")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Alginate oligosaccharide alleviates myocardial reperfusion injury by inhibiting nitrative and oxidative stress and endoplasmic reticulum stress-mediated apoptosis
Authors Guo JJ, Xu FQ, Li YH, Li J, Liu X, Wang XF, Hu LG, An Y
Received 18 May 2017
Accepted for publication 18 July 2017
Published 18 August 2017 Volume 2017:11 Pages 2387—2397
DOI https://doi.org/10.2147/DDDT.S142118
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Jun-Jie Guo,1,* Feng-Qiang Xu,2,* Yong-Hong Li,1,* Jian Li,1 Xin Liu,3 Xiao-Fan Wang,1 Long-Gang Hu,4 Yi An1
1Department of Cardiology, The Affiliated Hospital of Qingdao University, 2Department of Cardiology, Qingdao Municipal Hospital, 3Department of Geriatric Medicine, The Affiliated Hospital of Qingdao University, 4Department of Cardiology, The Affiliated Cardiovascular Hospital of Qingdao University, Qingdao, People’s Republic of China
*These authors contributed equally to this work
Abstract: Alginate oligosaccharide (AOS) has recently demonstrated the ability to protect against acute doxorubicin cardiotoxicity and neurodegenerative disorders by inhibiting oxidative stress and endoplasmic reticulum (ER) stress-mediated apoptosis, which are both involved in myocardial ischemia/reperfusion (I/R) injury. In the present study, we investigated whether pretreatment with AOS protects against myocardial I/R injury in mice and explored potential cardioprotective mechanisms. AOS pretreatment significantly decreased the infarct size, reduced the cardiac troponin-I concentration, and ameliorated the cardiac dysfunction. Accompanied with the reduced cardiac injury, AOS pretreatment clearly decreased I/R-induced myocardial apoptosis. With regard to mechanism, AOS pretreatment markedly attenuated nitrative/oxidative stress, as evidenced by decreases in 3-nitrotyrosine content and superoxide generation, and downregulated inducible nitric oxide synthase, NADPH oxidase2, and 4-hydroxynonenal. Moreover, AOS pretreatment decreased myocardial apoptosis by inhibiting the ER stress-mediated apoptosis pathway, which is reflected by the downregulation of C/EBP homologous protein, glucose-regulated protein 78, caspase-12, and Bcl-2-associated X protein, and by the upregulation of the anti-apoptotic protein B-cell lymphoma-2. Collectively, these findings demonstrate that AOS renders the heart resistant to I/R injury, at least in part, by inhibiting nitrative/oxidative stress and ER stress-mediated apoptosis.
Keywords: alginate oligosaccharide, myocardial ischemia/reperfusion injury, nitrative/oxidative stress, endoplasmic reticulum, cardioprotection
Introduction
During the past few decades, with the advancement of emergency percutaneous coronary intervention (PCI), more and more patients suffering from acute myocardial infarction have received timely PCI treatment.1 However, with the opening of the culprit artery, the myocardial reperfusion might trigger a more severe problem known as myocardial ischemia/reperfusion (I/R) injury.2,3 Many deleterious effects, such as excessive reactive oxygen species (ROS) production, lipid peroxidation, DNA/RNA damage, calcium overload, autophagy disorders, and endoplasmic reticulum (ER) stress-mediated apoptosis, have been linked to I/R injury. These effects increase myocardial apoptosis, which then leads to a high incidence of chronic heart failure.4–8 Therefore, cardiologists are increasingly interested in strategies to alleviate myocardial I/R injury. Among various protective strategies, antioxidant therapies, including the use of synthetic chemical compounds and transgenic mice that overexpress antioxidant enzymes, have proved to be cardioprotective in the face of I/R injury.5,9–11 Recent studies have demonstrated that the overproduction of ROS can activate ER stress-induced, C/EBP homologous protein (CHOP)-mediated myocardial apoptosis, while inhibition of ER stress, including using chemical ER chaperones or gene-deficient mice, can be cardioprotective in the setting of myocardial I/R injury.12,13
Alginate oligosaccharides (AOS) are produced by depolymerizing alginate, an acidic polysaccharide that is extracted from marine brown algae, using methods such as acid hydrolysis, oxidation, and enzymatic degradation. AOS is composed of 1,4-linked β-D-mannuronic acid (M) and α-L-guluronic acid (G) residues.14 It is attractive for biomedical applications because it is non-immunogenic and nontoxic, and has several pharmacological benefits including anti-inflammatory, anti-apoptotic, anti-proliferative, and antioxidant effects.15–19 A recent study demonstrated that the protective effect of AOS on neurodegenerative diseases such as Alzheimer’s disease is due to its inhibition of oxidative stress and ER stress-mediated apoptosis.17 Our previous study also observed the protective effect of AOS, acquired by an enzymatic degradation method, on acute doxorubicin cardiotoxicity.20 Nevertheless, the ability of AOS to protect against myocardial I/R injury is not yet clear. In this study, we sought to examine the effects of AOS, produced by an enzymatic degradation method, on myocardial I/R injury in vivo and to explore the potential mechanism of protection, specifically the effect of AOS on nitrative/oxidative stress and ER stress-mediated apoptosis.
Materials and methods
Preparation of AOS
AOS, a gift from Qingdao BZ Oligo Biotech Co. Ltd. (Qingdao, People’s Republic of China), was produced from alginate by enzymatic degradation as previously described,21 and the detailed information was exhibited in our previous study.20 The relative molecular weight of AOS is ~1.2 kDa. The chemical structure of AOS acquired through enzymatic degradation is shown in Figure 1.
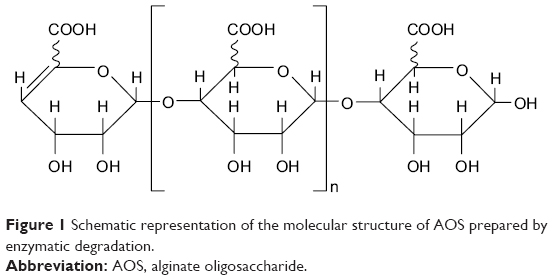 | Figure 1 Schematic representation of the molecular structure of AOS prepared by enzymatic degradation. |
Animals and drug administration
Eight-week-old male C57BL/6 mice (22–25 g) were purchased from the SLAC Laboratory Animal Company (Shanghai, People’s Republic of China). The mice were kept in cages under specific pathogen-free conditions at 24°C±2°C, with humidity of 40%±5%, a 12/12-hour dark/light cycle, and free access to food and water. Mice were allowed to adapt for 1 week before experiments began. All experiments were carried out in strict accordance with the approved guidelines of the China Council on Animal Management. The protocols were approved by the Animal Ethics Committee of Qingdao University. The mice were randomly assigned to the following groups: the sham group (SHAM), the vehicle group (I/R), the AOS (200 mg/kg/d, 7 days) + I/R group (A1+I/R), and the AOS (400 mg/kg/d, 7 days)+I/R group (A2+I/R). The doses used in the experiment were based on our previous study.20 Mice were pretreated with AOS dissolved in normal saline at a concentration of 2.0% w/v or with an equal volume of normal saline by gavage for 1 week. The final administration was given 1 hour before ischemia.
Surgical procedure
After administration of the last dose of AOS or normal saline, the myocardial I/R procedure was performed similarly to a previous study.22 Briefly, after initiating anesthesia with 2% isoflurane inhalation without intubation, a small oblique skin incision was made over the left chest, followed by separation of the pectoralis major from the pectoralis minor to clearly expose the fourth intercostal space, through which a small hole was made with a mosquito clamp. The heart was quickly exposed through the hole with the clamp slightly open. The left coronary artery (LCA) was ligated with a 6/0 silk suture slipknot, ~3–4 mm from its proximal origin, and the heart was gently backed to the thoracic cavity with one end of the silk suture outside of the chest. Successful myocardial ischemia appeared visually as a pale region on the anterior wall of the heart and was confirmed by electrocardiography, which presented the classic ischemic hallmark of ST segment elevation. Sham-operated animals were subjected to the same surgical procedure except that the ligation remained untied. After 40 minutes of ischemia, the ligation was opened in vitro by pulling the end of the silk suture, and the heart was reperfused for 3 or 24 hours. A total of 140 mice were used in these experiments. Eight mice died during the myocardial I/R injury procedure. Thirty-six mice were used to assess the myocardial infarct size (IS). Twenty-four mice were used to evaluate the cardiac function and measure the cardiac troponin-I concentration. Twenty-four mice were used for immunohistochemistry and myocardial apoptosis analysis. Twenty-four mice were used for measurement of ROS generation in the ischemic myocardium, and another 24 mice were used for evaluation of protein expression.
Assessment of myocardial IS
Evans blue-triphenyltetrazolium chloride (TTC) double staining was performed to determine the myocardial IS 24 hours after reperfusion as done in a previous study.4 In brief, the mice were re-anesthetized with 2% isoflurane inhalation, and the chest was opened. The same suture used for ligation around the LCA was retied. Evans blue dye (0.2 mL; 2%) was injected into the left ventricular via the apex and circulated throughout the body except in the ischemia area. Afterward, the heart was quickly extracted, frozen in liquid nitrogen for 10 seconds, and sliced into 1 mm sections from the apex to the ligation. The slices were then incubated separately with 1% TTC solution at 37°C for 15 minutes and digitally scanned. IS, area at risk (AAR), and left ventricle size (LV) were measured on the two sides of each section using ImageJ software.
Measurement of cardiac function
Twenty-four hours after reperfusion, the cardiac function was evaluated via transthoracic echocardiography (Vevo707B; Visual Sonics Inc., Toronto, ON, Canada) and hemodynamic measurement (Millar 1.4F, SPR 835; Millar Instruments, Houston, TX, USA) as previously described.23 Briefly, the mice were anesthetized using 2% isoflurane in oxygen gas, and the heart rates were maintained at 450–500 beats/minute. The B- and M-mode images were acquired via a 30-MHz high-frequency scanhead, and both left ventricular ejection fraction (EF) and left ventricular fractional shortening (FS) were measured. All measurements were carried out by three experienced technicians who were blinded to the animal groups. Thereafter, the right common carotid artery was isolated, and a 1.4F Millar micromanometer catheter was inserted and introduced into the left ventricle. The transducer was connected to a Power Laboratory System (AD Instruments, Castle Hill, NSW, Australia), and left ventricular end-systolic pressure (LVESP), left ventricular end-diastolic pressure (LVEDP), maximal slope of systolic pressure increment (dp/dtmax), and maximal slope of diastolic pressure decrement; (dp/dtmin) were obtained.
Measurement of cardiac troponin-I concentration
After the measurement of cardiac function, the blood was collected from the carotid artery with EDTA-containing syringes. Plasma was separated from the blood by centrifugation at 4,000 rpm for 30 minutes at 4°C, immediately frozen, and then stored at −80°C. The plasma was thawed only once for concentration of cardiac troponin-I (cTnI) measurement. A mouse cTnI ELISA kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, People’s Republic of China) was used to measure the cTnI concentration. The assay was performed according to the protocol provided by the manufacturer.
Detection of myocardial apoptosis
Three hours after reperfusion, the hearts were harvested and fixed in 4% formalin. Myocardial apoptosis was detected using a terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) technique. Paraffin-embedded myocardial slices were stained using an in situ Cell Apoptosis Detection Kit (POD; Roche Diagnostics Corp., Indianapolis, IN, USA) according to the manufacturer’s protocol. Five microscopic fields (400×) from each slice were acquired from the border of the ischemic area of the heart. The percentage of cell apoptosis was expressed as a ratio of TUNEL-positive nuclei (brown nuclei) over the total number of nuclei.
Determination of myocardial 3-nitrotyrosine
Myocardial 3-nitrotyrosine (3-NT) was detected by immunohistochemistry 3 hours after reperfusion. In brief, paraffin-embedded sections were stained with a primary antibody against 3-NT (Abcam). The immunostaining was performed using the Vectastain ABC kit (Vector Laboratories) according to the manufacturer’s protocol. After that, the sections were visualized and images were photographed using a light microscope.
Measurement of ROS generation
Myocardial ROS steady-state levels were detected by in situ dihydroethidium (DHE) staining 3 hours after reperfusion. In brief, unfixed frozen cross-sections were incubated with DHE staining (Sigma-Aldrich, St Louis, MO, USA) at 37°C for 30 minutes in a humidified chamber protected against light, followed by a wash in phosphate-buffered saline for 5 minutes. The images were confirmed and obtained with a Leica laser scanning confocal microscope. Myocardial superoxide production was measured 3 hours after reperfusion by a lucigenin-enhanced chemiluminescence method as previously described.24 Superoxide production was calculated as relative light units per mg of heart weight per second and presented as the fold change against the sham group.
Western blot analyses
Three hours after reperfusion, heart tissue from the AAR of I/R injury was harvested for Western blot analysis. The total protein content was extracted with complete protease inhibitor cocktail and RIPA lysis buffer (Beyotime Biotechnology, Nanjing, People’s Republic of China) from homogenized heart tissue. Subsequently, equal quantities of protein (50 μg) were added to 10% or 12% polyacrylamide gels based on the molecular weight of target proteins, and then transferred to a polyvinylidene fluoride membrane. Protein expression was detected by immunoblotting with antibodies against glucose-regulated protein 78 (GRP78; Abcam), NADPH oxidase2 (NOX2; Abcam), caspase-12 (Abcam), 4-HEN (Abcam), inducible nitric oxide synthase (iNOS; Cell Signaling Technology [CST]), CHOP (CST), Bcl-2-associated X protein (Bax; CST), and B-cell lymphoma-2 (Bcl-2; CST). GAPDH served as the loading control. After three washes, the blots were incubated with the corresponding secondary antibodies. The protein bands were visualized by chemiluminescence, and quantitation was carried out with a Bio-Rad Gel Imaging System equipped with Basic Quantity One imaging software.
Statistical analysis
Data were expressed as the mean ± standard deviation and compared by a one-way analysis of variance and the Student–Newman–Keuls post hoc test using GraphPad Prism 6 software (GraphPad Software, San Diego, CA, USA). For all data, a value of P<0.05 was considered statistically significant.
Results
AOS pretreatment limited the myocardial IS
The myocardial IS was assessed by Evans blue-TTC double staining at 24 hours after I/R injury. Representative images are shown in Figure 2A. No difference was observed in AAR/LV among the four groups (Figure 2B). Both doses of AOS pretreatment dramatically lessened the percentage of IS/AAR and IS/LV when compared with the I/R group (Figure 2C and D), but no difference was observed between the two AOS doses.
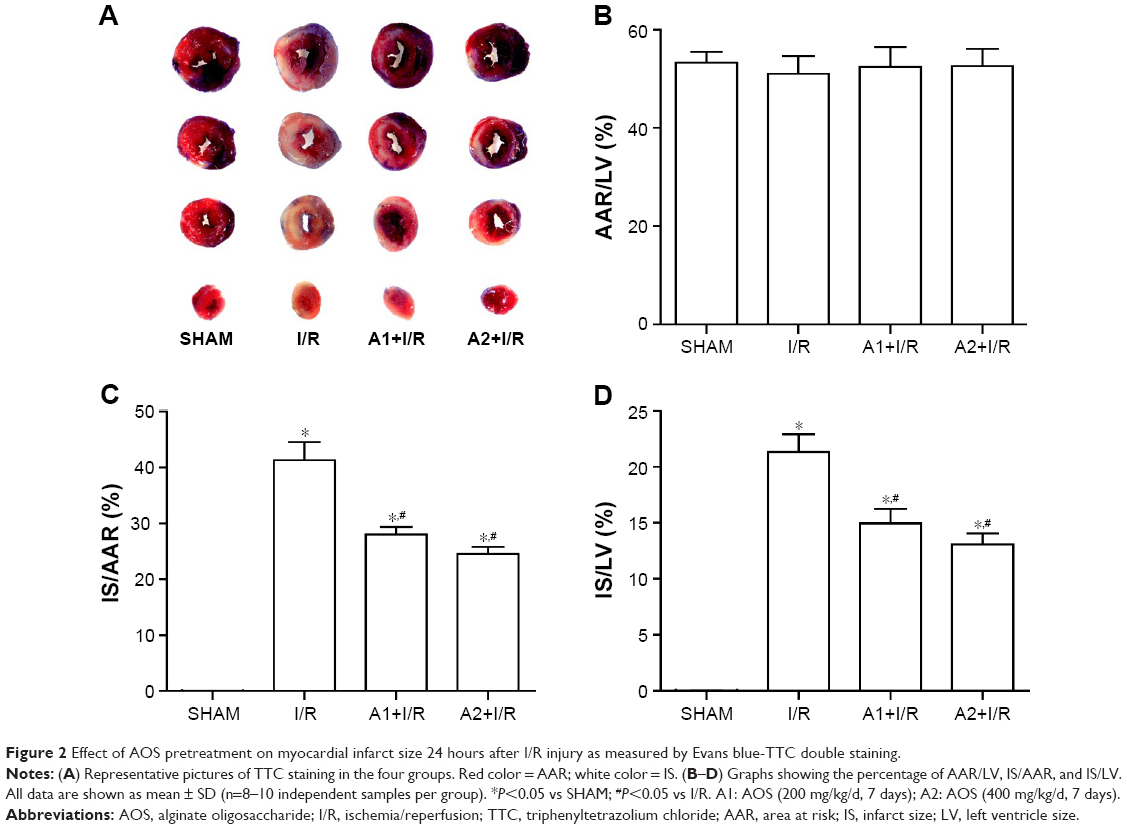 | Figure 2 Effect of AOS pretreatment on myocardial infarct size 24 hours after I/R injury as measured by Evans blue-TTC double staining. |
AOS pretreatment preserved cardiac function after I/R injury
Left ventricular function was evaluated 24 hours after I/R injury. I/R injury caused significant cardiac dysfunction when compared to the sham group, but both doses of AOS pretreatment significantly preserved the cardiac function that was impaired by the I/R injury (P<0.05), which is evidenced by the EF (I/R vs A1+I/R vs A2+I/R; 40.0±4.2 vs 50.7±5.1 vs 53.5%±6.4%), FS (I/R vs A1+I/R vs A2+I/R; 20.0±2.9 vs 26.5±3.4 vs 27.7%±3.6%), LVESP (I/R vs A1+I/R vs A2+I/R; 72.4±11.4 vs 86.7±11.2 vs 87.6±12.5 mmHg), LVEDP (I/R vs A1+I/R vs A2+I/R; 10.1±2.0 vs 5.5±1.6 vs 4.5±1.1 mmHg), dp/dtmax (I/R vs A1+I/R vs A2+I/R; 5,722±852 vs 8,600±953 vs 8,790±826 mmHg/s) and dp/dtmin (I/R vs A1+I/R vs A2+I/R; −4,716±837 vs −7,337±667 vs −8,028±886 mmHg/s) (Figure 3B–G). Although these indicators of cardiac function appeared to be better in the A2+I/R group compared to the A1+I/R group, this improvement was not statistically significant. Representative echocardiograms are shown in Figure 3A.
 | Figure 3 Effect of AOS pretreatment on cardiac function 24 hours after I/R injury. |
AOS pretreatment alleviated myocardial tissue injury after I/R injury
Myocardial tissue injury was analyzed by measuring the plasma cTnI, a specific biomarker of cardiac injury, which increases at 24 hours after I/R injury. Pretreatment with AOS for 1 week dramatically reduced the damage level when compared to the I/R group (Figure 4). Although the concentration of plasma cTnI was lower in the A2+I/R group than in the A1+I/R group, this difference did not reach statistical significance (P>0.05).
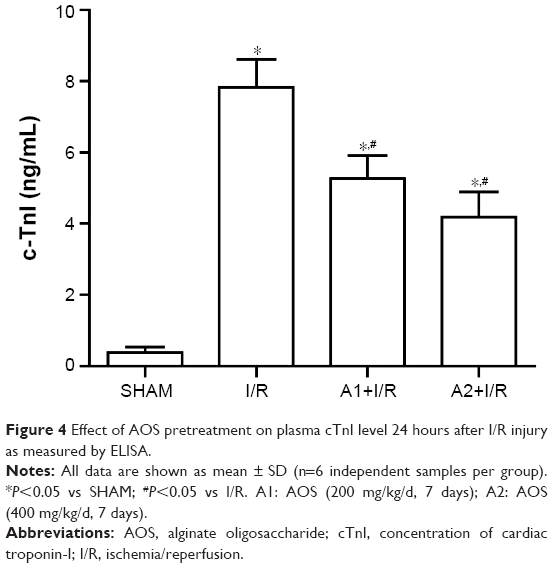 | Figure 4 Effect of AOS pretreatment on plasma cTnI level 24 hours after I/R injury as measured by ELISA. |
AOS pretreatment reduced myocardial apoptosis after I/R injury
To investigate the effect of AOS on myocardial apoptosis following I/R injury, we conducted a TUNEL assay to detect DNA fragmentation in the cardiac tissue 3 hours after reperfusion. As shown in Figure 5A and B, the I/R group showed more TUNEL-positive nuclei (expressed as a percentage of total nuclei) than the sham group. Conversely, pretreatment with AOS resulted in an obvious reduction of TUNEL-positive cells compared with the I/R group, but there was no significant difference between the two AOS doses.
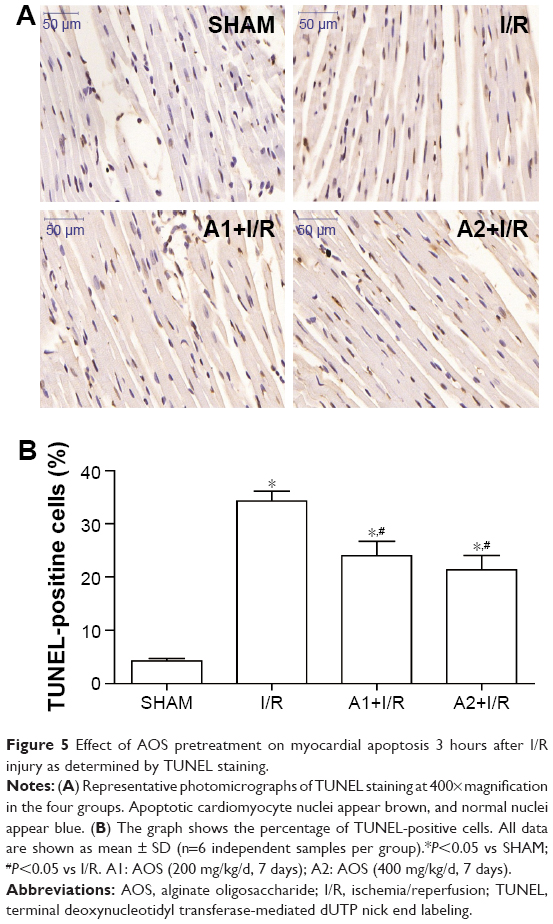 | Figure 5 Effect of AOS pretreatment on myocardial apoptosis 3 hours after I/R injury as determined by TUNEL staining. |
AOS pretreatment attenuated myocardial nitrative/oxidative stress after I/R injury
To examine the effect of AOS on myocardial nitrative/oxidative stress following I/R injury, we evaluated the generation of ROS and expression of specific proteins associated with nitrative/oxidative stress at 3 hours after reperfusion. Myocardial I/R injury markedly upregulated the expression of myocardial 3-NT and iNOS (Figure 6A and B), which were all downregulated by AOS pretreatment. Myocardial DHE fluorescence intensity, superoxide production (Figure 6C and D), and the expression of NOX2 (Figure 6E) and 4-hydroxynonenal (4-HNE; Figure 6F) were all upregulated in the face of I/R injury, but all were downregulated by AOS pretreatment. Similarly, although the expression levels of these proteins were lower in the A2+I/R group than in the A1+I/R group, this difference did not reach statistical significance.
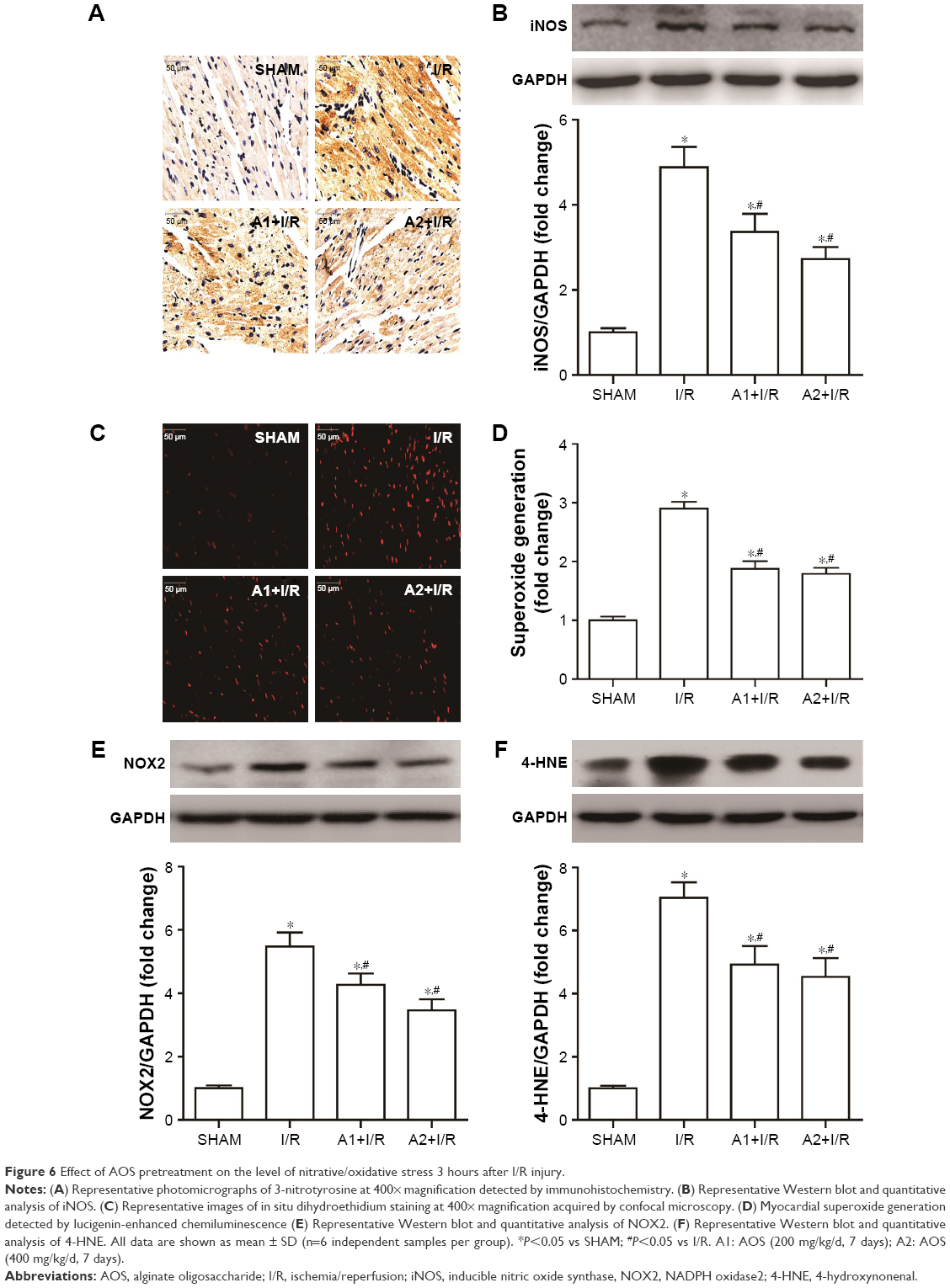 | Figure 6 Effect of AOS pretreatment on the level of nitrative/oxidative stress 3 hours after I/R injury. |
AOS pretreatment attenuated ER stress-mediated apoptosis after I/R injury
To examine the effect of AOS on the ER stress-mediated apoptosis pathway following myocardial I/R injury, we evaluated the associated protein expression 3 hours after reperfusion via Western blot analysis. As shown in Figure 7A and B, I/R injury upregulated the expression of Bax and downregulated the expression of B-cell lymphoma-2 (Bcl-2) compared with the sham group, while both doses of AOS pretreatment markedly reversed these changes. There was no difference between the two AOS dose groups. Representative Western blot images are shown in Figure 7C. The specific markers of ER stress (ie, CHOP, GRP78, and caspase-12 protein) were significantly elevated in the I/R group compared to the sham group (Figure 8A–C). Both doses of AOS pretreatment significantly lowered this increase in the three protein indicators of ER stress. No differences were detected between the two different AOS doses. Representative Western blot images are shown in Figure 8D.
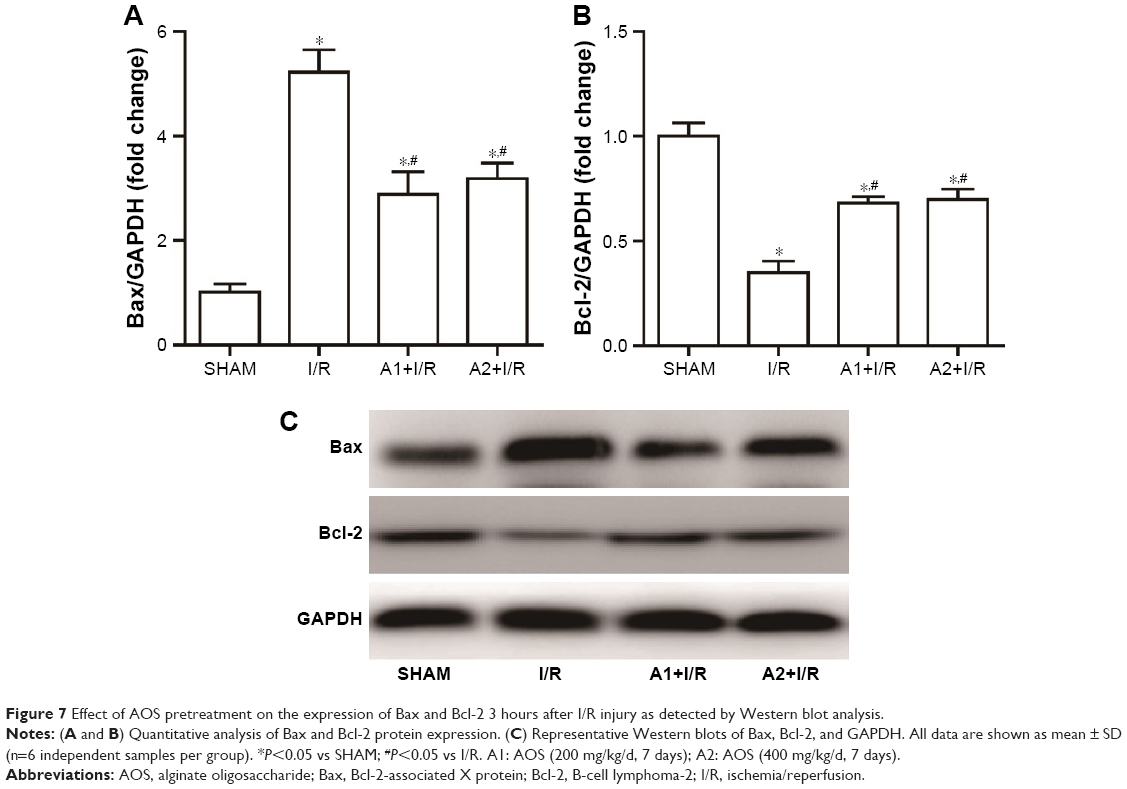 | Figure 7 Effect of AOS pretreatment on the expression of Bax and Bcl-2 3 hours after I/R injury as detected by Western blot analysis. |
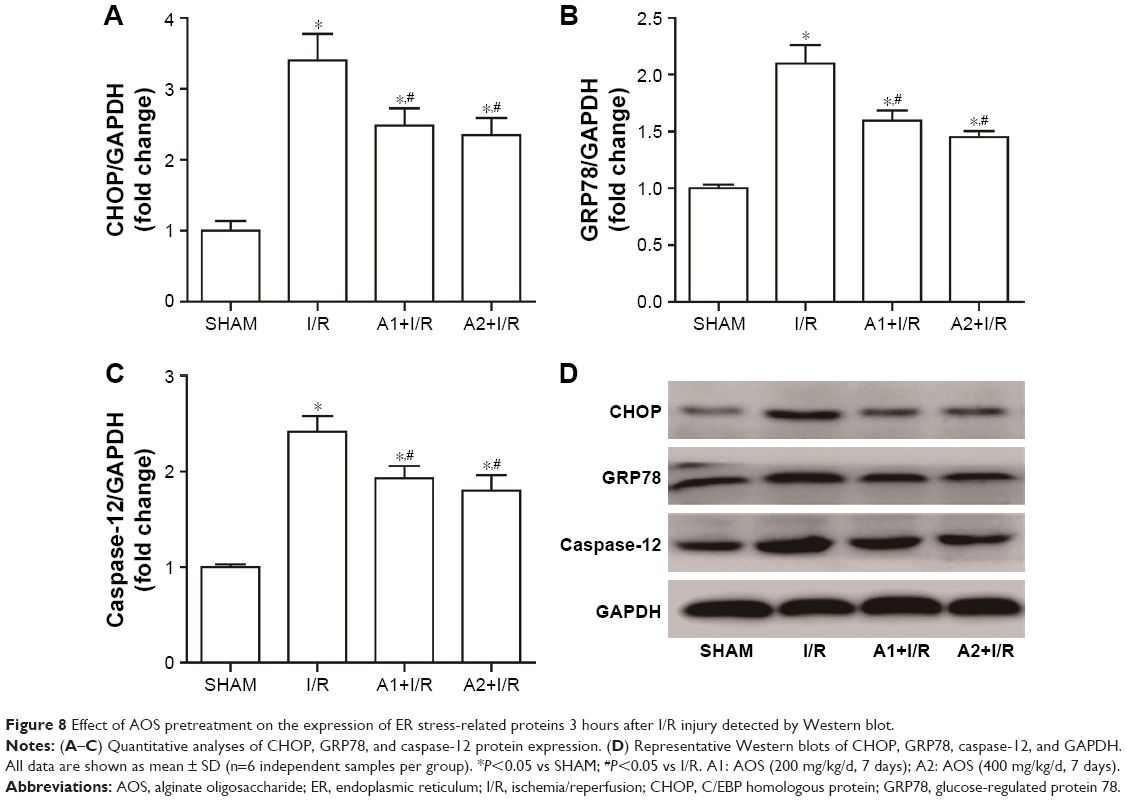 | Figure 8 Effect of AOS pretreatment on the expression of ER stress-related proteins 3 hours after I/R injury detected by Western blot. |
Discussion
Evidence suggests that nitrative/oxidative stress and ER stress-mediated apoptosis play critical roles in myocardial I/R injury, and they have also been verified to be pivotal targets for cardioprotection.4,10,13 Meanwhile, both ours and others’ studies have demonstrated that AOS, an antioxidant oligosaccharide, attenuates the level of oxidative stress and ER stress-mediated apoptosis in neurodegenerative diseases and acute doxorubicin cardiotoxicity.17,20 However, it is unknown whether AOS can protect against myocardial I/R injury and whether these inhibitory effects of AOS are involved in cardioprotection in the setting of I/R injury. In the current study, we found that AOS displayed cardioprotective effects in the face of I/R injury. Most importantly, we acquired the first direct evidence that inhibiting nitrative/oxidative stress and ER stress-mediated apoptosis was associated with the cardioprotection.
Increased IS, elevated myocardial enzyme concentration, and cardiac dysfunction are characteristics of myocardial I/R injury.24 In the present study, mice hearts insulted with I/R injury showed increased IS, plasma cTnI level, and cardiac dysfunction, which all agreed with previous studies.4,25 However, both low-dose AOS (200 mg/kg/d, 7 days) and high-dose AOS (400 mg/kg/d, 7 days) pretreatment for 1 week significantly decreased the IS and plasma cTnI level. These data hint that AOS may have cardioprotective effects on I/R injury. Meanwhile, we also found that AOS pretreatment could preserve cardiac function after I/R injury. To further explore the underlying mechanisms potentially contributing to the protective effect of AOS pretreatment, we assessed the levels of nitrative/oxidative stress and ER stress-mediated apoptosis.
Nitrative/oxidative stress plays an established deleterious role in the myocardial apoptosis observed in I/R injury.25,26 Nitrative stress, a crucial response that leads to I/R injury, is initiated and mediated by the highly toxic peroxynitrite. Previous studies have demonstrated that inhibition of peroxynitrite formation, either by detoxifying peroxynitrite or by inhibiting iNOS generation, reduces I/R injury.27–30 The results attained in our study affirmed that the levels of 3-NT and iNOS, markers of nitrosative stress, were significantly increased in the I/R group, which is consistent with previous studies. However, pretreatment with low-dose AOS (200 mg/kg/d, 7 days) and high-dose AOS (400 mg/kg/d, 7 days) for 1 week reduced this stress by downregulating the cardiac expression of 3-NT and iNOS. Overproduction of ROS has long been considered to be the main cause of oxidative stress and to serve as the vital mediator of myocardial I/R injury.31,32 Consistent with these studies, our study found that myocardial I/R injury induced the overproduction of ROS, reflected by increased DHE fluorescence intensity and superoxide production, which was attenuated by AOS pretreatment. Previous studies have demonstrated that NOX2, which has the primary function of ROS generation, is activated by various stimuli, including myocardial I/R injury.33,34 Meanwhile, the expression of the endogenous toxic aldehyde 4-HNE, a specific electrophilic reactive aldehyde that inhibits protein function by modifying key enzymes, is increased in the face of myocardial I/R injury.35,36 Both of these proteins are usually recognized as markers of oxidative stress, so we analyzed them directly by Western blot analysis. Our study presents results that I/R injury does cause excessive oxidative stress by upregulating the expression of NOX2 and 4-HNE, which were both inhibited by AOS pretreatment. These findings indicate that attenuation of nitrative/oxidative stress induced by myocardial I/R injury might contribute to the cardioprotective effect of AOS.
There is increasing evidence that excessive, prolonged ER stress plays a vital role in inducing cell death, especially apoptosis.13,37 The present study found that the significant increase in myocardial apoptosis of the I/R group was reduced by AOS pretreatment. In response to ER stress, GRP78, an ER chaperone protein, is activated. The level of GRP78 is considered to be a specific indicator of ER stress, and previous reports have demonstrated that myocardial I/R injury induces a significant upregulation of GRP78 level.38,39 This was consistent with our findings. However, pretreatment with low-dose AOS (200 mg/kg/d, 7 days) and high-dose AOS (400 mg/kg/d, 7 days) for 1 week downregulated GRP78 expression, indicating a decrease in ER stress. Meanwhile, CHOP and caspase-12 are two specific mediators of the ER stress-mediated apoptosis pathways.40 CHOP-deficient mice exhibited reduced cardiomyocyte apoptosis in response to ER stress after I/R injury.13 CHOP-mediated apoptosis is involved in the downregulation of the anti-apoptotic protein Bcl-2, the upregulation of the pro-apoptotic protein Bax, and the translocation of Bax protein from the cytosol to the mitochondria.41,42 Similarly, in response to ER stress, caspase-12, an ER membrane resident and pro-apoptotic molecule, activates caspase cascades that predominantly induce cell death.43 In accordance with previous studies,13,38 our results found that the expression levels of CHOP and caspase-12 significantly increased in the setting of I/R injury. In contrast, both low-dose AOS (200 mg/kg/d, 7 days) and high-dose AOS (400 mg/kg/d, 7 days) pretreatment markedly reversed these detrimental effects. Although we observed more beneficial effects on cardioprotection in the high-dose AOS pretreatment group than that in the low-dose AOS pretreatment group, these benefits did not reach statistical significance.
The current study is partly descriptive, and the limitations of this study are obvious. 1) Although the attenuation of nitrosative/oxidative stress can decrease ER stress-mediated apoptosis, we did not identify the exact mechanism through which AOS pretreatment inhibits ER stress-mediated apoptosis. 2) In this study, only two different doses of AOS were used to detect the cardioprotective effects, and no difference was observed between these two doses. It is possible that higher or lower doses of AOS could have significantly stronger or weaker cardioprotective effects.
In summary, the present study provides strong in vivo evidence, for the first time, that AOS effectively mitigates acute myocardial I/R injury in mice at least in part by reducing nitrosative/oxidative stress and ER stress-mediated apoptosis. Future studies will focus on elucidating the exact mechanisms governing these inhibitory effects and the pharmacokinetics of AOS. Our data indicate that AOS may serve clinically as a novel therapeutic strategy against myocardial I/R injury.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant number 81071246) and the Shandong Provincial Natural Science Foundation, China (ZR201702200205). We thank Qingdao BZ Oligo Biotech Co., Ltd., for preparation of the AOS.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Windecker S, Bax JJ, Myat A, Stone GW, Marber MS. Future treatment strategies in ST-segment elevation myocardial infarction. Lancet. 2013;382(9892):644–657. | ||
Skyschally A, Walter B, Heusch G. Coronary microembolization during early reperfusion: infarct extension, but protection by ischaemic postconditioning. Eur Heart J. 2013;34(42):3314–3321. | ||
Hausenloy DJ, Yellon DM. Targeting myocardial reperfusion injury – the search continues. N Engl J Med. 2015;373(11):1073–1075. | ||
Pei H, Yu Q, Xue Q, et al. Notch1 cardioprotection in myocardial ischemia/reperfusion involves reduction of oxidative/nitrative stress. Basic Res Cardiol. 2013;108(5):373. | ||
Koka S, Das A, Salloum FN, Kukreja RC. Phosphodiesterase-5 inhibitor tadalafil attenuates oxidative stress and protects against myocardial ischemia/reperfusion injury in type 2 diabetic mice. Free Radic Biol Med. 2013;60:80–88. | ||
Talukder MA, Zweier JL, Periasamy M. Targeting calcium transport in ischaemic heart disease. Cardiovasc Res. 2009;84(3):345–352. | ||
Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Diwan A. Autophagy is impaired in cardiac ischemia-reperfusion injury. Autophagy. 2012;8(9):1394–1396. | ||
Martindale JJ, Fernandez R, Thuerauf D, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98(9):1186–1193. | ||
Yoshida T, Watanabe M, Engelman DT, et al. Transgenic mice overexpressing glutathione peroxidase are resistant to myocardial ischemia reperfusion injury. J Mol Cell Cardiol. 1996;28(8):1759–1767. | ||
Tanaka M, Mokhtari GK, Terry RD, et al. Overexpression of human copper/zinc superoxide dismutase (SOD1) suppresses ischemia-reperfusion injury and subsequent development of graft coronary artery disease in murine cardiac grafts. Circulation. 2004;110(11 Suppl 1):II200–II206. | ||
Sener G, Sakarcan A, Yegen BC. Role of garlic in the prevention of ischemia-reperfusion injury. Mol Nutr Food Res. 2007;51(11):1345–1352. | ||
Jian L, Lu Y, Lu S, Lu C. Chemical chaperone 4-phenylbutyric acid protects H9c2 cardiomyocytes from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis. Mol Med Rep. 2016;13(5):4386–4392. | ||
Miyazaki Y, Kaikita K, Endo M, et al. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):1124–1132. | ||
Yang JH, Bang MA, Jang CH, Jo GH, Jung SK, Ki SH. Alginate oligosaccharide enhances LDL uptake via regulation of LDLR and PCSK9 expression. J Nutr Biochem. 2015;26(11):1393–1400. | ||
Wan LS, Heng PW, Chan LW. Drug encapsulation in alginate microspheres by emulsification. J Microencapsul. 1992;9(3):309–316. | ||
Zhou R, Shi X, Gao Y, Cai N, Jiang Z, Xu X. Anti-inflammatory activity of guluronate oligosaccharides obtained by oxidative degradation from alginate in lipopolysaccharide-activated murine macrophage RAW 264.7 cells. J Agric Food Chem. 2015;63(1):160–168. | ||
Tusi SK, Khalaj L, Ashabi G, Kiaei M, Khodagholi F. Alginate oligosaccharide protects against endoplasmic reticulum- and mitochondrial-mediated apoptotic cell death and oxidative stress. Biomaterials. 2011;32(23):5438–5458. | ||
Tajima S, Inoue H, Kawada A, Ishibashi A, Takahara H, Hiura N. Alginate oligosaccharides modulate cell morphology, cell proliferation and collagen expression in human skin fibroblasts in vitro. Arch Dermatol Res. 1999;291(7–8):432–436. | ||
Wang P, Jiang X, Jiang Y, et al. In vitro antioxidative activities of three marine oligosaccharides. Nat Prod Res. 2007;21(7):646–654. | ||
Guo JJ, Ma LL, Shi HT, et al. Alginate oligosaccharide prevents acute doxorubicin cardiotoxicity by suppressing oxidative stress and endoplasmic reticulum-mediated apoptosis. Mar Drugs. 2016;14(12):pii: E231. | ||
Li L, Jiang X, Guan H, Wang P. Preparation, purification and characterization of alginate oligosaccharides degraded by alginate lyase from Pseudomonas sp. HZJ 216. Carbohydr Res. 2011;346(6):794–800. | ||
Gao E, Lei YH, Shang X, et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107(12):1445–1453. | ||
Zhao G, Wang S, Wang Z, et al. CXCR6 deficiency ameliorated myocardial ischemia/reperfusion injury by inhibiting infiltration of monocytes and IFN-γ-dependent autophagy. Int J Cardiol. 2013;168(2):853–862. | ||
Wang Y, Wang X, Jasmin JF, et al. Essential role of caveolin-3 in adiponectin signalsome formation and adiponectin cardioprotection. Arterioscler Thromb Vasc Biol. 2012;32(4):934–942. | ||
Tao L, Gao E, Jiao X, et al. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation. 2007;115(11):1408–1416. | ||
Ji L, Fu F, Zhang L, et al. Insulin attenuates myocardial ischemia/reperfusion injury via reducing oxidative/nitrative stress. Am J Physiol Endocrinol Metab. 2010;298(4):E871–E880. | ||
Huang C, Cui Y, Ji L, et al. Catalpol decreases peroxynitrite formation and consequently exerts cardioprotective effects against ischemia/reperfusion insult. Pharm Biol. 2013;51(4):463–473. | ||
Liang F, Gao E, Tao L, et al. Critical timing of L-arginine treatment in post-ischemic myocardial apoptosis-role of NOS isoforms. Cardiovasc Res. 2004;62(3):568–577. | ||
Zweier JL, Fertmann J, Wei G. Nitric oxide and peroxynitrite in postischemic myocardium. Antioxid Redox Signal. 2001;3(1):11–22. | ||
Ferdinandy P, Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2003;138(4):532–543. | ||
Takano H, Zou Y, Hasegawa H, Akazawa H, Nagai T, Komuro I. Oxidative stress-induced signal transduction pathways in cardiac myocytes: involvement of ROS in heart diseases. Antioxid Redox Signal. 2003;5(6):789–794. | ||
Bagheri F, Khori V, Alizadeh AM, Khalighfard S, Khodayari S, Khodayari H. Reactive oxygen species-mediated cardiac-reperfusion injury: mechanisms and therapies. Life Sci. 2016;165:43–55. | ||
Braunersreuther V, Montecucco F, Asrih M, et al. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. 2013;64:99–107. | ||
Kasi V, Bodiga S, Kommuguri UN, Sankuru S, Bodiga VL. Zinc pyrithione salvages reperfusion injury by inhibiting NADPH oxidase activation in cardiomyocytes. Biochem Biophys Res Commun. 2011;410(2):270–275. | ||
Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J. 2011;32(8):1025–1038. | ||
Venkatachalam K, Prabhu SD, Reddy VS, Boylston WH, Valente AJ, Chandrasekar B. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J Biol Chem. 2009;284(12):7853–7865. | ||
Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115(10):2656–2664. | ||
Xia JG, Xu FF, Qu Y, Song DG, Shen H, Liu XH. Atorvastatin post-conditioning attenuates myocardial ischemia reperfusion injury via inhibiting endoplasmic reticulum stress-related apoptosis. Shock. 2014;42(4):365–371. | ||
Zhang GG, Teng X, Liu Y, et al. Inhibition of endoplasm reticulum stress by ghrelin protects against ischemia/reperfusion injury in rat heart. Peptides. 2009;30(6):1109–1116. | ||
Masciarelli S, Fra AM, Pengo N, et al. CHOP-independent apoptosis and pathway-selective induction of the UPR in developing plasma cells. Mol Immunol. 2010;47(6):1356–1365. | ||
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21(4):1249–1259. | ||
Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381–389. | ||
Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.