP3A4 inhibitors (fluconazole and itraconazole) could influence the pharmacokinetics of gilteritinib from a drug–drug interaction study in rats. Sample preparation was done by a simple protein crash with acetonitrile containing the internal standard (IS) pirfenidone, followed by UPLC-MS/MS quantification.
Results: The assay was successfully validated in a 1– 500 ng/mL calibration range for gilteritinib, where the lower limit of quantification (LLOQ) was set at 1 ng/mL. The intra-day and inter-day precisions for gilteritinib were less than 10.6%, and the accuracies were in the range of − 14.5% to 11.1%. Recovery and matrix effect of the analyte and IS were acceptable, and the analyte was stable during the assay and storage in plasma samples. The validated UPLC-MS/MS method was successfully applied to a drug–drug interaction study between gilteritinib and CYP3A4 inhibitors (fluconazole and itraconazole) in rats. Itraconazole significantly increased the exposure of gilteritinib, and affected the pharmacokinetics of gilteritinib in rats, not fluconazole.
Conclusion: A further clinical study should be conducted to investigate the effect of itraconazole on the metabolism of gilteritinib in subjects.
Keywords: gilteritinib, drug–drug interaction, pharmacokinetic, UPLC-MS/MS, CYP3A4 inhibitors
Introduction
Advances in the understanding of the molecular basis for acute myeloid leukemia (AML) have generated new potential targets for treatment. FMS-like tyrosine kinase 3 (FLT3) is one of the most frequently mutated genes in AML and mutations in this gene are associated with poor overall survival.1,2 AXL plays a role in the activation of FLT3 and has been implicated in the pathogenesis of AML. Gilteritinib (Figure 1A), a highly specific, novel, potent FLT3/AXL inhibitor, has demonstrated antileukemic activity in patients with AML.3–5 Recently, it was approved in Japan and USA for the treatment of adult patients who have relapsed or refractory AML with a FLT3 mutation.6 After oral administration, gilteritinib is primarily metabolized in the liver by the activity of CYP3A4.7 It is also reported that strong CYP3A4 inhibitors may increase gilteritinib exposure and the frequency of adverse reactions, even serious or life-threatening toxicity.6 Therefore, it is suggested that co-administration of gilteritinib with strong or moderate CYP3A4 inhibitors should be avoided. However, the possibility of pharmacokinetic interaction between gilteritinib and CYP3A4 inhibitors (such as fluconazole, and itraconazole) have not been investigated in detail.
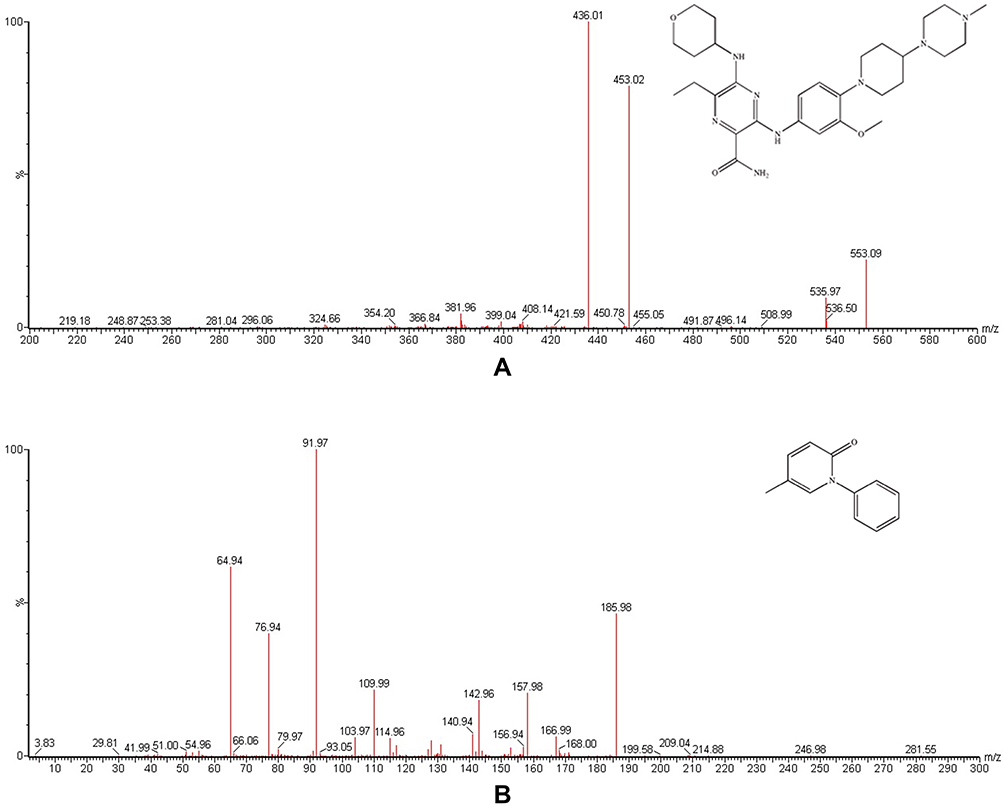 |
Figure 1 Mass spectra of gilteritinib (A) and pirfenidone IS, (B) in this study. |
As pharmacokinetic and drug–drug interaction information are important for dosing optimization, and therefore maximizing treatment effectivity and minimizing the risk of emergence of adverse reactions, it is necessary to develop and validate a sensitive analytical method for the quantification of gilteritinib in biological fluids. Until now, only one paper assessed the pharmacokinetic profile of gilteritinib in patients with AML.5 However, this analytical method did not offer enough data for repeating in other laboratories (e.g. plasma extraction procedure, chromatography conditions, parameters of the method, etc). Thus, this method does not meet the requirement of high sample throughput in bioanalysis for pharmacokinetic or drug–drug interaction study.
Therefore, we designed, developed and validated a sensitive and quick ultra performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) method allowing the determination of gilteritinib in plasma with simple sample preparation, and investigated the effects of fluconazole and itraconazole on the exposure and pharmacokinetic changes of gilteritinib in rats by comparing the plasma concentrations and pharmacokinetic parameters of gilteritinib.
Purpose and Methods
Chemicals Materials
Gilteritinib, fluconazole, itraconazole (all purity >98%) and pirfenidone (IS, purity >98%, Figure 1B) were supplied by Beijing Sunflower and Technology Development Co., Ltd (Beijing, China). Acetonitrile and methanol were HPLC grade and offered from Merck Company (Darmstadt, Germany). Analytical grade of formic acid was purchased from Beijing Sunflower and Technology Development Co., Ltd. Ultrapure water was prepared using a Milli-Q Water Purification System (EMD Millipore).
Animal Experiments
Healthy male Sprague Dawley rats with body weight of 180–220 g, were used and obtained from Laboratory Animal Center of Wenzhou Medical University (Zhejiang, China). Rats were raised under standard temperature, humidity, and light conditions, and fed standard rodent diet and water. This experiment was approved by the Animal Care and Use Committee of Wenzhou Medical University, according to National Institutes of Health (NIH) Guidelines for the welfare and use of animals.8
Gilteritinib, fluconazole, and itraconazole were all suspended in 0.5% carboxymethyl cellulose sodium (CMC-Na). Thirty Sprague Dawley rats were randomly divided into five groups (n=6) and orally administered the approximate equivalent volume solutions: Group A (the control group, 0.5% CMC-Na); Group B (single dose administration of 20 mg/kg fluconazole half an hour before experiment); Group C (20 mg/kg fluconazole once daily for seven days before experiment); Group D (single dose administration of 20 mg/kg itraconazole half an hour before experiment); Group E (20 mg/kg itraconazole once daily for seven days before experiment). Thirty minutes later, 10 mg/kg gilteritinib was orally administered to each rat. At the time points of 0, 0.333, 0.667, 1, 2, 4, 9, 12, 24, 36 and 48 h, approximately 0.3 mL of blood samples were withdrawn from the tail vein into heparinized 1.5 mL polythene tubes. Subsequently, after centrifugation at 4000 g for 10 min at room temperature, 100 µL plasma was taken after the separation and stored at −20°C until analysis.
Instrumentations and Analytical Conditions
LC-MS/MS method was conducted by a Waters Acquity ultra performance liquid chromatography (UPLC) system coupled with a Waters Xevo TQ-S triple quadrupole tandem mass spectrometer (Milford, MA, USA). The Masslynx 4.1 software (Waters Corp., Milford, MA, USA) was used for data acquisition, processing and instrument control.
The chromatographic separation of gilteritinib and IS was carried out on an Acquity BEH C18 column (2.1 mm × 50 mm, 1.7 μm). Meanwhile, the mobile phase used for the analysis was acetonitrile (solvent A) and 0.1% formic acid in water (solvent B) delivered at a flow rate of 0.40 mL/min. The procedures for the linear gradient elution were conducted as follows: 0–0.5 min, 10% A; 0.5–1.0 min, 10–90% A; 1.0–2.0 min, 90% A; and 2.0–2.1 min, 90–10% A. Then, 10% A from 2.1 to 3.0 min was maintained for equilibration. The entire run time was 3.0 min for an injection volume of 2.0 µL. The samples were maintained at 10°C in an autosampler, while the column oven temperature was set at 40°C.
Mass spectrometric determination was performed by an electrospray ionization (ESI) source set in the positive ion mode. General parameters, such as gas, voltage and temperature, were optimized as follows: cone gas, 150 L/h, desolvation gas, 1000 L/h, collision gas, 0.15 mL/min, capillary voltage, 2.0 kV, desolvation temperature, 800°C. Multiple reaction monitoring (MRM) mode was selected and used, in which the ion transitions for gilteritinib and IS were m/z 553.1 → 436.0 and m/z 186.0 → 92.0, respectively. The cone voltage and collision energy were 30 V and 30 eV for gilteritinib and were 20 V and 25 eV for IS, respectively.
Standard Solutions, Calibration Standards and Quality Control (QC) Samples
Standard stock solutions of 1.00 mg/mL of both gilteritinib and IS were prepared by dissolving the compounds in an appropriate amount of methanol for calibration curve and quality control (QC) samples. Two series of working solutions were obtained by diluting the stock solutions with methanol so as to prepare the plasma standard points of the calibration curve and the plasma QC samples with concentrations of 1, 5, 10, 20, 50, 100, 200, 500 ng/mL and 2, 40, 400 ng/mL, respectively. The IS working solution was prepared at 100 ng/mL by dilution of its stock solution with methanol. All stock and working solutions were stored at −20°C for further use.
Sample Preparation
A volume of 100 µL of the actual sample, 20 µL IS working solution was added and mixed for 1.0 min. Then, the plasma samples were mixed with 300 µL of acetonitrile for protein precipitation, and each tube was thoroughly vortexed for 1.0 min and centrifugated at 13,000 g for 10 min at nominally 4°C. Finally, 100 µL of the obtained supernatant was transferred to an autosampler vial and 2.0 µL was injected into the UPLC-MS/MS system for analysis.
Method Validation
This study was conducted in compliance with the FDA principle on the bioanalytical method validation,9 which needs to examine the following parameters: the selectivity, calibration curve, lower limit of quantification (LLOQ), accuracy and precision, matrix effect, recovery and stability at various test conditions.
Statistical Analysis
The mean plasma gilteritinib concentration-time curve was explored by Origin 8.0 (Originlab Company, Northampton, MA, USA), and the pharmacokinetic parameters of gilteritinib were calculated by DAS (Drug and Statistics) software (Version 2.0, Shanghai University of Traditional Chinese Medicine, China) in noncompartmental mode. Statistical comparisons of the main pharmacokinetic parameters within each group were carried out with the Statistical Package for the Social Sciences (version 17.0; SPSS Inc., Chicago, IL, USA) by one-way analysis of variance coupled with the Dunnett’s test. In all cases, a P-value below 0.05 was deemed statistically significant.
Results and Discussion
Method Development and Optimization
Gilteritinib and IS (200 ng/mL in methanol: water 50:50, v/v for each substance) were directly infused into the mass spectrometer, and the conditions of product ion mass spectra of gilteritinib and IS were optimized. Different settings that produced the highest sensitivity by monitoring the product ion transition were compared. In positive full mass scan (as shown in Figure 1), gilteritinib and IS generated a protonated molecular ion [M+H]+, and the most abundant fragment ions for MRM were m/z 553.1 → 436.0 and m/z 186.0 → 92.0, with collision energy values of 30 eV and 25 eV for gilteritinib and IS, respectively.
Different analytical columns were evaluated and compared, and BEH C18 (2.1 mm × 50 mm, 1.7 μm) column displayed efficient separation, short chromatographic retention time and peak symmetry. Considering the addition of formic acid might enhance the chromatograms and signals of the analyte and IS in the mobile phase, thus, the optimized mobile phase comprised acetonitrile and 0.1% formic acid in water. In addition, the excessive numbers of plasma samples generated in pharmacokinetic or drug–drug interaction study, a simple and quick sample preparation method is always preferable. Although plasma protein precipitation may introduce endogenous components that cause matrix effects during LC-MS/MS analysis, an appropriate retention time and gradient elution could overcome matrix effects to some extent. Therefore, acetonitrile was employed for sample preparation in this study.
Method Validation
Selectivity
The chromatograms of blank plasma from six separate batches, blank plasma spiked with gilteritinib at LLOQ (1 ng/mL) and IS, and the real plasma sample of rat were shown in Figure 2. The retention times of gilteritinib and IS were 1.14 and 1.27 min, respectively. No apparent interferences from endogenous substance were observed at the retention times of the analyte and IS.
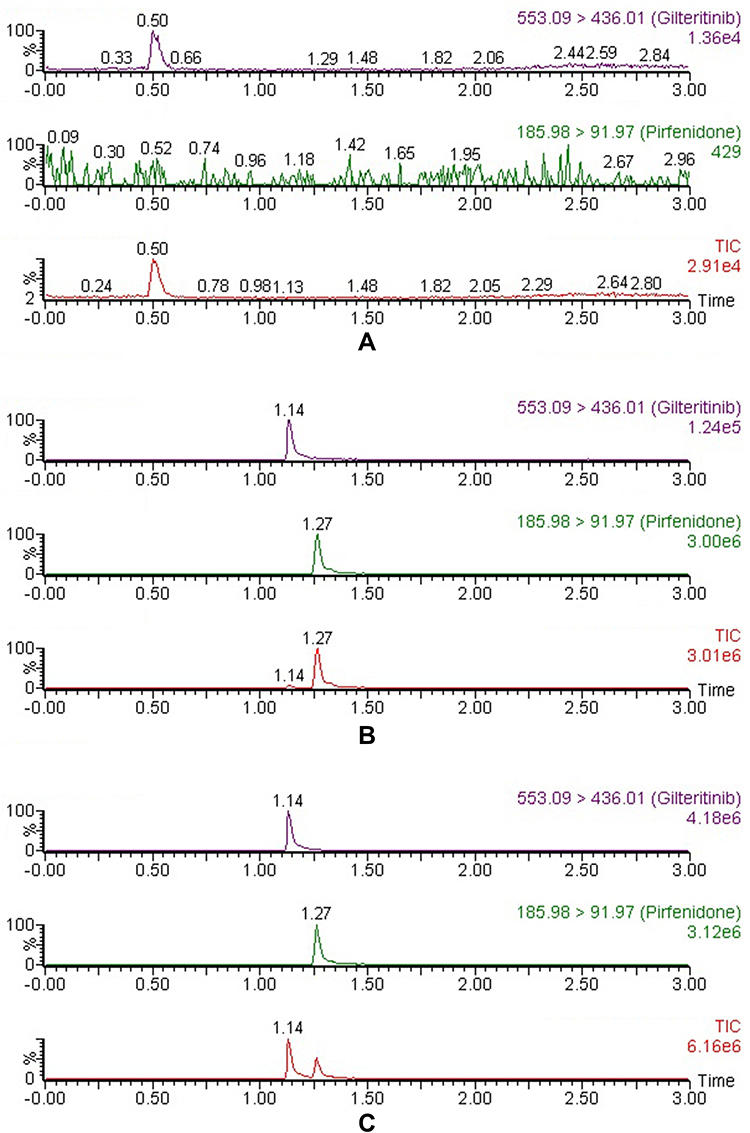 |
Figure 2 Representative chromatograms of gilteritinib and IS in rat plasma: (A) blank plasma; (B) blank plasma spiked with standard solution at LLOQ (1 ng/mL) and IS; (C) sample obtained from a rat at 1.0 h after oral administration of 10 mg/kg gilteritinib. |
Calibration Curve and LLOQ
In the range of 1–500 ng/mL gilteritinib, the calibration curve exhibited an excellent linearity. The sensitivity of the method was measured by LLOQ and established as 1 ng/mL, with sufficient accuracy and precision (Table 1).
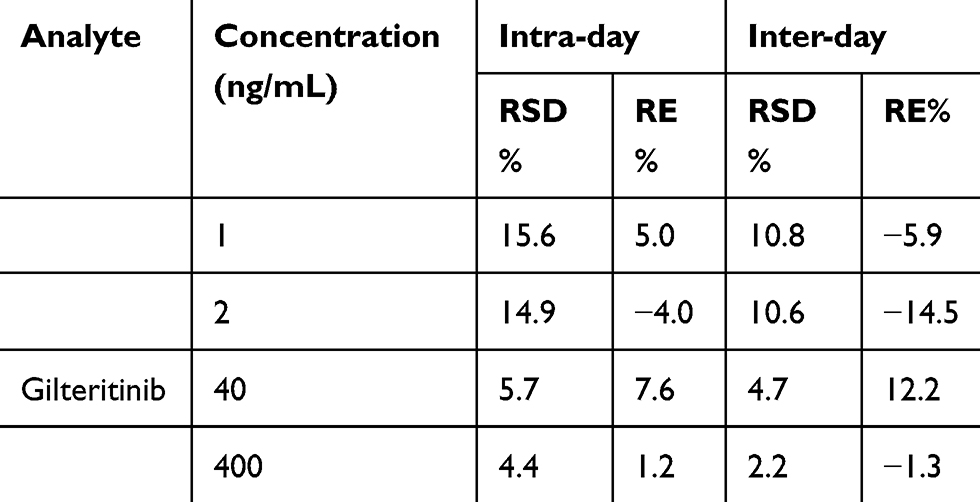 |
Table 1 The Precision and Accuracy of Gilteritinib in Rat Plasma (n=6) |
Accuracy and Precision
The accuracy and precision of the developed LC-MS/MS assay were investigated on three separate days. As presented in Table 1, the intra- and inter-day accuracy for gilteritinib was within −4.0 to 4.3% and −14.5 to 11.1%, respectively. The intra- and inter-day precision for gilteritinib was ≤9.2% and ≤10.6%, respectively.
Recovery and Matrix Effect
At three QC levels, the recovery of gilteritinib in rat plasma ranged from 84.6% to 91.9% and the mean recovery of the IS was 90.6 ± 6.6%. Moreover, the matrix effect was evaluated and indicated within the acceptable limit (96.1–102.4%) for gilteritinib, while the matrix effect of the IS was 99.8 ± 5.9%. All the results were shown in Table 2. Therefore, the matrix effect on the ionization of the analyte appeared to be insignificant and did not affect the accuracy of the optimized LC-MS/MS method.
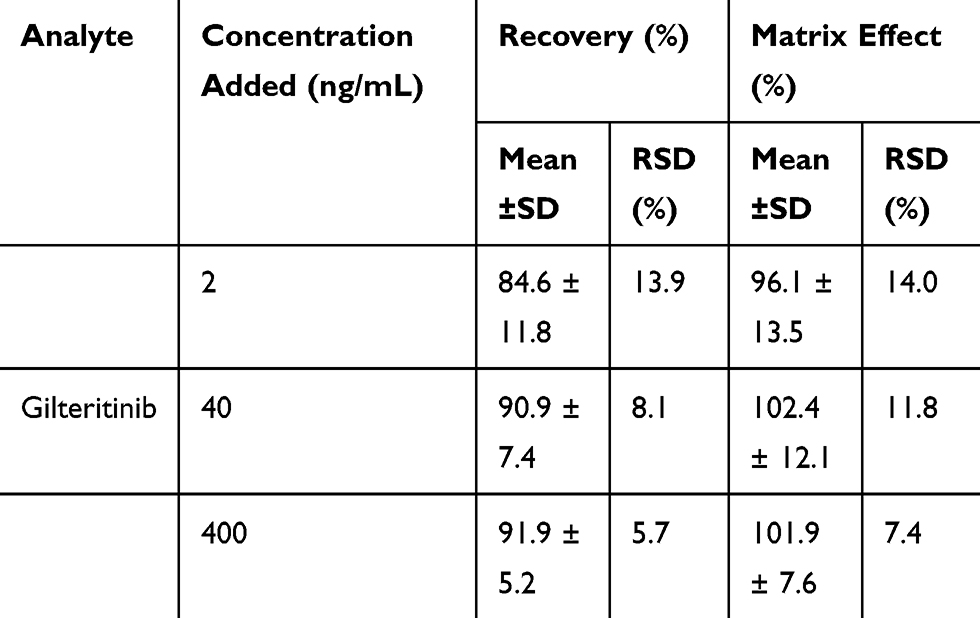 |
Table 2 Recovery and Matrix Effect of Gilteritinib in Rat Plasma (n=6) |
Stability
Stability experiments were performed to determine whether gilteritinib was stable under typical sample storage and analysis conditions in rat plasma. It was found to be stable when plasma gilteritinib samples were in the auto-sampler (10°C) for at least four hours and at room temperature for at least 24 h, three complete freeze (−20°C)/thaw (RT) cycles and at −20°C for at least 31 days.
Animal Study
The established method was successfully employed for measuring the plasma concentrations of gilteritinib in a drug–drug interaction study between gilteritinib and CYP3A4 inhibitors (fluconazole and itraconazole) in rats. After a single oral administration of 10 mg/kg gilteritinib, the average plasma concentration-time curves of gilteritinib in different treatment groups of rats were demonstrated in Figure 3, and the main pharmacokinetic parameters by noncompartment model analysis were summarized in Table 3.
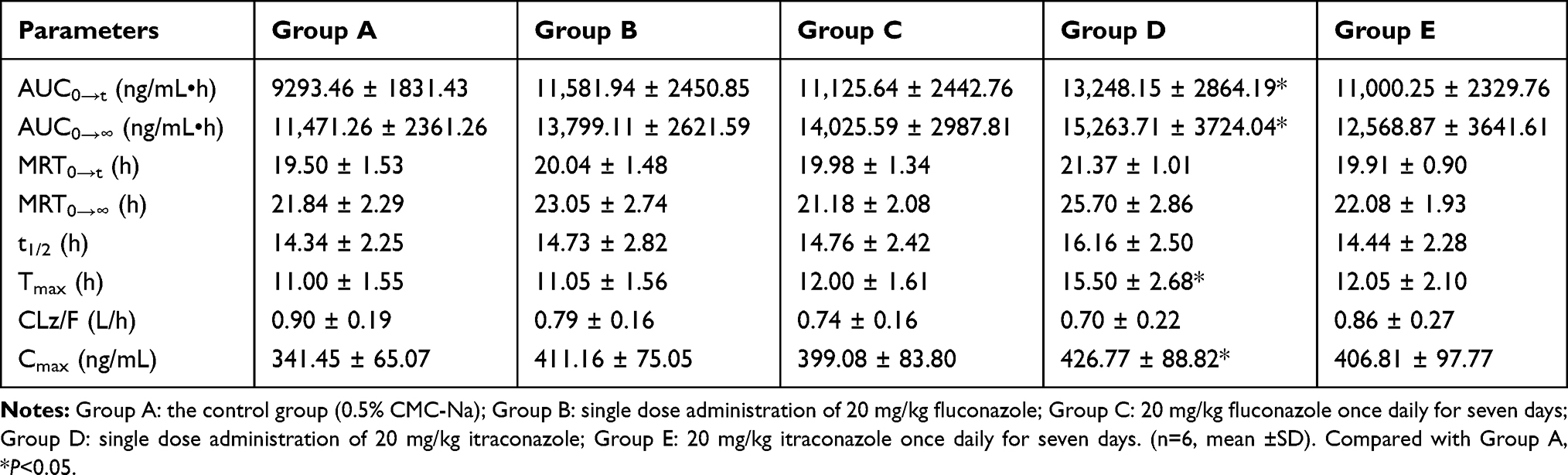 |
Table 3 The Main Pharmacokinetic Parameters of Gilteritinib in Different Treatment Groups of Rats |
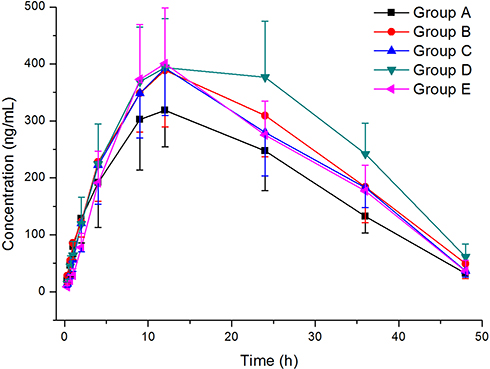 |
Figure 3 Mean plasma concentration-time curves of gilteritinib in different treatment groups of rats. Group A: the control group (0.5% CMC-Na); Group B: single dose administration of 20 mg/kg fluconazole; Group C: 20 mg/kg fluconazole once daily for seven days; Group D: single dose administration of 20 mg/kg itraconazole; Group E: 20 mg/kg itraconazole once daily for seven days. (n=6). |
Fluconazole and itraconazole are widely used in the treatment of opportunistic fungal infections, and, as shown by interaction studies in humans, itraconazole is a strong CYP3A4 inhibitor,10–12 whereas fluconazole inhibits CYP3A4, CYP2C9, and CYP2C19.13,14 In this study, an dose of 20 mg/kg of fluconazole and itraconazole was administered only once in Group B and Group D, and once daily for seven days in Group C and Group E, respectively. On the seventh day, 10 mg/kg gilteritinib was co-administered half an hour after fluconazole and itraconazole administration. When compared with Group A, although co-administered with fluconazole increased the AUC and Cmax of gilteritinib (P>0.05), it had no relevant effect on the exposure of gilteritinib in Group B and Group C. These results were consistent with the literature,15 where there was no meaningful pharmacokinetic drug–drug interaction between vismodegib and fluconazole. However, itraconazole produced an increase in total gilteritinib systemic exposure in Group D, where the changes in exposure parameters of gilteritinib were considered relevant, but not in Group E. Therefore, concomitant use of gilteritinib with strong CYP3A4 inhibitors should be avoided as the combined use increases gilteritinib exposure. If concomitant use of a strong CYP3A4 inhibitor is unavoidable, the results of this study support a dose reduction of gilteritinib. As the study of the effects of CYP3A4 inhibitors on gilteritinib metabolism were performed in rats with a few, further research should be done.
Conclusions
In conclusion, a simple, sensitive and reliable UPLC-MS/MS method was fully developed and validated to determine the plasma concentration of gilteritinib in rat plasma for the first time. The applicability of the optimized UPLC-MS/MS method was demonstrated in a drug–drug interaction study between gilteritinib and CYP3A4 inhibitors (fluconazole and itraconazole), where itraconazole increased the exposure of gilteritinib in rats. Therefore, adaptation of the dose of gilteritinib might be required when administered concomitantly with itraconazole.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Cancer Genome Atlas Research N, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi:10.1056/NEJMoa1301689
2. Levis M. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematol Am Soc Hemat. 2013;2013:220–226. doi:10.1182/asheducation-2013.1.220
3. Usuki K, Sakura T, Kobayashi Y, et al. Clinical profile of gilteritinib in Japanese patients with relapsed/refractory acute myeloid leukemia: an open-label Phase 1 study. Cancer Sci. 2018;109(10):3235–3244. doi:10.1111/cas.13749
4. Ueno Y, Mori M, Kamiyama Y, et al. Evaluation of gilteritinib in combination with chemotherapy in preclinical models of FLT3-ITD(+) acute myeloid leukemia. Oncotarget. 2019;10(26):2530–2545. doi:10.18632/oncotarget.26811
5. Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017;18(8):1061–1075. doi:10.1016/S1470-2045(17)30416-3
6. Dhillon S. Gilteritinib: first global approval. Drugs. 2019;79(3):331–339. doi:10.1007/s40265-019-1062-3
7. Astellas Pharma. XOSPATA (gilteritinib): US prescribing infor- mation. 2018. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211349s000lbl.pdf.
8. Clark JD, Gebhart GF, Gonder JC, Keeling ME, Kohn DF Special report: the 1996 guide for the care and use of laboratory animals. ILAR J. 1997;38(1):41–48. doi:10.1093/ilar.38.1.41
9. Center for Drug Evaluation and Research of the U.S. Department of Health and Human Services Food and Drug Administration, Guidance for industry; Bioanalytical method validation. 2018. Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm064964.htm.
10. Zhou X, Pant S, Nemunaitis J, et al. Effects of rifampin, itraconazole and esomeprazole on the pharmacokinetics of alisertib, an investigational aurora a kinase inhibitor in patients with advanced malignancies. Invest New Drugs. 2018;36(2):248–258. doi:10.1007/s10637-017-0499-z
11. Barbour AM, Punwani N, Epstein N, et al. Effect of Itraconazole or rifampin on itacitinib pharmacokinetics when administered orally in healthy subjects. J Clin Pharmacol. 2019;59(12):1641–1647. doi:10.1002/jcph.1484
12. Dai D, Yang H, Nabhan S, et al. Effect of itraconazole, food, and ethnic origin on the pharmacokinetics of ivosidenib in healthy subjects. Eur J Clin Pharmacol. 2019;75(8):1099–1108. doi:10.1007/s00228-019-02673-6
13. Kaukonen KM, Olkkola KT, Neuvonen PJ. Fluconazole but not itraconazole decreases the metabolism of losartan to E-3174. Eur J Clin Pharmacol. 1998;53(6):445–449. doi:10.1007/s002280050405
14. Yang J, Atkins WM, Isoherranen N, Paine MF, Thummel KE. Evidence of CYP3A allosterism in vivo: analysis of interaction between fluconazole and midazolam. Clin Pharmacol Ther. 2012;91(3):442–449. doi:10.1038/clpt.2011.178
15. Malhi V, Colburn D, Williams SJ, et al. A clinical drug-drug interaction study to evaluate the effect of a proton-pump inhibitor, a combined P-glycoprotein/cytochrome 450 enzyme (CYP)3A4 inhibitor, and a CYP2C9 inhibitor on the pharmacokinetics of vismodegib. Cancer Chemother Pharmacol. 2016;78(1):41–49. doi:10.1007/s00280-016-3020-z
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.